

Abstract
Modification of lipid A with the 4-amino-4-deoxy-L-arabi-nose (L-Ara4N) moiety is required for resistance to polymyxin and cationic antimicrobial peptides in Escherichia coli and Salmonella typhimurium. An operon of seven genes (designated pmrHFIJKLM in S. typhimurium), which is regulated by the PmrA transcription factor and is also present in E. coli, is necessary for the maintenance of polymyxin resistance. We previously elucidated the roles of pmrHFIJK in the biosynthesis and attachment of L-Ara4N to lipid A and renamed these genes arn-BCADT, respectively. We now propose functions for the last two genes of the operon, pmrL and pmrM. Chromosomal inactivation of each of these genes in an E. coli pmrAc parent switched its phenotype from polymyxin-resistant to polymyxin-sensitive. Lipid A was no longer modified with L-Ara4N, even though the levels of the lipid-linked donor of the L-Ara4N moiety, undecaprenyl phosphate-α-L-Ara4N, were not reduced in the mutants. However, the undecaprenyl phosphate-α-L-Ara4N present in the mutants was less concentrated on the periplasmic surface of the inner membrane, as judged by 4–5-fold reduced labeling with the inner membrane-impermeable amine reagent N-hydroxysulfosuccin-imidobiotin. In an arnT mutant of the same pmrAc parent, which lacks the enzyme that transfers the L-Ara4N unit to lipid A but retains the same high levels of undecaprenyl phosphate-α-L-Ara4N as the parent, N-hydroxysulfosuccinimidobiotin labeling was not reduced. These results implicate pmrL and pmrM, but not arnT, in transporting undecaprenyl phosphate-α-L-Ara4N across the inner membrane. PmrM and PmrL, now renamed ArnE and ArnF because of their involvement in L-Ara4N modification of lipid A, may be subunits of an undecaprenyl phosphate-α-L-Ara4N flippase.
The lipid A moiety of lipopolysaccharide (LPS)4 in the outer membranes of Gram-negative bacteria typically consists of a β,1′–6-linked disaccharide of glucosamine, modified with phosphate groups at the 1- and 4′-positions, and with R-3-hydroxyacyl chains at the 2-, 3-, 2′-, and 3′-positions (Fig. 1) (1, 2). Lipid A plays an important role in the pathogenesis of Gram-negative bacterial infections. It potently activates the innate immune system (3, 4), triggering the synthesis of cationic antimicrobial peptides, cytokines, clotting factors, and other immunostimulatory molecules (5–8).
FIGURE 1. Regulated covalent modifications of lipid A in E. coli and S. typhimurium.

As indicated by the dashed bonds, modifications of the phosphate groups and/or acyl chains of lipid A are observed under certain growth conditions or in some mutants (2). The phosphates can be modified with L-Ara4N and/or pEtN groups, both of which are regulated by pmrA (26). The latter gene, encoding the PmrA transcription factor (73), is constitutively active in polymyxin-resistant mutants, and the relevant modifying enzymes ArnT (31) and EptA (22, 23) are induced under these conditions. In some instances, lipid A species are made in which the locations of the L-Ara4N and pEtN groups are reversed (20, 88), or in which both phosphates are modified with the same group, as is the case with pEtN when L-Ara4N synthesis is blocked (21). The outer membrane enzyme PagP (89) can add an additional palmitate unit at position 2, whereas PagL (90) can remove the hydroxymyristoyl chain at position 3. These pag genes are regulated by the PhoP/PhoQ system (91), which can be activated by low Mg2+ in Salmonella. PagL (90), LpxO (92), and LpxR (93) are present in Salmonella but not E. coli K-12. However, they can function in E. coli K-12 when expressed from plasmids.
Polymyxin B, a cationic antibiotic secreted by certain Gram-positive bacteria, binds to lipid A with high affinity and damages the cell envelope (9, 10). Mutants of Gram-negative bacteria resistant to polymyxin B usually acquire constitutive mutations in the transcription factor PmrA (11–13). The lipid A molecules isolated from these pmrAc strains are extensively modified with phosphoethanolamine (pEtN) and 4-amino-4-deoxy-L-arabinose (L-Ara4N) moieties (Fig. 1), which reduce the net negative charge of lipid A and limit its interaction with polymyxin (14–21). Transcription of the genes encoding the enzymes responsible for lipid A modification with pEtN and L-Ara4N are under the direct control of PmrA (2, 22, 23), which can also be activated in wild-type Escherichia coli by external stimuli, such as low pH in phagolysosomes or exposure to metavanadate (24–26). Attachment of the L-Ara4N moiety (27) to lipid A is essential for resistance to polymyxin B and other cationic antimicrobial peptides (11, 28).
A Salmonella typhimurium operon of seven genes, designated pmrHFIJKLM, is co-transcribed under the control of the transcription factor PmrA (16). The Pmr gene products are required for the maintenance of polymyxin resistance and the attachment of the L-Ara4N moiety to lipid A (16). The same operon is present in E. coli. Based upon the predicted sequences of the Pmr protein products, we proposed a biosynthetic pathway for generating the L-Ara4N moiety and transferring it to lipid A (25). We have previously assigned enzymatic functions to five (PmrHFIJK) of the seven gene products of the operon and have renamed them ArnB (29), ArnC (28), ArnA (28, 30), ArnD (28), and ArnT (13, 31), respectively, to reflect their roles in L-Ara4N production (Fig. 2, A and B). The pathway starts with the conversion of UDP-glucose to UDP-glucuronic acid (Fig. 2A) (30). The C-terminal domain of ArnA then catalyzes the oxidative decarboxylation of UDP-glucuronic acid to generate a novel UDP-4-ketopentose (28, 30, 32, 33). This substance is transaminated by ArnB (29, 34), using glutamic acid as the amine donor, to generate UDP-β-L-Ara4N, which is N-formylated by the N-terminal domain of ArnA (Fig. 2A) (28). ArnC next transfers the N-formylated L-Ara4N moiety to undecaprenyl phosphate, the product of which is rapidly deformylated by ArnD (Fig. 2A) (28). After being transported to the outer surface of the inner membrane by an unknown mechanism, the L-Ara4N group of undecaprenyl phosphate-L-Ara4N is transferred to lipid A by ArnT (13, 31) (Fig. 2A). Further evidence that L-Ara4N transfer to lipid A occurs on the outer surface of the inner membrane is provided by the observation that L-Ara4N modification of lipid A is dependent upon the essential core-lipid A flippase, MsbA (35, 36).
FIGURE 2. Biosynthesis of undecaprenyl phosphate-α-L-Ara4N and transfer of the L-Ara4N moiety to lipid A.

A, in polymyxin-resistant E. coli and S. typhimurium, biosynthesis of the L-Ara4N moiety begins with oxidation of UDP-glucose to UDP-glucuronic acid (2). Next, the C-terminal domain of ArnA catalyzes the NAD+-dependent oxidative decarboxylation of UDP-glucuronic acid to yield an unusual UDP-4-ketopentose (30, 32), which is converted by the transaminase ArnB to UDP-β-L-Ara4N (29). Subsequently, the N-terminal domain of ArnA uses N-10-formyltetrahydrofolate to N-formylate UDP-β-L-Ara4N (28, 30, 32). ArnC (a distant orthologue of dolichyl phosphate-mannose synthase) selectively transfers the L-Ara4-formyL-N residue to undecaprenyl phosphate (28). Next, ArnD catalyzes deformylation of this substance to undecaprenyl phosphate-α-L-Ara4N (13), preventing the reversal of the ArnC-catalyzed reaction (28). After transport to the outer surface of the inner membrane, presumably by ArnE (PmrL) and ArnF (PmrM) as shown in the present study, the membrane enzyme ArnT (31) transfers the L-Ara4N moiety to lipid A. B, order of genes and direction of transcription (left to right) of the pmr operon (16). The preferred arn terminology (2, 45), which is consistent with the generalized bacterial polysaccharide gene nomenclature (94), is shown in red. We suggest that the older pmr nomenclature be retained for the regulatory genes pmrA, pmrB, and pmrD, because their products have many other functions besides regulating the expression of enzymes needed for L-Ara4N biosynthesis.
We now report on the functions of the two remaining gene products of the operon, PmrL and PmrM (Fig. 2B) (16), which display distant sequence similarity to the multidrug efflux pump exporter EmrE (37–39). In-frame chromosomal deletion mutants of pmrL and pmrM, generated in a polymyxin-resistant pmrAc parental strain, regain polymyxin sensitivity. Despite the presence of the same high levels of undecaprenyl phosphate-α-L-Ara4N in these mutants as in their parent, lipid A modification with L-Ara4N is almost completely abolished. Using the inner membrane-impermeable amine reagent N-hydroxysulfosuccin-imidobiotin (sulfo-NHS-biotin) (36), in conjunction with mass spectrometry, we show that undecaprenyl phosphate-α-L-Ara4N is 4–5-fold less accessible to chemical modification in pmrL/pmrAc and pmrM/pmrAc mutants than in the pmrAc parent, presumably reflecting the reduced ability of undecaprenyl phosphate-α-L-Ara4N to gain access to the periplasmic side of the inner membrane. Reduced chemical modification of undecaprenyl phosphate-α-L-Ara4N is not observed in an arnT mutant derived from the same pmrAc parent, in which undecaprenyl phosphate-α-L-Ara4N levels also remain as high as they are in the parent. We suggest that PmrL and PmrM, renamed ArnE and ArnF, respectively (see Fig. 2B for the old and new nomenclature), function as a heterodimer to flip undecaprenyl phosphate-α-L-Ara4N from the cytosolic side to the periplasmic side of the inner membrane, where the active site of ArnT is located (Fig. 2A).
EXPERIMENTAL PROCEDURES
Materials
Glass-backed 0.25-mm Silica Gel 60 TLC plates were purchased from Merck. Chloroform was obtained from EM Science, whereas pyridine, methanol, and formic acid were from Mallinckrodt. Trypticase soy broth, yeast extract, and tryptone were purchased from Difco. Pfu PCR reagents were purchased from Stratagene, and Taq PCR reagents were from Roche Applied Science. The spin miniprep and gel extraction kits were from Qiagen. Sulfo-NHS-biotin, a membrane-impermeable amine reagent (40), and N-hydroxysuccinimidobiotin (NHS-biotin), a membrane-permeable analogue, were the products of Pierce.
Bacterial Strains
The bacterial strains used in this study are described in Table 1. Typically, bacteria were grown at 37 °C in LB medium, which consists of 10 g of NaCl, 10 g of tryptone, and 5 g of yeast extract per liter (41). When required for selection of plasmids or mutations, cells were grown in the presence of 100 μg/ml ampicillin, 10 μg/ml polymyxin, or 20 μg/ml kanamycin.
TABLE 1.
Bacterial strains and plasmids
| Description | Source or Ref. | |
|---|---|---|
| Strains | ||
| E. coli | ||
| W3110 | Wild type, F−, λ− | E. coli Genetic Stock Center, Yale University |
| DY330 | W3110, ΔlacU169 gal490 λcl857 Δ(cro-bioA) | 47 |
| MST100 | DY330, pmrAc | 28 |
| SDB200 | MST100, ΔarnD::kan, Kanr | S. Breazeale, manuscript in preparation |
| AY100 | MST100, ΔpmrL::kan, Kanr | This work |
| AY101 | MST100, ΔpmrM::kan, Kanr | This work |
| Y102 | MST100, ΔpmrLM::kan, Kanr | This work |
| AY103 | MST100, ΔarnT::kan, Kanr | This work |
| NovaBlue (DE3) | Δ(srL-recA)30::Tn10(DE3), Tetr | Novagen |
| XL-1 Blue-MR | ΔmcrABC, recA1, lac | Stratagene |
|
| ||
| Plasmids | ||
| pET24b | Vector containing a T7lac promoter, Kanr | Novagen |
| pET28b | Vector containing a T7lac promoter, Kanr | Novagen |
| pWSK29 | Low copy number vector, Ampr | 48 |
| pET-PmrL | pET24b expressing E. coli pmrL | This work |
| pET-PmrM | pET24b expressing E. coli pmrM | This work |
| pET-PmrLM | pET24b expressing E. coli pmrL & pmrM | This work |
| pET-ArnT | pET21 expressing E. coli arnT | 31 |
| pWSK29-PmrL | pWSK29 expressing E. coli pmrL | This work |
| pWSK29-PmrM | pWSK29 expressing E. coli pmrM | This work |
| pWSK29-PmrLM | pWSK29 expressing E. coli pmrL and pmrM | This work |
| pWSK29-ArnT | pWSK29 expressing E. coli arnT | This work |
Molecular Biology Applications
Protocols for handling of DNA samples were those of Sambrook and Russell (42). Transformation-competent cells of E. coli were prepared by the method of Inoue et al. (43). When required, E. coli cells were prepared for electroporation by the method of Sambrook and Russell (42). Plasmids were prepared using the Qiagen spin prep kit. DNA fragments were isolated from agarose gels using the QIAquick gel extraction kit. Genomic DNA was isolated using the protocol for bacterial cultures in the Easy-DNA™ kit (Invitrogen). The rapid DNA ligation kit (Roche Applied Science), restriction endonucleases (New England Biolabs), and shrimp alkaline phosphatase (U. S. Biochemical Corp.) were used according to the manufacturer’s instructions. Double-stranded DNA sequencing was performed with an ABI Prism 377 instrument at the Duke University DNA Analysis Facility. Primers were purchased from MWG Biotec.
In-frame Replacements of pmrL, pmrM, pmrLM, or arnT with Kanamycin Resistance Cassettes in a pmrAc Derivative of E. coli DY330
The old and new gene nomenclature is summarized in Fig. 2B and Table 2. The pmr designations (11, 12, 16) were originally used to describe genes required for the maintenance of polymyxin resistance in S. typhimurium (Fig. 2B). A similar group of genes, present in E. coli, was originally identified using the “Yfb,” “Orf,” or “b” nomenclatures (Table 2) (44, 45). The Arn system (28–30) should now be adopted, given that the functions of the Pmr operon gene products in aminoarabinose (L-Ara4N) biosynthesis are now well defined (Fig. 2A), including PmrL and PmrM, as described below.
TABLE 2.
Alternative names of gene products involved in L-Ara4N biosynthesis and transfer to lipid A
The construction of an in-frame replacement of pmrL with a kanamycin resistance cassette (kan) was based on the method of Derbise et al. (46). A pmrAc derivative of E. coli strain DY330 (47), designated MST100 (28), which contains a λ prophage harboring the recombination genes exo, bet, and gam under control of a temperature-sensitive λ cI-repressor, was utilized as the parental strain.
A linear piece of DNA consisting of a kan cassette flanked by 500 bp of E. coli DNA upstream and downstream of pmrL was generated by a three-step overlap PCR procedure. First, E. coli K-12 W3110 genomic DNA was used as the template to amplify the 500-bp upstream and downstream regions of pmrL. The upstream segment included on its 3′ end the first 20 bp of the kan gene. The downstream region included on its 5′ end the last 20 bp of the kan gene plus a ribosomal binding site. The forward and reverse primers used for amplifying the upstream (PmrL-uf and PmrL-ur) and downstream 500-bp regions (PmrL-df and PmrL-dr) are provided in supplemental Table 1.
In parallel, the kanamycin resistance gene (Tn903) from plasmid pWKS130 (48) was utilized as the template to amplify the kan cassette, flanked on its 5′ end by 20 bp of chromosomal DNA immediately upstream of pmrL, and flanked on its 3′ end by a ribosome-binding site plus 20 bp of chromosomal DNA located immediately downstream of pmrL. The primers for amplifying this DNA fragment (Kan-PmrL-f and Kan-PmrL-r) are likewise provided in supplemental Table 1.
In the second step, a mixture of 100 ng of each of the upstream and downstream PCR products, together with 100 ng of the kan cassette PCR fragment, served as the templates to amplify a large piece of DNA containing the desired segment, consisting of the 500 bp upstream of pmrL, the kan cassette, and the 500 bp downstream of pmrL. The primers (PmrL-uf and PmrL-dr) used for this purpose are provided in supplemental Table 1.
Because multiple bands were generated during this PCR procedure, a third step was introduced. The DNA fragment of the correct size was recovered from an agarose gel, and 50 ng of the gel-purified material was used as the template DNA for a second PCR with the same pair of the primers. The resulting PCR product was resolved on a 1.0% agarose gel and purified. The product was then electroporated into MST100 cells. Colonies were selected on LB agar containing kanamycin (20 μg/ml) after incubation overnight at 30 °C. Kanamycin-resistant colonies with in-frame pmrL replacements were initially confirmed by colony PCR, using external primers located 100 bp upstream and 100 downstream of pmrL. The genomic DNA from a colony yielding a fragment of the correct size was subsequently prepared and used as the template to amplify larger amounts of the same fragment, using the same pair of the external primers as in the colony PCR. A single PCR product was resolved on a 1% agarose gel, purified, and sequenced using the same primers to confirm the gene replacement. The resulting strain was designated as AY100 (pmrL::kan) (Table 1).
The same general methods were used to construct in-frame replacements of pmrM (AY101), of pmrLM (AY102), and of arnT (AY103) (Table 1). In each case, the same kanamycin resistance cassette was introduced into the pmrAc parental strain MST100. However, in the pmrM and the pmrLM deletion strains, no ribosomal binding site was introduced at the 3′ end of the kanamycin resistance cassette, because the pmrD regulatory gene (Fig. 2B), which is immediately downstream of pmrM, is transcribed in the opposite direction (44, 45). All constructs were verified by colony PCR and full DNA sequencing. The sequences of the primers used for generating all of these constructs are provided in supplemental Table 1.
Construction of Covering Plasmids
The pmrL gene of E. coli was initially cloned into pET24b (Novagen) behind the T7lac promoter. The coding region for pmrL was amplified by PCR from E. coli W3110 genomic DNA. The forward primer contained a clamp region, an NdeI site (supplemental Table 1, underlined), and the pmrL-coding region with its start codon (boldface). The reverse primer contained a clamp region, an XhoI site (supplemental Table 1, underlined), and the coding region with its stop codon (boldface). Sequences of the forward and reverse primers are shown in supplemental Table 1. The PCR product and the pET24b vector were both digested with NdeI and XhoI. The linearized vector was treated with shrimp alkaline phosphatase (U. S. Biochemical Corp.) for 0.5 h at 37 °C, followed by inactivation of the phosphatase at 65 °C for 15 min. The vector and the PCR product were then ligated together using the rapid DNA ligation kit (Roche Applied Science) and transformed into XL-1 Blue cells (Stratagene) for propagation of the desired plasmid, designated pET-PmrL. The insertion was further verified by enzyme digestion and DNA sequencing. The same approach was used to construct pETPmrM and pET-PmrLM (Table 1). The primers for construction of pET-PmrM and pET-PmrLM are also shown in supplemental Table 1.
The pmrL, pmrM, and contiguous pair of pmrLM genes were moved from the pET vector into the covering plasmid pWSK29 (48), a lac-inducible, low copy expression vector useful for complementation experiments. The XbaI/XhoI-digested fragments, consisting of the either pmrL, pmrM, or the contiguous pair of pmrLM genes downstream of a pET24b-derived ribosome-binding site, were ligated into the corresponding restriction sites of pWSK29. These plasmids, designated as pWSK29-PmrL, pWSK29-PmrM, or pWSK29-PmrLM (Table 1), were then electroporated into the appropriate pmrL or pmrM deletion strains to test for complementation.
Extraction and Separation of Phospholipids from the pmrAC Parent MST100 or Various Mutants
Typically, 1 ml of overnight culture from a freshly streaked single colony was inoculated into 100 ml of LB broth (for MST100) or into LB broth containing 20 μg/ml kanamycin (for the various mutant strains). The cells were grown until A600 reached ∼1.0. They were collected by centrifugation and washed with 25 ml of phosphate-buffered saline, pH 7.4. To extract the phospholipids, the cell pellet was resuspended in 19 ml of a single-phase Bligh/Dyer mixture (49), consisting of chloroform/methanol/water (1:2:0.8, v/v). After mixing and incubating for 60 min at room temperature, the insoluble debris pellet was removed by centrifugation, and the supernatant containing the phospholipids was transferred to a clean glass tube. The supernatant was converted to a two-phase Bligh/Dyer system consisting of chloroform/methanol/water (2:2:1.8 v/v) by adding appropriate amounts of chloroform and water. The phases were separated by low speed centrifugation (4000 × g), and the lower phase was collected. The sample was dried under a stream of N2 and redissolved in a small volume of chloroform/methanol (2:1, v/v). Typically, 20 μl of this sample was further diluted 10-fold with chloroform/methanol (1:1, v/v) for electrospray ionization-time of flight mass spectrometry (ESI/MS) analysis. The rest of the sample was dried under a stream of N2 and was subjected to mild alkaline hydrolysis.
Mild Alkaline Hydrolysis of Lipids
Mild alkaline hydrolysis to deacylate the glycerophospholipids was performed as described (50). Typically, an alkaline methanol solution was prepared by adding 0.4 ml of 5 M NaOH to 5 ml of methanol (final NaOH concentration of 0.37 M). After mixing, 5 ml of chloroform was added. The N2-dried phospholipids, prepared as described above, were redissolved in 1 ml of this mixture and incubated at room temperature for 1 h. The NaOH was neutralized with 3–4 drops of concentrated HCl, as judged by pH paper, and the system was converted to a two-phase Bligh-Dyer mixture (49). The lower phase was retrieved and dried under a stream of N2. The sample was then redissolved in 50 μl of chloroform/methanol (2:1, v/v), and 40 μl of this solution was spotted onto a Silica Gel 60 TLC plate, which was developed in the solvent chloroform/methanol/water/acetic acid (25:15:4:4, v/v). The remaining 10 μl was further diluted 10-fold with chloroform/methanol (1:1, v/v) for ESI/MS analysis.
Extraction of Lipid A from the pmrAC Parental Strain MST100 or Various Mutants
Cells were grown, harvested, and washed as described above. Each cell pellet was extracted with 120 ml of a single phase Bligh/Dyer mixture (49). After 60 min at room temperature, the mixture was subjected to centrifugation (4,000 × g for 20 min). The resulting cell debris pellet was extracted two more times with 120 ml of a single phase Bligh/Dyer mixture. The final insoluble residue, which contains lipopolysaccharide, was subjected to hydrolysis at 100 °C in 25 mM sodium acetate buffer, pH 4.5, in the presence of 1% SDS to cleave the Kdo-lipid A linkage (51). The released lipid A molecular species were extracted with a two-phase Bligh/Dyer system by adding appropriate amounts of chloroform and methanol. The lower phase was saved, and the upper phase was washed once with a pre-equilibrated acidic lower phase. The pooled lower phases were dried under a stream of N2. The isolated lipid A was redissolved in chloroform/methanol (4:1, v/v). A portion of the sample was subjected to ESI/MS analysis, and a portion of the remainder was spotted onto a Silica Gel 60 TLC plate, which was developed in the solvent chloroform, pyridine, 88% formic acid, water (50:50:16:5, v/v). The plate was dried, sprayed with 10% sulfuric acid in ethanol, and then charred on a hot plate to visualize the lipid A species.
Sulfo-NHS-Biotin and NHS-Biotin Modification of Undecaprenyl Phosphate L-Ara4N in the Parent MST100 or in Mutant Strains
Overnight cultures of the pmrAc parental strain MST100, and its derivatives AY100 (pmrL), AY101 (pmrM), or AY103 (arnT) were diluted 1:100 into 120 ml of LB broth, pH 7.0. Cells were grown with shaking at 30 °C until the density (A600) reached 1.0. Cells were harvested by centrifugation at 4,000 × g for 20 min and washed once with 50 ml of phosphate-buffered saline, pH 7.4 (52). For optimal labeling with the biotin reagents, the intact cells were subsequently washed twice with 50 ml of modified phosphate-buffered saline, pH 7.9, as recommended by the manufacturer. Cells were then resuspended in 12 ml of phosphate-buffered saline, pH 7.9, and divided into three portions (no labeling control, labeled with sulfo-NHS-biotin, or labeled with NHS-biotin). Next, 40 μl of 200 mM sulfo-NHS-biotin dissolved in water or, alternatively, 40 μl of 200 mM NHS-biotin dissolved in Me2SO were added to the cell suspensions, which were incubated at 4 °C for various times. The final concentrations of the labeling reagents were 2 mM. Reactions were quenched by the addition of 25 ml of 50 mM glycine, and the cells were collected by centrifugation. The cell pellets were extracted to recover the lipids (49), which were subjected to mild alkaline hydrolysis, as described above (50). The samples were then dissolved in the appropriate solvent for TLC or ESI/MS analysis.
ESI/MS of Lipid Preparations
All mass spectra were acquired on a QSTAR XL quadrupole time-of-flight tandem mass spectrometer (ABI/MDS-Sciex, Toronto, Canada), equipped with an electrospray ionization source. Spectra were acquired in the negative ion mode, and typically were the summation of 60 scans from 200 to 2000 atomic mass units. For MS analysis, the lipid A or mild alkali stable lipid preparations were dissolved in 200 μl of chloroform/methanol (4:1, v/v) or chloroform/methanol (2:1, v/v). Typically, 20 μl of this material was further diluted into 200 μl of chloroform/methanol (1:1, v/v), containing 1% piperidine, and immediately infused into the ion source at 5–10 μl/min. The negative ion ESI/MS was carried out at −4200 V. In the MS/MS mode, collision-induced dissociation tandem mass spectra were obtained using a collision energy of −80 V (laboratory frame of energy). Nitrogen was used as the collision gas. Data acquisition and analysis were performed using Analyst QS software.
RESULTS
Relationship of PmrL and PmrM to the Small Multidrug Resistance Efflux Pump Exporters
A BLASTp analysis of PmrL and PmrM shows that both are small integral membrane proteins with four putative trans-membrane helices. The PmrL and PmrM sequences (111 and 128 amino acid residues respectively) are distantly related to each other and to the drug/metabolite transporter tabolite transporter superfamily (37, 38, 53). The hydropathy plots of the two proteins resemble that of EmrE, an exporter of positively charged hydrophobic drugs in E. coli (54). X-ray structures of EmrE with or without the bound substrate tetraphenylphosphonium were reported by Chang and co-workers (55, 56) but were recently retracted (57). Orthologues of PmrL and PmrM exist in many bacteria, but their functions are poorly characterized. Orthologues with strong sequence similarity to PmrL and PmrM are present in all strains of E. coli, Salmonella, Yersinia pestis, Yersinia pseudotuberculosis, and Pseudomonas aeruginosa that modify their lipid A with the L-Ara4N moiety (2, 58). Given their sequences and genomic context (Fig. 2B), PmrL and PmrM might function as part of an inner membrane transporter or flippase system for undecaprenyl phosphate-α-L-Ara4N (Fig. 2A).
Deletion Mutants of pmrL and pmrM in MST100 Regain Poly-myxin Sensitivity
To investigate the roles of the contiguous pmrL and pmrM genes in the polymyxin operon (Fig. 2B), we generated chromosomal deletions of pmrL and/or pmrM in the polymyxin-resistant strain MST100 (pmrAc), using in-frame kanamycin cassette replacements. In the case of the pmrL knock-out, a ribosome-binding site was inserted between the stop codon of kan gene and the start codon of the downstream pmrM gene to ensure proper expression of PmrM. In the pmrM-deleted construct, an extra sequence of nucleotides corresponding to the overlapping region between pmrM and the downstream regulatory pmrD gene (Fig. 2B), which is transcribed in the opposite direction (45), was inserted behind the stop codon of the kan gene in the primer (supplemental Table 1) to avoid altered regulation of pmrD expression. We also constructed a kan replacement of both the pmrL and pmrM genes in MST100. These isogenic mutants and the parental strain were then tested for polymyxin resistance. An arnD deletion mutant (Fig. 2, A and B) of MST100, previously shown to regain polymyxin sensitivity, was utilized as another control.5 As shown in Fig. 3, the pmrL and the pmrM deletion mutants (as well as the double deletion mutant) were sensitive to 15 μg/ml polymyxin, indicating that both genes participate in the maintenance of polymyxin resistance in E. coli. The growth of the mutants was not impaired on LB agar in the absence of polymyxin, and both were resistant to kanamycin (Fig. 3).
FIGURE 3. The ΔpmrL::kan and ΔpmrM::kan mutations confer polymyxin sensitivity on the pmrAc strain MST100.
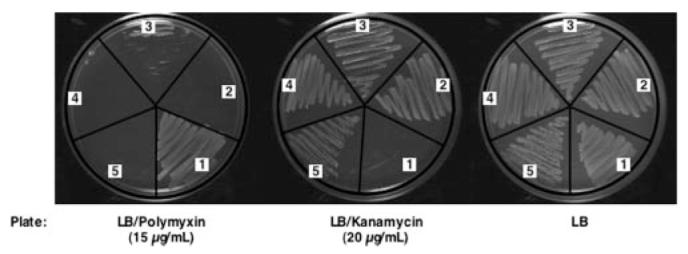
The in-frame replacements of pmrL and pmrM with the kan cassette were constructed as described under “Experimental Procedures.” Strains were streaked on LB agar or on LB agar containing either 15 μg/ml polymyxin or 20 μg/ml kanamycin. Cells were then grown overnight at 30 °C. Numbers indicate the following strains: 1, MST100 (pmrAc); 2, SDB200 (pmrAc ΔarnD::kan); 3, AY100 (pmrAc ΔpmrL::kan); 4, AY101 (pmrAc ΔpmrM::kan); and 5, AY102 (pmrAc ΔpmrLM::kan).
Polymyxin resistance was specifically restored to the pmrL deletion mutant AY100 by transformation with pWSK29-PmrL (Table 1) but not with pWSK29-PmrM (data not shown). Conversely, polymyxin resistance was recovered in the pmrM deletion mutant AY101 by transforming with pWSK29-PmrM (Table 1), but not with pWSK29-PmrL (data not shown), demonstrating that both pmrL and pmrM play indispensable roles in the maintenance of polymyxin resistance in MST100.
The pmrL and pmrM Deletion Mutants Lack L-Ara4N-modified Lipid A Species
Because the L-Ara4N modification of lipid A is required for polymyxin resistance (2), the status of lipid A modification with the L-Ara4N moiety was examined in each of the mutants. Crude lipid A, obtained by pH 4.5 hydrolysis of a chloroform/methanol-extracted cell residue, was subjected to TLC (data not shown) and ESI/MS analysis (Fig. 4). A significant fraction of the lipid A molecular species present in the parental strain MST100 was modified with the L-Ara4N unit (Fig. 4, peaks with red numbers, and Table 3). In addition, lipid A molecules bearing one or two pEtN units (but no L-Ara4N moiety) were also present in the parental strain (Fig. 4A, peaks with black numbers, and Table 3). In the pmrL or pmrM deletion mutants, over 95% of each of the L-Ara4N-modified lipid A species was missing (Fig. 4, B and C). In contrast, the pEtN and/or palmitate-modified lipid A species (Figs. 1 and 4 and Table 3) were present at normal or elevated levels. As seen previously with S. typhimurium arnC (pmrF) mutants (Fig. 2), the relative amounts of lipid A species bearing two pEtN units (Fig. 4, peaks 5 and 7) were actually elevated in the mutants unable to add the L-Ara4N unit to lipid A (21). These results imply that the PmrL and PmrM proteins are involved in the biosynthesis, trafficking, or attachment of the L-Ara4N unit to lipid A (Fig. 2).
FIGURE 4. Loss of L-Ara4N-modified lipid A species in ΔpmrL::kan and ΔpmrM::kan mutants of pmrAc strain MST100.

Lipid A was isolated from the strains indicated and was subjected to ESI/MS analysis in the negative ion mode. Compared with the parental strain MST100, shown in A, the ΔpmrL and ΔpmrM strains (B and C, respectively) lacked a series of L-Ara4N modified lipid A species (red numbers in A). The proposed compositions of the numbered peaks and their expected exact masses are listed in Table 3. amu, atomic mass units.
TABLE 3. Exact masses of modified lipid A species and assignments of the major peaks.
Observed masses are taken from Fig. 4A. In the pmrL and pmrM deletion mutants (Fig. 4, B and C) an additional peak (not shown) at m/z 1150.7 is attributed to a lipid A species modified with two pEtN and one palmitoyl substituent.
| Covalent group(s) | Peak no. | Exact mass | Predicted [M — 2H]2− | Observed [M — 2H]2− | Predicted [M + Na-3H]2− | Observed [M + Na-3H]2− |
|---|---|---|---|---|---|---|
| L-Ara4N | 2 | 1928.278 | 963.131 | 963.114 | ||
| L-Ara4N | 4 | 1928.278 | 974.122 | 974.106 | ||
| L-Ara4N | 6 | 2051.286 | 1024.635 | 1024.619 | ||
| pEtN | ||||||
| L-Ara4N | 8 | 2051.286 | 1035.626 | 1035.612 | ||
| pEtN | ||||||
| L-Ara4N | 9 | 2166.507 | 1082.246 | 1082.228 | ||
| C16:0 | ||||||
| pEtN | 1 | 1920.228 | 959.106 | 959.089 | ||
| pEtN | 3 | 1920.228 | 970.097 | 970.078 | ||
| (pEtN)2 | 5 | 2043.236 | 1020.610 | 1020.593 | ||
| (pEtN)2 | 7 | 2043.236 | 1031.601 | 1031.580 | ||
| pEtN | 10 | 2158.458 | 1089.212 | 1089.188 | ||
| C16:0 |
Undecaprenyl Phosphate-α-L-Ara4N Levels in pmrL and pmrM Mutants of MST100
The expression of the other gene products in the polymyxin resistance operon (Fig. 2, A and B) should not be affected by the pmrL or pmrM deletions. Accordingly, the intermediates of the pathway (Fig. 2A) should be present in normal amounts. Given that the modification of lipid A with L-Ara4N was largely abolished upon disruption of either the pmrL or the pmrM gene, it was conceivable that undecaprenyl phosphate-α-L-Ara4N might actually accumulate to higher levels than seen in the parental strain. We therefore extracted the phospholipids from the parent and mutant strains and subjected them to mild alkaline hydrolysis to remove the glycerophospholipids. TLC (Fig. 5) and ESI/MS analysis (see below) demonstrated that undecaprenyl phosphate-α-L-Ara4N levels were the same or slightly higher in the mutants than in the parent, indicating that appropriate amounts of the prenol lipid donor of the L-Ara4N moiety are still synthesized in these mutants.
FIGURE 5. Undecaprenyl phosphate-α-L-Ara4N levels in pmrAc strain MST100 and its ΔpmrL::kan and ΔpmrM::kan derivatives.
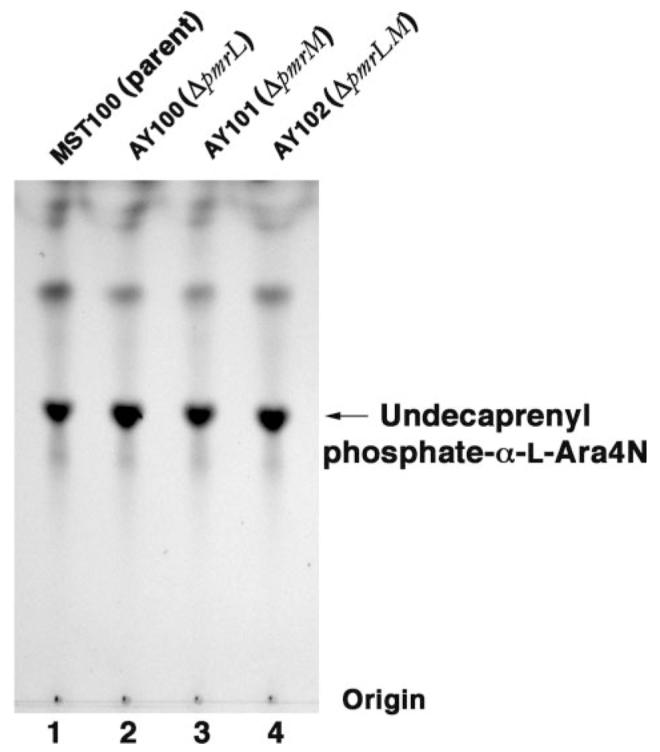
The pmrA constitutive parental strain, MST100, as well as the derived mutants AY100 (ΔpmrL), AY101 (ΔpmrM), and AY102 (ΔpmrLM) were grown to mid-log phase. The lipids were extracted and subjected to mild alkaline hydrolysis, as described under “Experimental Procedures.” The hydrolysis products were separated by silica TLC, using the solvent chloroform/methanol/acetic acid/ H2O (25:15:4:4, v/v), and visualized by charring on a hot plate after spraying with 10% sulfuric acid in ethanol.
The active site of ArnT, which transfers the L-Ara4N moiety from undecaprenyl phosphate-α-L-Ara4N to lipid A, is thought to be located on the periplasmic surface of the inner membrane (13, 31). The failure to attach the L-Ara4N moiety to lipid A in our mutants is therefore consistent with a defect in the translocation of undecaprenyl phosphate-α-L-Ara4N from its site of biosynthesis on the inner surface of the inner membrane to the outer surface of the inner membrane (Fig. 2A).
Reduced Labeling of Undecaprenyl Phosphate-α-L-Ara4N by Sulfo-NHS-Biotin in pmrL and pmrM Mutants
To evaluate the roles of PmrL and PmrM in undecaprenyl phosphate-α-LAra4N transport across the inner membrane, cells were treated with the inner membrane-impermeable amine reagent, sulfo-NHS-biotin (Scheme 1). Sulfo-NHS-biotin is a hydrophilic, low molecular weight biotinylation reagent that reacts with any primary amine. It is able to diffuse through the outer membrane porins of E. coli to reach the periplasmic space (36), but it cannot enter the cytoplasm because it does not cross the phospholipid bilayer of the inner membrane (40). Accordingly, if undecaprenyl phosphate-α-L-Ara4N were trapped on the inner surface of the inner membrane in the PmrL- and/or PmrM-deficient strains, sulfo-NHS-biotin labeling of the endogenous undecaprenyl phosphate-α-L-Ara4N should be reduced, when compared with the parental strain or to a comparable arnT (Fig. 2) deletion mutant.
SCHEME 1. Biotinylation of undecaprenyl phosphate-α-L-Ara4N by sulfo-NHS-biotin or NHS-biotin.
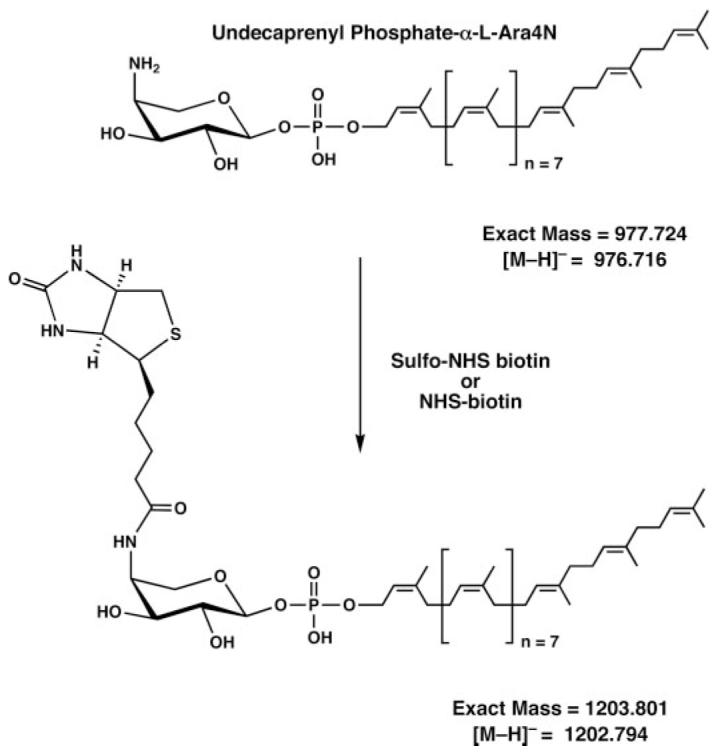
The same lipid product is generated with both reagents.
To evaluate undecaprenyl phosphate-α-L-Ara4N accessibility, we labeled MST100 and the mutant cells for various times with sulfo-NHS-biotin. We then analyzed the extent of undecaprenyl phosphate-α-L-Ara4N modification by TLC (Fig. 6) or ESI/MS (Fig. 7). As shown in Fig. 6A for MST100, a new compound, proposed to be biotinylated undecaprenyl phosphate-α-L-Ara4N (Scheme 1), was generated over the course of 4 h. This correlated with the appearance of a singly charged peak at a mass-to-charge ratio (m/z) of 1202.785 in the negative ion ESI mass spectrum (Fig. 7, A versus B), consistent with the structure shown in Scheme 1. The monoisotopic peak area ratio of unmodified undecaprenyl phosphate-α-L-Ara4N (m/z 976.707) to biotinylated undecaprenyl phosphate-α-L-Ara4N (m/z 1202.785) was 0.069 (Fig. 7B) at this time point (±0.001 for technical replicates). The actual molar ratio may be higher, as the ionization efficiency of the biotinylated undecaprenyl phosphate-α-L-Ara4N (Scheme 1) is likely to be somewhat lower than that of the unmodified material under the electrospray conditions employed.
FIGURE 6. Reduced labeling of undecaprenyl phosphate-α-L-Ara4N by sulfo-NHS-biotin in PmrL and PmrM mutants of MST100.
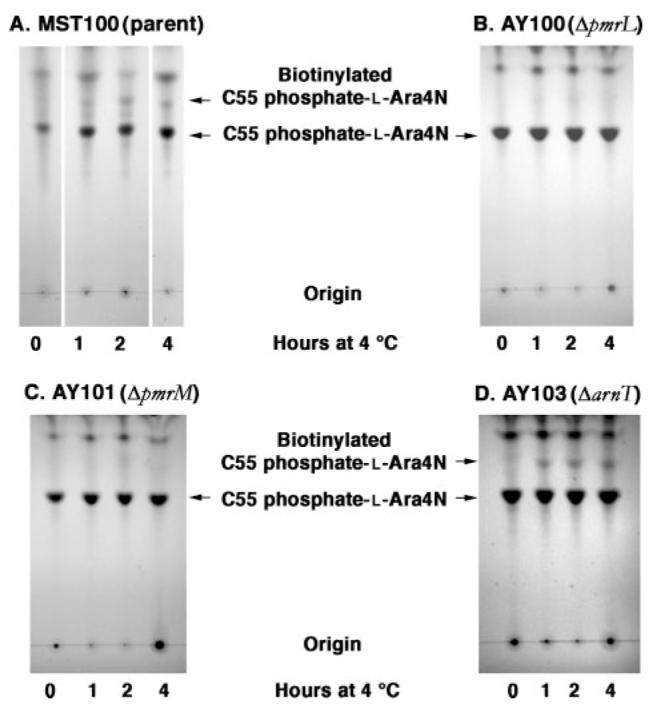
The pmrA constitutive parental strain MST100 (A), as well as the derived mutants AY100 (ΔpmrL)(B), AY101 (ΔpmrM)(C), and AY103 (ΔarnT)(D) were grown to mid-log phase. The washed cells were treated with sulfo-NHS-biotin at a final concentration of 2 mM for 0,1,2, or 4 h at 4°C. Reactions were quenched with 50 mM glycine. Phospholipids were extracted and were subjected to mild base hydrolysis (50). The base-stable prenol lipids were separated by TLC, using the solvent chloroform/methanol/acetic acid/H2O (25:15:4:4, v/v), and visualized by charring as in Fig. 5.
FIGURE 7. ESI/MS of undecaprenyl phosphate-α-L-Ara4N and its sulfo-NHS-biotin-modified derivative in parental and mutant strains.

The pmrA constitutive parent MST100 and the mutants AY100, AY101, and AY103 were grown to mid-log phase. The washed cells were treated with sulfo-NHS-biotin at 2mM for 4 h at 4°C. Reactions were quenched with 50 mM glycine. Lipids were extracted and subjected to mild base hydrolysis (50). The base-stable prenol lipids were analyzed by negative ion ESI/MS. A, MST100 lipids without sulfo-NHS-biotin treatment; B, MST100 lipids with sulfo-NHS-biotin for 4 h; C, AY100 lipids with sulfo-NHS-biotin for 4 h; D, AY101 lipids with sulfo-NHS-biotin for 4 h; and E, AY103 lipids with sulfo-NHS-biotin for 4 h. The insets highlight the peaks attributed to the biotinylated derivative of undecaprenyl phosphate-α-L-Ara4N, with the MST100 parental spectrum in blue and the indicated control or mutant spectra overlaid in red. The peak area ratios are derived from the monoisotopic peak area of the derivatized lipid near m/z 1202.79 divided by the monoisotopic peak area of the underivatized lipid near m/z 976.71 for each lipid sample (see Scheme 1). amu, atomic mass units.
In the pmrL and pmrM mutants, significantly less biotinylated undecaprenyl phosphate-α-L-Ara4N was generated, as judged by both TLC (Fig. 6, B and C) and ESI/MS (Fig. 7, C and D). At the 4-h time point, there was a 4–5-fold reduction in ratio of the monoisotopic peak areas for biotinylated undecaprenyl phosphate-α-L-Ara4N compared with unmodified undecaprenyl phosphate-α-L-Ara4N (Fig. 7, C and D), despite comparable levels of undecaprenyl phosphate-α-L-Ara4N in all strains (Fig. 5 and Fig. 7, B versus C and D). A liquid chromatography ESI/MS analysis of all the samples gave similar results (data not shown). The data imply that both PmrL and PmrM are involved in the translocation of undecaprenyl phosphate-α-LAra4N across the inner membrane.
Labeling of Undecaprenyl Phosphate-α-L-Ara4N by the Membrane-permeable Reagent NHS-Biotin
To exclude other possibilities, such as sequestration by protein binding, for the reduced modification of undecaprenyl phosphate-α-L-Ara4N by sulfo-NHS-biotin in pmrL and pmrM mutants, we examined undecaprenyl phosphate-α-L-Ara4N reactivity toward the hydrophobic amine reagent, NHS-biotin, under otherwise identical conditions. This compound is able to cross both the outer and the inner membrane of E. coli, and it should label undecaprenyl phosphate-α-L-Ara4N with equal efficiency, irrespective of its membrane localization. Use of NHS-biotin should generate similar amounts of biotinylated undecaprenyl phosphate-α-L-Ara4N in the parental and mutant strains. Similar levels of biotinylated undecaprenyl phosphate-α-L-Ara4N were indeed observed in all the strains with NHS-biotin, as demonstrated by TLC (Fig. 8, lanes 1–3) and ESI/MS analysis (not shown), although the rate of amine group labeling with NHS-biotin was slightly slower than with sulfo-NHS-biotin.
FIGURE 8. Modification of undecaprenyl phosphate-α-L-Ara4N by membrane-permeable NHS-biotin in parental and mutant strains.
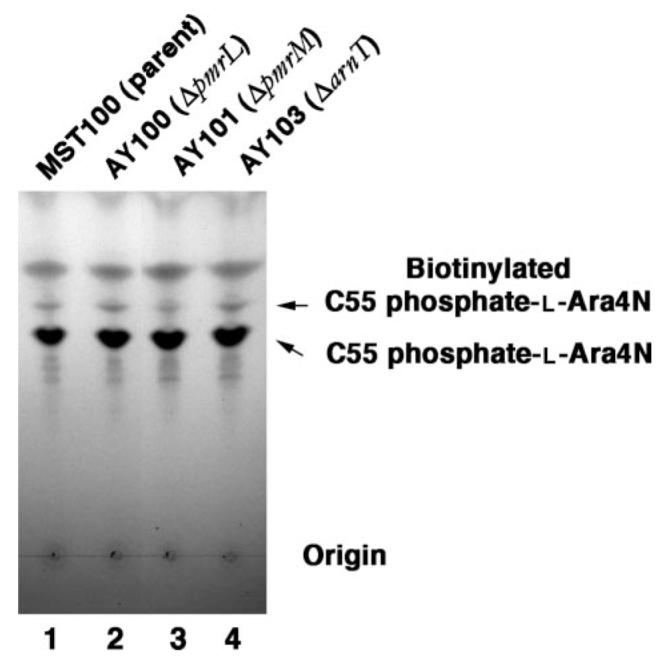
The pmrA constitutive parental strain MST100 (1), as well as AY100 (ΔpmrL) (2), AY101(ΔpmrM)(3), and AY103 (ΔarnT)(4) were grown to mid-log phase. The washed cells were treated with NHS-biotin at 2 mM for 4 h at 4°C. Reactions were quenched with 50 mM glycine. Lipids were extracted and subjected to mild alkaline hydrolysis (50). The hydrolysis products were separated by TLC, using the solvent chloroform/methanol/acetic acid/H2O (25:15:4:2, v/v), and visualized by charring as in Fig. 5.
ArnT Is Not Involved in the Transport of Undecaprenyl Phosphate-α-L-Ara4N
Given its 12 predicted trans-membrane helices (31), the enzyme ArnT (Fig. 2) might function not only to attach the L-Ara4N unit to lipid A but also to translocate undecaprenyl phosphate-α-L-Ara4N across the inner membrane. Accordingly, we constructed an in-frame kan replacement of the arnT gene, using the same MST100 background strain as for the pmrL and pmrM deletions. Lipid A modification with L-Ara4N was abolished in the arnT knock-out (data not shown), as in the pmrL and pmrM deletions (Fig. 4, B and C). The high levels of undecaprenyl phosphate-α-L-Ara4N seen in MST100 were unaffected by the disruption of the arnT gene (Fig. 6, A versus D, and Fig. 7, B versus E). In contrast to the pmrL and pmrM mutants, however, the undecaprenyl phosphate-α-L-Ara4N present in the arnT mutant reacted with the hydrophilic reagent sulfo-NHS-biotin exactly as it did in the parental strain (Fig. 7, B versus E), yielding a peak area ratio of 0.068 (Fig. 7E). These results are consistent with the idea that the undecaprenyl phosphate-α-L-Ara4N gains access to the periplasmic side of the inner membrane independently of arnT function. When the arnT mutant cells were labeled with the hydrophobic reagent NHS-biotin, modified undecaprenyl phosphate-α-L-Ara4N (Fig. 8, lane 4) was formed as efficiently as in MST100 (Fig. 8, lane 1) and in the pmrL or pmrM deletions (Fig. 8, lanes 2 and 3). These data suggest that ArnT does not participate in translocating its donor substrate, undecaprenyl phosphate-α-L-Ara4N, to its active site. Instead, PmrL and PmrM could specifically function to flip undecaprenyl phosphate-α-L-Ara4N from the cytosolic to the periplasmic side of the inner membrane, possibly functioning as a heterodimer. Given their likely roles as transporters in the L-Ara4N pathway, we suggest that PmrL and PmrM be renamed ArnE and ArnF (Fig. 2B).
DISCUSSION
Phospholipid flip-flop across biological membranes is an important yet poorly characterized process. The slow rate of phospholipid flip-flop in model lipid bilayers compared with biological membranes first suggested that specific proteins might be required to facilitate this process (59–62). The best characterized lipid transporters are members of the ATP-binding cassette (ABC) transporter superfamily (63, 64), which hydrolyze ATP to drive the unidirectional transport of specific compounds, including many lipids, across biological membranes. For instance, mouse mutants lacking the ABC transporter Mdr2 lack phosphatidylcholine in their bile (65). Patients with sitosterolemia lack the ABC transporters that pump absorbed sterols, including the plant-derived sitosterol, back into the gut or the bile (66). In bacteria, point mutants in the essential ABC transporter MsbA are defective in flipping nascent LPS across the inner membranes at elevated temperatures (35, 36, 67, 68), as judged by accessibility to periplasmic lipid A modification enzymes. Such mutants are also defective in glycerophospholipid export to the outer membrane (35), but this phenomenon might be secondary to LPS accumulation within the inner membrane. Despite the compelling genetic and physiological evidence for their roles in lipid trafficking, no simple, generally applicable, assays have been devised for demonstrating ABC transporter-dependent flipping of specific lipids in vitro.
Bacteria possess other types of transporters that use the energy from proton or electrochemical gradients to pump chemicals or ions cross the cytoplasmic membrane (37, 38, 53). For instance, the small multidrug resistance transporters are typically 105–121 amino acids long, contain four predicted trans-membrane helices, and function as homo- or heterooligomers (37, 38, 53, 54). Examples include heterodimeric complexes, such as EbrA/EbrB of Bacillus subtilis (39), as well as the homodimeric transporter EmrE of E. coli (54). The EmrE transporter confers resistance to lipophilic cations, such as tetraphenylphosphonium, methyl viologen, and ethidium (69), as well as to certain antibiotics (70). Although a published crystal structure of EmrE (55, 56) was recently retracted (57), and its proposed mechanism (55, 56) must be therefore questioned, there is no doubt that EmrE functions to export lipophilic cations from living bacteria (71). In vitro transport assays dependent upon purified reconstituted EmrE have been described (72).
PmrL and PmrM are small, hydrophobic, inner membrane proteins with many of the characteristics of small multidrug resistance transporters. Both PmrL and PmrM are predicted to have four membrane-spanning helices. These proteins are the distal genes of an operon required for the maintenance of polymyxin resistance (16) (Fig. 2B), which is present in both E. coli and Salmonella, and is regulated by the PmrA-(BasR) transcription factor (73), in conjunction with the PmrB and PmrD proteins (19, 74). We have now demonstrated that in-frame chromosomal deletions of pmrL, pmrM, or both restore polymyxin sensitivity to a PmrA-constitutive, polymyxin-resistant parental strain (Fig. 3). Concomitantly, the L-Ara4N-modified lipid A species (Fig. 4) are no longer synthesized in large amounts. However, careful inspection of the spectrum (Fig. 4, B and C) suggests that small amounts of L-Ara4N-modified lipid A species are still present, whereas they are completely absent in the ArnT or ArnA deletion mutants (data not shown). The pmrL and pmrM mutations do not inhibit the biosynthesis of undecaprenyl phosphate-α-L-Ara4N (Fig. 5), which is the immediate donor of the L-Ara4N unit to lipid A. However, the apparent trans-location of undecaprenyl phosphate-α-L-Ara4N from the cytoplasmic to the periplasmic side of the inner membrane is reduced by 4–5-fold, as judged by accessibility to labeling with the impermeable reagent sulfo-NHS-biotin (Figs. 6 and 7). We propose that PmrL and PmrM be renamed ArnE and ArnF, respectively (Fig. 2B), given their function in generating L-Ara4N modified lipid A species (Fig. 4).
It is possible that ArnE and ArnF, like some of the multidrug efflux pumps (39), form a heterodimer in vivo. The systematic studies by von Heijne and co-workers (75–77) on the topology of E. coli membrane proteins are consistent with the ArnE/ArnF model shown in Fig. 9. This proposal could be tested further by immunoprecipitation or purification of ArnE and/or ArnF. ArnE and ArnF share low sequence similarity to each other and to E. coli EmrE. All three proteins display similar overall hydropathy profiles. It will also be important to determine whether or not metabolic energy is required for the functioning of ArnE/F, for instance, the proton motive force across the inner membrane or the availability of ATP.
FIGURE 9. Proposed topography of the ArnE/ArnF heterodimer.
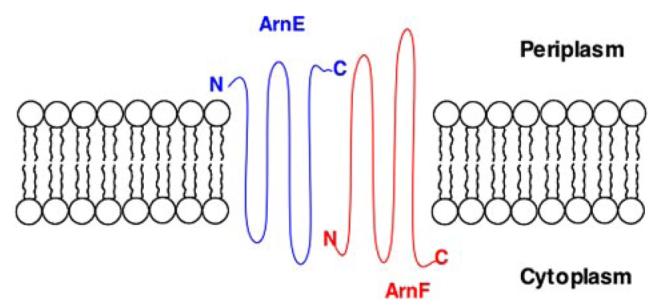
This model is based on the systematic studies of E. coli membrane proteins by von Heijne and co-workers (75–77, 95).
The origin of the ∼20% residual sulfo-NHS-biotin-labeled material in the prmL- and pmrM-deficient strains (Fig. 7, C and D) is uncertain. It may be that under our conditions of labeling some sulfo-NHS-biotin does reach the cytoplasm or some of the cells have become leaky. Alternatively, there may be other membrane proteins that can flip undecaprenyl phosphate-α-L-Ara4N across the inner membrane at a slower rate.
Membrane-impermeable reagents, such as sulfo-NHS-biotin or 2,4,6-trinitrobenzene sulfonic acid, have previously been utilized to characterize phosphatidylethanolamine flip-flop in whole cells (36, 40). We have successfully utilized sulfo-NHS-biotin (Figs. 6 and 7) and its membrane-permeable analogue NHS-biotin (Fig. 8) to measure the accessibility of undecaprenyl phosphate-α-L-Ara4N to covalent modification. Negative ion mode ESI/MS was used for the first time as an unambiguous criterion of undecaprenyl phosphate-α-L-Ara4N modification. Our results clearly show that ESI/MS is an efficient and definitive tool for monitoring lipid modification with membrane-impermeable reagents. It may be possible to adapt this approach to assay the flip-flop of other amine-containing lipids in whole cells or in isolated membrane vesicles.
Many other biogenic processes employ polyisoprene phosphate-oligosaccharides as precursors for extracellular glycosylation reactions. These systems include N-linked protein glycosylation in eukaryotic (78–80) and bacterial cells (81, 82), the generation of LPS O-antigen (1), the assembly of enterobacterial common antigen (83), and the polymerization of peptidoglycan (84). Although the sugar compositions and linkages of these polyisoprene phosphate-oligosaccharides differ greatly, two common principles emerge. First, a polyisoprene derivative (dolichyl phosphate in eukaryotic cells or undecaprenyl phosphate in E. coli) functions as the acceptor for one or more sugars residues, which are donated by cytoplasmic sugar nucleotides. Second, the polyisoprene phosphate-sugar intermediates are translocated (flipped) to the lumen of the endoplasmic reticulum in eukaryotic cells or to the periplasmic surface of the inner membrane in Gram-negative bacteria.
Recently, several specific membrane proteins have been implicated as polyisoprene phosphate-oligosaccharide flip-pases in yeast and bacteria. The yeast membrane protein Rft1p is proposed to flip dolichyl diphosphate-GlcNAc2Man5 from the cytoplasmic to the luminal side of the endoplasmic reticulum (78). The ABC transporter PglK (WlaB) in the human pathogen Campylobacter jejuni translocates an undecaprenyldiphosphate-linked heptasaccharide from the cytoplasmic to the periplasmic surface of the inner membrane (82). The Wzx proteins of Gram-negative bacteria appear to translocate O-antigen subunits attached to undecaprenyL-diphosphate across the inner membrane (85, 86) prior to polymerization and attachment to core-lipid A. None of these transporters are related in sequence to ArnE or ArnF. The flippases for the peptidoglycan precursors remain unknown. Generally applicable in vitro assays for these polyisoprene phosphate-sugar flippases have not been reported, although the use of short chain isoprenes is a very promising approach in some instances (87).
Although our genetic and biochemical studies implicate ArnE and ArnF as flippases for undecaprenyl phosphate-α-L-Ara4N, many questions remain. For example, the amount of undecaprenyl phosphate-α-L-Ara4N in arnE, arnF, and arnT deletion mutants is about the same as in the parental strain. Regulatory mechanisms may limit undecaprenyl phosphate-α-L-Ara4N accumulation when it is not being used for lipid A modification. Alternatively, there may be other pathways that consume undecaprenyl phosphate-α-L-Ara4N. Additional genetic and biochemical studies will be required to address these questions. Furthermore, the mechanism of ArnE and ArnF action as transporters remains to be characterized with in vitro assays, using purified, reconstituted preparations. A crystal structure of ArnE/ArnF with bound undecaprenyl phosphate-α-L-Ara4N would be especially informative. Despite the technical problems encountered recently with the EmrE crystal structure (71), we are optimistic that the high resolution structure of ArnE/ArnF can be solved and will provide molecular insights into its function as the undecaprenyl phosphate-α-L-Ara4N flippase.
Supplementary Material
Acknowledgments
We are grateful to all members of the Raetz laboratory for their help with experimental techniques, inspiring discussion, and friendship. The mass spectrometry facility in the Department of Biochemistry, Duke University Medical Center, was supported by the LIPID MAPS Large Scale Collaborative Grant GM-069338 from the National Institutes of Health.
Footnotes
This work was supported in part by National Institutes of Health Grant GM-51310 (to C. R. H. R.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1.
- L-Ara4N
- 4-amino-4-deoxy-L-arabinose
- ESI/MS
- electrospray ionization/mass spectrometry
- LPS
- lipopolysaccharide
- NHS-biotin
- N-hydroxysuccinimidobiotin
- pEtN
- phosphoethanolamine
- Sulfo-NHS-biotin
- N-hydroxysulfosuccinimidobiotin
- ABC
- ATP-binding cassette
S. D. Breazeale and C. R. H. Raetz, manuscript in preparation.
REFERENCES
- 1.Raetz CRH, Whitfield C. Annu. Rev. Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raetz CRH, Reynolds CM, Trent MS, Bishop RE. Annu. Rev. Biochem. 2007;76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lien E, Ingalls RR. Crit. Care Med. 2002;30:S1–S11. [PubMed] [Google Scholar]
- 4.Beutler B, Rietschel ET. Nat. Rev. Immunol. 2003;3:169–176. doi: 10.1038/nri1004. [DOI] [PubMed] [Google Scholar]
- 5.Esmon CT. Biochim. Biophys. Acta. 2000;1477:349–360. doi: 10.1016/s0167-4838(99)00266-6. [DOI] [PubMed] [Google Scholar]
- 6.Janeway CA, Jr., Medzhitov R. Annu. Rev. Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 7.Akira S, Uematsu S, Takeuchi O. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 8.Gay NJ, Gangloff M. Annu. Rev. Biochem. 2007;76:141–165. doi: 10.1146/annurev.biochem.76.060305.151318. [DOI] [PubMed] [Google Scholar]
- 9.Nikaido H, Vaara M. Microbiol. Rev. 1985;49:1–32. doi: 10.1128/mr.49.1.1-32.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaara M. Microbiol. Rev. 1992;56:395–411. doi: 10.1128/mr.56.3.395-411.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaara M, Vaara T, Jensen M, Helander I, Nurminen M, Rietschel ET, Makela PH. FEBS Lett. 1981;129:145–149. doi: 10.1016/0014-5793(81)80777-6. [DOI] [PubMed] [Google Scholar]
- 12.Groisman EA, Kayser J, Soncini FC. J. Bacteriol. 1997;179:7040–7045. doi: 10.1128/jb.179.22.7040-7045.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trent MS, Ribeiro AA, Doerrler WT, Lin S, Cotter RJ, Raetz CRH. J. Biol. Chem. 2001;276:43132–43144. doi: 10.1074/jbc.M106962200. [DOI] [PubMed] [Google Scholar]
- 14.Helander IM, Kilpeläinen I, Vaara M. Mol. Microbiol. 1994;11:481–487. doi: 10.1111/j.1365-2958.1994.tb00329.x. [DOI] [PubMed] [Google Scholar]
- 15.Nummila K, Kilpeläinen I, Zähringer U, Vaara M, Helander IM. Mol. Microbiol. 1995;16:271–278. doi: 10.1111/j.1365-2958.1995.tb02299.x. [DOI] [PubMed] [Google Scholar]
- 16.Gunn JS, Lim KB, Krueger J, Kim K, Guo L, Hackett M, Miller SI. Mol. Microbiol. 1998;27:1171–1182. doi: 10.1046/j.1365-2958.1998.00757.x. [DOI] [PubMed] [Google Scholar]
- 17.Guo L, Lim KB, Gunn JS, Bainbridge B, Darveau RP, Hackett M, Miller SI. Science. 1997;276:250–253. doi: 10.1126/science.276.5310.250. [DOI] [PubMed] [Google Scholar]
- 18.Gunn JS, Miller SI. J. Bacteriol. 1996;178:6857–6864. doi: 10.1128/jb.178.23.6857-6864.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gunn JS, Ryan SS, Van Velkinburgh JC, Ernst RK, Miller SI. Infect. Immun. 2000;68:6139–6146. doi: 10.1128/iai.68.11.6139-6146.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou Z, Ribeiro AA, Raetz CRH. J. Biol. Chem. 2000;275:13542–13551. doi: 10.1074/jbc.275.18.13542. [DOI] [PubMed] [Google Scholar]
- 21.Zhou Z, Ribeiro AA, Lin S, Cotter RJ, Miller SI, Raetz CRH. J. Biol. Chem. 2001;276:43111–43121. doi: 10.1074/jbc.M106960200. [DOI] [PubMed] [Google Scholar]
- 22.Trent MS, Raetz CRH. J. Endotoxin Res. 2002;8:158. [Google Scholar]
- 23.Lee H, Hsu FF, Turk J, Groisman EA. J. Bacteriol. 2004;186:4124–4133. doi: 10.1128/JB.186.13.4124-4133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Groisman EA. J. Bacteriol. 2001;183:1835–1842. doi: 10.1128/JB.183.6.1835-1842.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou Z, Lin S, Cotter RJ, Raetz CRH. J. Biol. Chem. 1999;274:18503–18514. doi: 10.1074/jbc.274.26.18503. [DOI] [PubMed] [Google Scholar]
- 26.Gibbons HS, Kalb SR, Cotter RJ, Raetz CRH. Mol. Microbiol. 2005;55:425–440. doi: 10.1111/j.1365-2958.2004.04409.x. [DOI] [PubMed] [Google Scholar]
- 27.Volk WA, Galanos C, Lüderitz O. Eur. J. Biochem. 1970;17:223–229. doi: 10.1111/j.1432-1033.1970.tb01157.x. [DOI] [PubMed] [Google Scholar]
- 28.Breazeale SD, Ribeiro AA, McClerren AL, Raetz CRH. J. Biol. Chem. 2005;280:14154–14167. doi: 10.1074/jbc.M414265200. [DOI] [PubMed] [Google Scholar]
- 29.Breazeale SD, Ribeiro AA, Raetz CRH. J. Biol. Chem. 2003;279:24731–24739. doi: 10.1074/jbc.M304043200. [DOI] [PubMed] [Google Scholar]
- 30.Breazeale SD, Ribeiro AA, Raetz CRH. J. Biol. Chem. 2002;277:2886–2896. doi: 10.1074/jbc.M109377200. [DOI] [PubMed] [Google Scholar]
- 31.Trent MS, Ribeiro AA, Lin S, Cotter RJ, Raetz CRH. J. Biol. Chem. 2001;276:43122–43131. doi: 10.1074/jbc.M106961200. [DOI] [PubMed] [Google Scholar]
- 32.Williams GJ, Breazeale SD, Raetz CRH, Naismith JH. J. Biol. Chem. 2005;280:23000–23008. doi: 10.1074/jbc.M501534200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gatzeva-Topalova PZ, May AP, Sousa MC. Structure (Camb.) 2005;13:929–942. doi: 10.1016/j.str.2005.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Noland BW, Newman JM, Hendle J, Badger J, Christopher JA, Tresser J, Buchanan MD, Wright TA, Rutter ME, Sanderson WE, Muller-Dieckmann HJ, Gajiwala KS, Buchanan SG. Structure (Camb.) 2002;10:1569–1580. doi: 10.1016/s0969-2126(02)00879-1. [DOI] [PubMed] [Google Scholar]
- 35.Doerrler WT, Reedy MC, Raetz CRH. J. Biol. Chem. 2001;276:11461–11464. doi: 10.1074/jbc.C100091200. [DOI] [PubMed] [Google Scholar]
- 36.Doerrler WT, Gibbons HS, Raetz CRH. J. Biol. Chem. 2004;279:45102–45109. doi: 10.1074/jbc.M408106200. [DOI] [PubMed] [Google Scholar]
- 37.Paulsen IT, Skurray RA, Tam R, Saier MH, Jr., Turner RJ, Weiner JH, Goldberg EB, Grinius LL. Mol. Microbiol. 1996;19:1167–1175. doi: 10.1111/j.1365-2958.1996.tb02462.x. [DOI] [PubMed] [Google Scholar]
- 38.Chung YJ, Saier MH., Jr. Curr. Opin. Drug Discovery Dev. 2001;4:237–245. [PubMed] [Google Scholar]
- 39.Zhang Z, Ma C, Pornillos O, Xiu X, Chang G, Saier MH., Jr. Biochemistry. 2007;46:5218–5225. doi: 10.1021/bi7001604. [DOI] [PubMed] [Google Scholar]
- 40.Muller WA. Methods Mol. Biol. 1994;27:31–42. doi: 10.1385/0-89603-250-7:31. [DOI] [PubMed] [Google Scholar]
- 41.Miller JR. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1972. [Google Scholar]
- 42.Sambrook JG, Russell DW. Molecular Cloning: A Laboratory Manual. 3rd Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2001. [Google Scholar]
- 43.Inoue H, Nojima H, Okayama H. Gene (Amst.) 1990;96:23–28. doi: 10.1016/0378-1119(90)90336-p. [DOI] [PubMed] [Google Scholar]
- 44.Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 45.Riley M, Abe T, Arnaud MB, Berlyn MK, Blattner FR, Chaudhuri RR, Glasner JD, Horiuchi T, Keseler IM, Kosuge T, Mori H, Perna NT, Plunkett G, III, Rudd KE, Serres MH, Thomas GH, Thomson NR, Wishart D, Wanner BL. Nucleic Acids Res. 2006;34:1–9. doi: 10.1093/nar/gkj405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Derbise A, Lesic B, Dacheux D, Ghigo JM, Carniel E. FEMS Immunol. Med. Microbiol. 2003;38:113–116. doi: 10.1016/S0928-8244(03)00181-0. [DOI] [PubMed] [Google Scholar]
- 47.Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. Proc. Natl. Acad. Sci. U. S. A. 2000;97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang RF, Kushner SR. Gene (Amst.) 1991;100:195–199. [PubMed] [Google Scholar]
- 49.Bligh EG, Dyer JJ. Can. J. Biochem. Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 50.KanjilaL-Kolar S, Raetz CRH. J. Biol. Chem. 2006;281:12879–12887. doi: 10.1074/jbc.M513865200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Caroff M, Tacken A, Szabó L. Carbohydr. Res. 1988;175:273–282. doi: 10.1016/0008-6215(88)84149-1. [DOI] [PubMed] [Google Scholar]
- 52.Dulbecco R, Vogt M. J. Exp. Med. 1954;99:167–182. doi: 10.1084/jem.99.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hvorup RN, Winnen B, Chang AB, Jiang Y, Zhou XF, Saier MH., Jr. Eur. J. Biochem. 2003;270:799–813. doi: 10.1046/j.1432-1033.2003.03418.x. [DOI] [PubMed] [Google Scholar]
- 54.Tate CG. Curr. Opin. Struct. Biol. 2006;16:457–464. doi: 10.1016/j.sbi.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 55.Pornillos O, Chen YJ, Chen AP, Chang G. Science. 2005;310:1950–1953. doi: 10.1126/science.1119776. [DOI] [PubMed] [Google Scholar]
- 56.Ma C, Chang G. Proc. Natl. Acad. Sci. U. S. A. 2004;101:2852–2857. doi: 10.1073/pnas.0400137101. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 57.Ma C, Chang G. Proc. Natl. Acad. Sci. U. S. A. 2007;104:3668. doi: 10.1073/pnas.0700711104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zähringer U, Lindner B, Rietschel ET. In: Endotoxin in Health and Disease. Brade H, Opal SM, Vogel SN, Morrison DC, editors. Marcel Dekker, Inc.; New York: 1999. pp. 93–114. [Google Scholar]
- 59.Kornberg RD, McConnell HM. Biochemistry. 1971;10:1111–1120. doi: 10.1021/bi00783a003. [DOI] [PubMed] [Google Scholar]
- 60.Bretscher MS. J. Mol. Biol. 1972;71:523–528. doi: 10.1016/s0022-2836(72)80020-2. [DOI] [PubMed] [Google Scholar]
- 61.Rothman JE, Kennedy EP. Proc. Natl. Acad. Sci. U. S. A. 1977;74:1821–1825. doi: 10.1073/pnas.74.5.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gummadi SN, Menon AK. J. Biol. Chem. 2002;277:25337–25343. doi: 10.1074/jbc.M203809200. [DOI] [PubMed] [Google Scholar]
- 63.Borst P, Elferink RO. Annu. Rev. Biochem. 2002;71:537–592. doi: 10.1146/annurev.biochem.71.102301.093055. [DOI] [PubMed] [Google Scholar]
- 64.Schmitz G, Liebisch G, Langmann T. FEBS Lett. 2006;580:5597–5610. doi: 10.1016/j.febslet.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 65.Smit JJM, Schinkel AH, Oude Elferink RPJ, Groen AK, Wagenaar E, van Deemter L, Mol CAAM, Ottenhoff R, van der Lugt NMT, van Roon MA, van der Valk MA, Offerhaus GJA, Berns AJM, Borst P. Cell. 1993;75:451–462. doi: 10.1016/0092-8674(93)90380-9. [DOI] [PubMed] [Google Scholar]
- 66.Berge KE, Tian H, Graf GA, Yu L, Grishin NV, Schultz J, Kwiterovich P, Shan B, Barnes R, Hobbs HH. Science. 2000;290:1771–1775. doi: 10.1126/science.290.5497.1771. [DOI] [PubMed] [Google Scholar]
- 67.Doerrler WT, Raetz CRH. J. Biol. Chem. 2002;277:36697–36705. doi: 10.1074/jbc.M205857200. [DOI] [PubMed] [Google Scholar]
- 68.Wang X, Karbarz MJ, McGrath SC, Cotter RJ, Raetz CRH. J. Biol. Chem. 2004;279:49470–49478. doi: 10.1074/jbc.M409078200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rotem D, Schuldiner S. J. Biol. Chem. 2004;279:48787–48793. doi: 10.1074/jbc.M408187200. [DOI] [PubMed] [Google Scholar]
- 70.Li XZ, Poole K, Nikaido H. Antimicrob. Agents Chemother. 2003;47:27–33. doi: 10.1128/AAC.47.1.27-33.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schuldiner S. Trends Biochem. Sci. 2007;32:252–258. doi: 10.1016/j.tibs.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 72.Curnow P, Lorch M, Charalambous K, Booth PJ. J. Mol. Biol. 2004;343:213–222. doi: 10.1016/j.jmb.2004.08.032. [DOI] [PubMed] [Google Scholar]
- 73.Roland KL, Martin LE, Esther CR, Spitznagel JK. J. Bacteriol. 1993;175:4154–4164. doi: 10.1128/jb.175.13.4154-4164.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Winfield MD, Groisman EA. Proc. Natl. Acad. Sci. U. S. A. 2004;101:17162–17167. doi: 10.1073/pnas.0406038101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Daley DO, Rapp M, Granseth E, Melen K, Drew D, von Heijne G. Science. 2005;308:1321–1323. doi: 10.1126/science.1109730. [DOI] [PubMed] [Google Scholar]
- 76.Granseth E, Daley DO, Rapp M, Melen K, von Heijne G. J. Mol. Biol. 2005;352:489–494. doi: 10.1016/j.jmb.2005.07.053. [DOI] [PubMed] [Google Scholar]
- 77.Rapp M, Granseth E, Seppala S, von Heijne G. Nat. Struct. Mol. Biol. 2006;13:112–116. doi: 10.1038/nsmb1057. [DOI] [PubMed] [Google Scholar]
- 78.Helenius J, Ng DT, Marolda CL, Walter P, Valvano MA, Aebi M. Nature. 2002;415:447–450. doi: 10.1038/415447a. [DOI] [PubMed] [Google Scholar]
- 79.Yan A, Lennarz WJ. Glycobiology. 2005;15:1407–1415. doi: 10.1093/glycob/cwj026. [DOI] [PubMed] [Google Scholar]
- 80.Helenius A, Aebi M. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 81.Wacker M, Linton D, Hitchen PG, Nita-Lazar M, Haslam SM, North SJ, Panico M, Morris HR, Dell A, Wren BW, Aebi M. Science. 2002;298:1790–1793. doi: 10.1126/science.298.5599.1790. [DOI] [PubMed] [Google Scholar]
- 82.Alaimo C, Catrein I, Morf L, Marolda CL, Callewaert N, Valvano MA, Feldman MF, Aebi M. EMBO J. 2006;25:967–976. doi: 10.1038/sj.emboj.7601024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rick PD, Hubbard GL, Kitaoka M, Nagaki H, Kinoshita T, Dowd S, Simplaceanu V, Ho C. Glycobiology. 1998;8:557–567. doi: 10.1093/glycob/8.6.557. [DOI] [PubMed] [Google Scholar]
- 84.Lazar K, Walker S. Curr. Opin. Chem. Biol. 2002;6:786–793. doi: 10.1016/s1367-5931(02)00355-1. [DOI] [PubMed] [Google Scholar]
- 85.Liu D, Cole RA, Reeves PR. J. Bacteriol. 1996;178:2102–2107. doi: 10.1128/jb.178.7.2102-2107.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Feldman MF, Marolda CL, Monteiro MA, Perry MB, Parodi AJ, Valvano MA. J. Biol. Chem. 1999;274:35129–35138. doi: 10.1074/jbc.274.49.35129. [DOI] [PubMed] [Google Scholar]
- 87.Rick PD, Barr K, Sankaran K, Kajimura J, Rush JS, Waechter CJ. J. Biol. Chem. 2003;278:16534–16542. doi: 10.1074/jbc.M301750200. [DOI] [PubMed] [Google Scholar]
- 88.Strain SM, Armitage IM, Anderson L, Takayama K, Qureshi N, Raetz CRH. J. Biol. Chem. 1985;260:16089–16098. [PubMed] [Google Scholar]
- 89.Bishop RE, Gibbons HS, Guina T, Trent MS, Miller SI, Raetz CRH. EMBO J. 2000;19:5071–5080. doi: 10.1093/emboj/cdd507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Trent MS, Pabich W, Raetz CRH, Miller SI. J. Biol. Chem. 2001;276:9083–9092. doi: 10.1074/jbc.M010730200. [DOI] [PubMed] [Google Scholar]
- 91.Ohl ME, Miller SI. Annu. Rev. Med. 2001;52:259–274. doi: 10.1146/annurev.med.52.1.259. [DOI] [PubMed] [Google Scholar]
- 92.Gibbons HS, Lin S, Cotter RJ, Raetz CRH. J. Biol. Chem. 2000;275:32940–32949. doi: 10.1074/jbc.M005779200. [DOI] [PubMed] [Google Scholar]
- 93.Reynolds CM, Ribeiro AA, McGrath SC, Cotter RJ, Raetz CRH, Trent MS. J. Biol. Chem. 2006;281:21974–21987. doi: 10.1074/jbc.M603527200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Reeves PR, Hobbs M, Valvano MA, Skurnik M, Whitfield C, Coplin D, Kido N, Klena J, Maskell D, Raetz CRH, Rick PD. Trends Microbiol. 1996;4:495–503. doi: 10.1016/s0966-842x(97)82912-5. [DOI] [PubMed] [Google Scholar]
- 95.Rapp M, Seppala S, Granseth E, von Heijne G. Science. 2007;315:1282–1284. doi: 10.1126/science.1135406. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.