

Summary
In the present study, we investigated the mechanism of cytochrome c release from isolated brain mitochondria induced by recombinant oligomeric BAX (BAXoligo). We found that BAXoligo caused a complete release of cytochrome c in a concentration- and time-dependent manner. The release was similar to those induced by alamethicin, which causes maximal mitochondrial swelling and eliminates barrier properties of the OMM. BAXoligo also produced large amplitude mitochondrial swelling as judged by light scattering assay and transmission electron microscopy. In addition, BAXoligo resulted in a strong mitochondrial depolarization. ATP or a combination of cyclosporin A and ADP, inhibitors of the mPT, suppressed BAXoligo-induced mitochondrial swelling and depolarization as well as cytochrome c release but did not influence BAXoligo insertion into the OMM. Both BAXoligo- and alamethicin-induced cytochrome c releases were accompanied by inhibition of ROS generation, which was assessed by measuring mitochondrial H2O2 release with an Amplex Red assay. The mPT inhibitors antagonized suppression of ROS generation caused by BAXoligo but not by alamethicin. Thus, BAXoligo resulted in a complete cytochrome c release from isolated brain mitochondria in the mPT-dependent manner without involvement of oxidative stress by the mechanism requiring mitochondrial remodeling and permeabilization of the OMM.
Keywords: brain, mitochondria, apoptosis, ROS, calcium, BAX
In apoptosis, a release of mitochondrial apoptogenic proteins occurs due to the interaction of mitochondria with pro-apoptotic members of the Bcl-2 family such as activated (truncated) BID (tBID) and BAX [1,2]. Monomeric BAX resides in the cytosol and remains inactive until tBID causes its oligomerization and incorporation into the OMM [3,4]. This leads to permeabilization of the OMM and escape of mitochondrial apoptogenic proteins from mitochondrial intermembrane space [5]. In the experimental conditions, an oligomerization of BAX can be enforced by a low concentration of mild detergents such as octyl glucoside [1,6,7]. This oligomerized BAX (BAXoligo) also permeabilizes the OMM and releases cytochrome c [1,7].
In early studies, the mitochondrial permeability transition (mPT) was implicated in protein-induced cytochrome c release as an essential mechanism leading to mitochondrial swelling and rupture of the OMM [8-12]. However, in our previous study with isolated brain mitochondria, recombinant tBID alone, or in combination either with monomeric BAX lacking C-terminal segment (BAXΔC) or with a full-length monomeric BAX, caused cytochrome c release, which was not sensitive to inhibitors of the mPT [13,14]. This suggested an mPT-independent release of cytochrome c. This conclusion is consistent with numerous observations from different laboratories, indicating that protein-induced cytochrome c release may occur without involvement of the mPT [15-20]. However, it still remains unknown whether BAXoligo causes a release of cytochrome c from brain mitochondria in an mPT-dependent or mPT-independent manner.
The massive cytochrome c release induced by pro-apoptotic proteins was proposed to occur in two steps including (i) cristae remodeling, which eliminates the diffusion barrier for cytochrome c and (ii) cytochrome c escape from the intermembrane space following either pore formation in the OMM or the rupture of the OMM due to matrix swelling [21]. Alternatively, the release of cytochrome c induced by BAXoligo from liver mitochondria was hypothesized to occur also in two steps involving loosening of cytochrome c binding to the inner mitochondrial membrane (IMM) due to oxidative stress and lipid peroxidation followed by its dissociation from the membrane and escape through the permeabilized OMM [22]. Later, it was proposed that cytochrome c release during apoptotic events might occur in a single step requiring only permeabilization of the OMM [23]. In our study, we addressed a question whether mitochondrial remodeling and oxidative stress play an essential role in the BAXoligo-induced cytochrome c release from brain mitochondria.
In the present paper, we demonstrate that in isolated brain mitochondria, recombinant BAXoligo induces massive cytochrome c release sensitive to a combination of cyclosporin A (CsA) and ADP, the inhibitors of the mPT [24-26]. Moreover, we found that BAXoligo caused large amplitude mitochondrial swelling and depolarization of organelles, which could be suppressed by mPT inhibitors. In addition, we found that an oxidative stress was not required for a complete cytochrome c release produced by BAXoligo or by antibiotic alamethicin, which eliminated barrier properties of the OMM [27]. Thus, our data are consistent with the hypothesis that BAXoligo produces complete cytochrome c release from isolated brain mitochondria in the mPT-dependent manner by the mechanism involving mitochondrial remodeling but not oxidative stress.
Materials and Methods
Recombinant BAX
Recombinant BAX was prepared and olgomerized in the dialysis buffer containing 25 mM HEPES-NaOH, pH 7.5, 1% (w/v) octyl glucoside, 0.2 mM dithiothreitol, 30% glycerol (v/v) as described previously [6].
Isolation and purification of brain mitochondria
Mitochondria from the brains or livers of male Sprague-Dawley rats, 200–250 g (Harlan, Indianapolis, IN, USA) were isolated in mannitol-sucrose medium according to an IACUC approved protocol and purified on a discontinuous Percoll gradient as described previously [27]. Mitochondrial protein was measured by the Bradford method [28], using BSA as a standard.
Measurements of mitochondrial respiration
Mitochondrial respiration was measured in the standard incubation medium at 37°C under continuous stirring. The standard incubation medium contained 125 mM KCl, 10 mM HEPES, pH 7.4, 0.5 mM MgCl2, 3 mM KH2PO4, 10 μM EGTA, 0.1% bovine serum albumin (free from fatty acids) and was supplemented either with 3 mM succinate plus 3 mM glutamate, or with 3 mM succinate plus 1μM rotenone, or with 3 mM pyruvate plus 1 mM malate. The 0.3 ml incubation chamber was equipped with a Clark-type oxygen electrode and a tightly closed lid. The slope of the O2 electrode trace corresponded to the respiratory rate. All data traces shown are representative of at least three separate experiments.
Monitoring of mitochondrial membrane potential
The mitochondrial membrane potential (Δψ) was monitored by following the distribution of TPP+ between the external medium (initially 1.8 μM TPP+-Cl) and the mitochondrial matrix with a TPP+-sensitive electrode [29] in the standard incubation medium supplemented with 3 mM succinate plus 3 mM glutamate unless stated otherwise. A decline in the external TPP+ concentration in the medium corresponded to mitochondrial polarization, whereas a rise in the TPP+ concentration in the medium corresponded to depolarization. In all experiments with isolated mitochondria, the concentration of mitochondrial protein in the chamber was 0.2 mg/ml. All data traces shown are representative of at least three separate experiments.
Measurements of mitochondrial light scattering
Mitochondrial swelling was evaluated in the standard incubation medium by monitoring the scattering of light directed on mitochondrial suspension under 90° to the axis of the photodetector at 525 nm in a 0.4-ml cuvette under continuous stirring using a PerkinElmer LS-55 luminescence spectrometer unless stated otherwise.
Measurements of ROS Generation
Production of ROS by isolated brain mitochondria incubated in the standard incubation medium was assessed using the Amplex Red assay for H2O2 (Invitrogen), as described previously [14].
Transmission electron microscopy
Electron microscopy of isolated brain mitochondria was performed as described previously [30]. Mitochondria were incubated in the standard 125 mM KCl-based medium supplemented with 3 mM succinate plus 3 mM glutamate at 37°C prior to fixation in 2% paraformaldehyde and 2% glutaraldehyde in 0.05 M phosphate buffer in the same incubation medium at room temperature for 15 minutes. Samples for transmission electron microscopy (TEM) were taken using a Tecnai G12 BioTwin electron microscope (FEI, Hillsboro, OR) equipped with an AMT 2.6×2.6K digital CCD camera. To quantitatively assess the morphological changes, we used the morphometric analysis described previously [12]. Total mitochondrial population was categorized into three groups according to their morphology as follows: (i) condensed, (ii) mitochondria with tubular cristae, and (iii) swollen. Mitochondria with characteristics bridging morphologic groups were assigned to the lower category. Mitochondria were counted in a blind fashion, and morphological distribution was statistically analyzed using a one-way analysis of variance followed by Bonferroni's posttest (GraphPad Prism, 4.0).
BAX insertion
To determine alkali-resistant fraction of BAX inserted into the OMM the earlier described method was used (Roucou et al. 2002a). Briefly, mitochondria treated with BAXoligo (10.8 μg/ml) at 37°C for 30 min were pelleted at 15,800 g for 5 min, and supernatant was used for the cytochrome c release measurements. Mitochondrial pellets were re-suspended in 0.2 ml of 0.1 Na2CO3, pH 11.5, and incubated for 20 min on ice. Samples were centrifuged for 25 min at 100,000 g in Sorvall Ultra Pro® 80 ultracentrifuge. The pellets were solubilized using 2% 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate (CHAPS) and analyzed by western blotting against BAX and cytochrome oxidase subunit IV (COX IV).
Immunoblotting
The release of cytochrome c from isolated brain mitochondria was assessed using western blotting in supernatants obtained through incubation of mitochondria in the standard 125 mM KCl-based incubation medium for 30 min at 37 °C. For electrophoresis, we used 4-12% Bis-Tris MOPS gels (Invitrogen, Carlsbad, CA). Western blotting was performed as previously described [14]. The release of cytochrome c from mitochondria treated with alamethicin (30μg/ml) was used as a control for maximal cytochrome c release. For detection of Smac/DIABLO, AIF, Omi/HtrA2, and Endo G the supernatants were concentrated 6 fold by using Microcon YM-10 filtering devices (Millipore Corporation, Bedford, MA). Mitochondrial voltage-dependent anion channel (VDAC) or COX IV were used as a loading control for the pellet samples. VDAC was detected with goat polyclonal anti-VDAC antibody, dilution 1:200 (Santa Cruz Biotechnology, Santa Cruz, CA). COX IV was detected with mouse monoclonal anti-COX IV antibody, dilution 1:5000 (Invitrogen, Carlsbad, CA). Following electrophoresis proteins were transferred to Hybond™-ECL™ nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ) and blots were incubated with primary mouse anti-cytochrome c antibody (7H8.2C12, PharMingen, San Diego, CA) at 1:1000, rabbit anti-Smac/DIABLO antibody (Alexis Biochemicals, San Diego, CA) at 1:500, anti-EndoG antibody (Oncogene, San Diego, CA) at 1:1000, rabbit anti-Omi/HtrA2 antibody (R&D Systems, Minneapolis, MN) at 1:1000 or rabbit anti-AIF antibody (R&D Systems, Minneapolis, MN) at 1:2000 dilution for an hour at room temperature in 5% non-fat milk, phosphate-buffered saline, pH 7.2, and 0.15% Triton X-100. In the BAXoligo insertion experiments, BAX was detected with rabbit anti-BAX antibody (Upstate, Lake Placid, NY) used at 1:2000 dilution. Blots were developed using goat anti-rabbit and anti-mouse IgG (1:20000) coupled to horseradish peroxidase (Jackson ImmunoResearch Laboratories, West Grove, PA) and Supersignal West chemiluminescent reagents (Pierce, Rockford, IL). Molecular weight marker SeeBlue® Plus 2 Standards (5 μl), (Invitrogen, Carlsbad, CA) were used to determine molecular weights of the bands.
Statistics
Statistical analyses of experimental data consisted of a one-way analysis of variance followed by Bonferroni's posttest (GraphPad Prism, version 4.0; GraphPad Software, San Diego, CA). The data represent the mean ± S.E. of at least three separate, independent experiments.
Results
Since during isolation and purification procedure the outer mitochondrial membrane (OMM) could be damaged, we first analyzed the intactness of the OMM in our mitochondrial preparations. Figure 1 shows measurements of mitochondrial respiration performed with and without exogenous cytochrome c. The lack of cytochrome c effect in these experiments indicated intactness of the OMM in the majority of mitochondria. This was also supported by the fact that isolated brain mitochondria used in our study retained their structural integrity and released minimal amounts of cytochrome c during incubation in the standard incubation medium at 37°C for 30 minutes (Fig. 2). An addition of BAXoligo to mitochondria caused cytochrome c release in a concentration- and time-dependent fashion. The release of cytochrome c became evident with as low as 1.8μg/ml BAXoligo (Fig. 2a). Higher concentrations of BAXoligo produced larger cytochrome c release, culminating at 10.8μg/ml of BAXoligo. At this concentration, BAXoligo released the entire cytochrome c similar to alamethicin, an antibiotic which completely eliminated barrier properties of the OMM [27] and induced maximal cytochrome c release (Fig. 2a). Consistent with this, the amount of cytochrome c remaining in the corresponding mitochondrial pellets appeared to be below the detection limit of western blotting. Here and in other similar experiments, detection of VDAC in the pellets with anti-VDAC antibody ensured equal sample loading. Cytochrome c release induced by 10.8μg/ml of BAXoligo occurred in a time-dependent manner and was completed within 30 minutes (Fig. 2b). Figures 2c and 2f show statistical analyses of cytochrome c release induced by BAXoligo.
Figure 1. Exogenous cytochrome c did not influence respiration of isolated brain mitochondria.
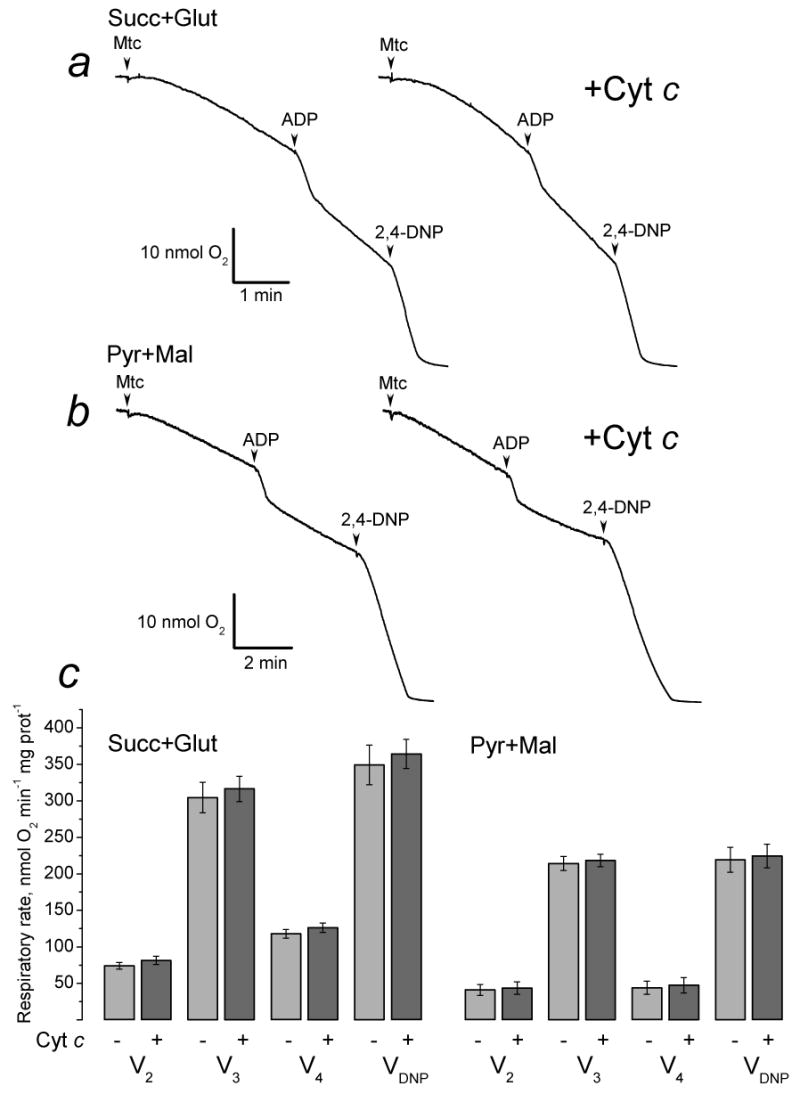
Mitochondria were incubated in the standard incubation medium supplemented either with 3 mM succinate plus 3 mM glutamate (a) or with 3 mM pyruvate plus 1 mM malate (b). Cytochrome c (+Cyt c, 50μM) was present in the media prior to addition of mitochondria. Where indicated, 200μM ADP or 60μM 2,4-dinitrophenol (2,4-DNP) were added. Panel c shows statistical analysis of respiratory rates obtained with and without 50μM cytochrome c.
Figure 2. Recombinant oligomeric BAX (BAXoligo) triggered cytochrome c release from isolated brain mitochondria in a time- and concentration-dependent manner.
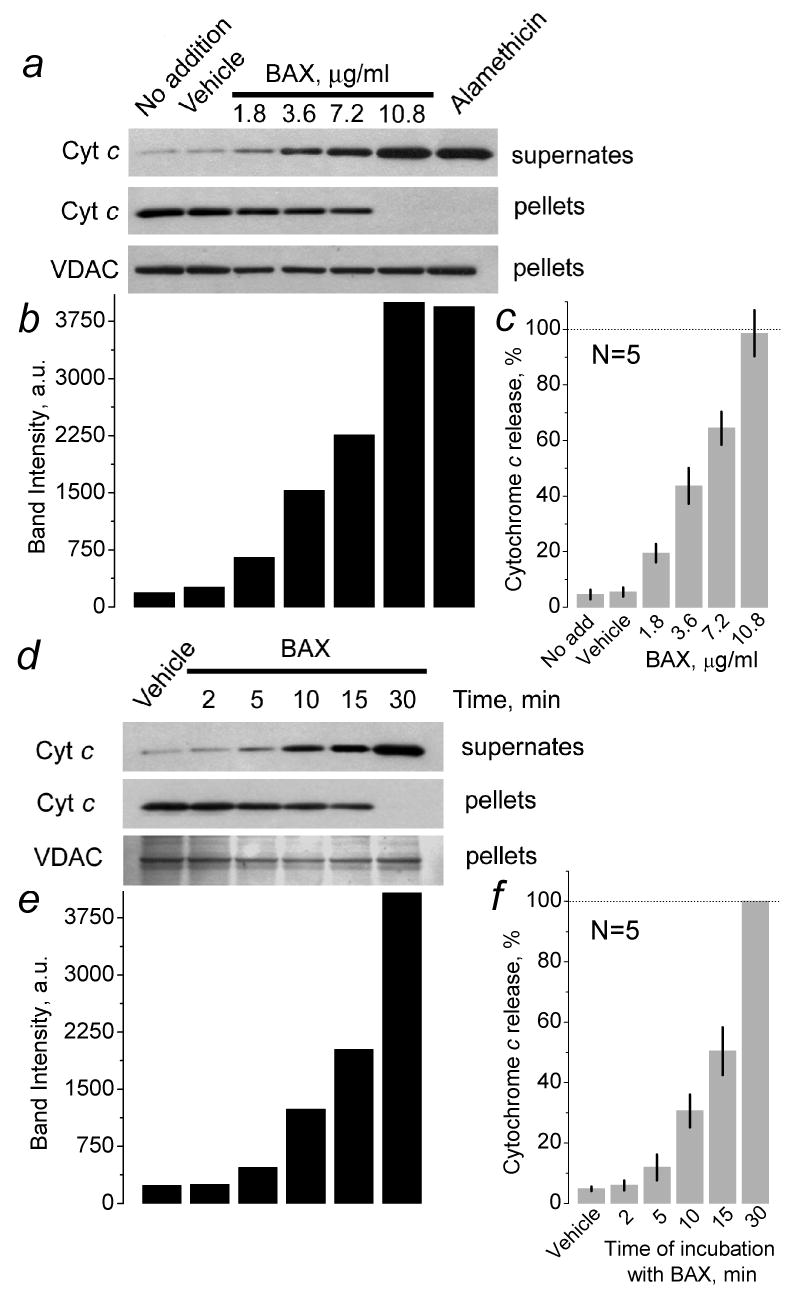
In a, a concentration-dependence of BAXoligo-induced cytochrome c release is shown. Mitochondria were incubated under very gentle shaking for 30 minutes at 37°C with the indicated concentrations of BAXoligo. Here and in other experiments, western blots obtained with solubilized pellets and anti-VDAC antibody were used as loading controls. Here and in other similar experiments, an aliquot (6μl added into 300μl of the incubation medium) of the dialysis buffer containing 1% octyl glucoside was used as a vehicle. In d, a time-dependence of BAXoligo-induced cytochrome c release is shown. 10.8μg/ml BAXoligo was applied to isolated brain mitochondria for the time indicated. In b and e, columns show the results of densitometry produced with Quantity One® software (Bio-Rad Laboratories, Hercules, CA) and expressed in arbitrary units (a.u.) for the corresponding bands. In c and f, the statistical analyses of densitometry results are shown as percentage of cytochrome c release. In c, cytochrome c release with alamethicin was taken as 100%. In f, cytochrome c release after 30 minutes of incubation with BAXoligo was taken as 100%.
In parallel with cytochrome c release, BAXoligo induced a massive release of Smac/DIABLO while Endo G was released neither after BAXoligo nor after alamethicin treatment (Fig. 3). With anti-Omi/HtrA2 antibody we detected faint bands in the supernatants obtained after incubation of mitochondria with BAXoligo or alamethicin (Fig. 3). With anti-AIF antibody, we detected two bands in the supernatants obtained after incubation of mitochondria with BAXoligo and three bands after incubation with alamethicin (Fig. 3). In the experiments with AIF release measurements we incubated mitochondria without BSA because BSA interferes with AIF detection [30]. While the major, thick band detected with the supernatant sample after alamethicin treatment might belong to AIF, the two faint bands detected with the supernatants obtained after incubation of mitochondria with BAXoligo or alamethicin might represent products of AIF cleavage. To estimate the extent of the protein release, the same amount of brain mitochondria used in the “release” experiments was solubilized and analyzed by western blotting. These estimations revealed that the total amount of AIF and Omi/HtrA2 significantly exceeded the amount of these proteins detected in the supernatants after incubation of mitochondria with BAXoligo. Thus, the release of AIF and Omi/HtrA2 induced by BAXoligo was miniscule in comparison with a complete release of cytochrome c and Smac/DIABLO.
Figure 3. Detection of Smac/DIABLO, AIF, Omi/HtrA2 and Endonuclease G (Endo G) in the supernatants obtained after incubation of brain mitochondria with BAXoligo.
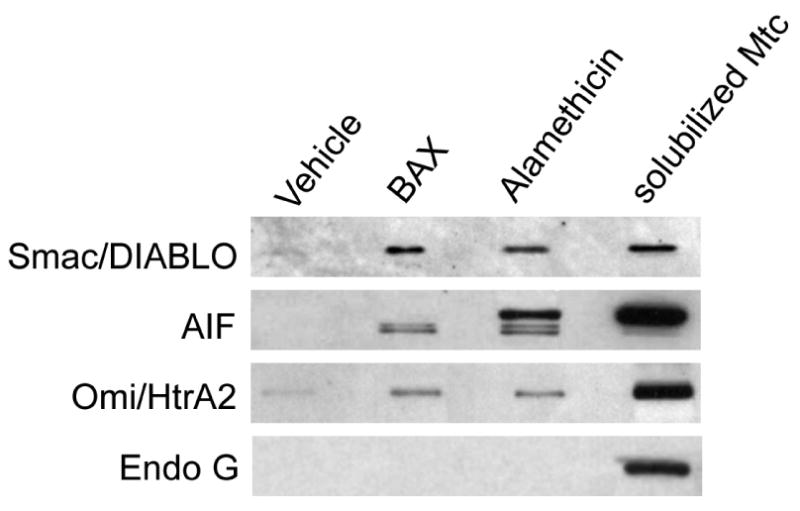
Mitochondria were incubated under very gentle shaking for 30 minutes at 37°C with 10.8μg/ml BAXoligo or with 30μg/ml alamethicin. Endo G was not detected in the supernatant even after incubation of mitochondria with alamethicin. The total amount of Smac/DIABLO, AIF, Omi/HtrA2 and Endo G was determined in the same amount of mitochondria (solubilized Mtc) used in the “release” experiments.
Replacement of the standard KCl-based incubation medium for the low ionic strength, mannitol-sucrose (MS) medium completely prevented BAXoligo-induced cytochrome c release (Fig. 4). Similar results were obtained with alamethicin (not shown). In mannitol-sucrose medium BAX induced mitochondrial swelling and depolarization in CsA, ADP-sensitive manner (Fig. 4b,c). Thus, the lack of cytochrome c release in mannitol-sucrose medium cannot be explained by the absence of the mPT under these conditions. These observations suggest that in brain mitochondria, cytochrome c attachment to the IMM appears to be primarily due to an electrostatic interaction between cytochrome c and the inner mitochondrial membrane (IMM) in agreement with early reports [31,32].
Figure 4. BAXoligo caused complete cytochrome c release in high-ionic strength KCl-based medium but not in low-ionic strength mannitol-sucrose incubation medium. BAXoligo produced mitochondrial swelling and depolarization in mannitol-sucrose medium.
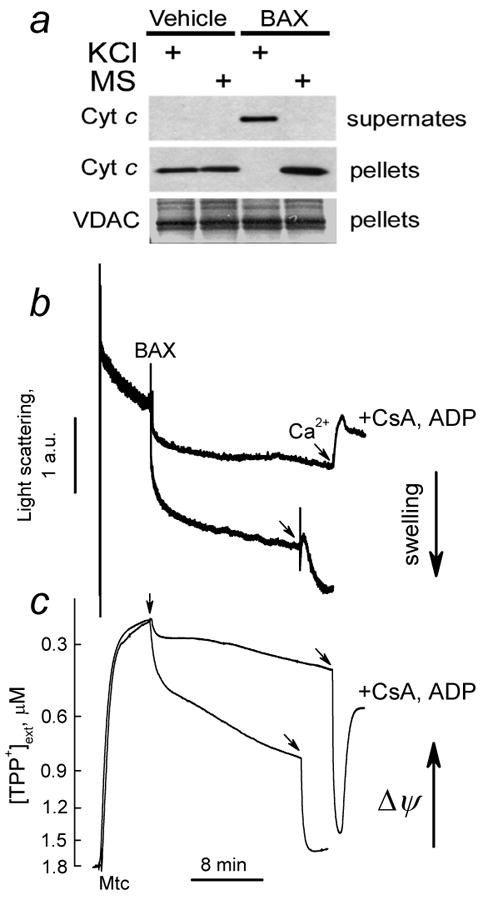
In a, the results of western blotting. Brain mitochondria were incubated with 10.8μg/ml BAXoligo for 30 minutes at 37°C either in the standard 125 mM KCl-based incubation medium or in the incubation medium where KCl was replaced for 215 mM mannitol plus 70 mM sucrose (MS). In b and c, in mannitol-sucrose medium BAXoligo (10.8μg/ml) induced mitochondrial swelling and depolarization in CsA, ADP-sensitive manner. In these experiments mitochondrial swelling and membrane potential were monitored simultaneously by following changes in light scattering at 180° (thick traces) and distribution of TPP+ between external medium and mitochondrial matrix (thin traces). Two pairs of light scattering traces, indicative of changes in mitochondrial volume, and TPP+ traces, indicative of changes in mitochondrial membrane potential, obtained with and without 1μM CsA and 100μM ADP (plus 1μM oligomycin) are overlapped for comparison. At the end of the experiments 267μM Ca2+ was added to induce Ca2+-dependent permeability transition.
The release of cytochrome c from mitochondria incubated with succinate plus rotenone appeared to be smaller than the release from mitochondria fueled with succinate plus glutamate (Fig. 5a). It is known that rotenone suppresses the mPT by maintaining mitochondrial pyridine nucleotides in the reduced state [24,33-35]. Therefore, the mPT might be involved in BAXoligo-induced cytochrome c release from brain mitochondria as it was demonstrated earlier for liver mitochondria [9,36]. Indeed, a combination of CsA and ADP, inhibitors of the mPT [24-26], significantly diminished cytochrome c release induced by BAXoligo (Fig. 5a). Here and in all other experiments, ADP was used in the presence of 1μM oligomycin to prevent ADP phosphorylation. The time-dependence of BAXoligo-induced cytochrome c release in the presence of CsA and ADP is shown in Fig. 5e. In the parallel experiments, CsA and ADP did not influence BAXoligo insertion into the OMM (Fig. 5g) suggesting that BAXoligo insertion did not require the mPT. Of note, the amount of endogenous BAX in naïve brain mitochondria was below the detection limit of western blot (Fig. 5g). ATP, another inhibitor of the mPT [37], also attenuated cytochrome c release induced by BAXoligo (Fig. 5b) but did not influence BAXoligo insertion (not shown). Alamethicin-induced release of cytochrome c was insensitive to mPT inhibitors (Fig. 5a-d). Figures 5b, d, and f show statistical analyses of cytochrome c release. Thus, suppression of BAXoligo-induced cytochrome c release by inhibitors of the mPT suggested involvement of the mPT in this process.
Figure 5. Rotenone, a combination of cyclosporin A and ADP, or ATP inhibited cytochrome c release induced by BAXoligo but did not influence BAX insertion into the outer membrane.
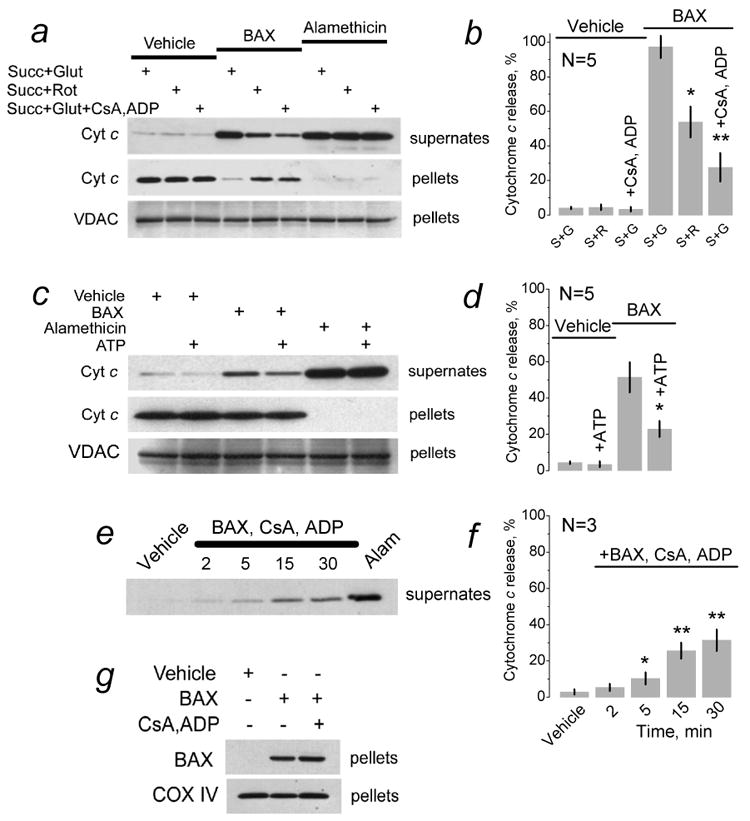
In a, c, and e, brain mitochondria were treated with vehicle (6μl of dialysis buffer for BAXoligo solubilization added into 300μl of the standard incubation medium), with 10.8 (a,e) or 7.2 μg/ml BAXoligo (c), or with 30μg/ml alamethicin (Alam) under very gentle shaking for 30 minutes at 37°C. The incubation medium was supplemented either with 3 mM succinate and 3 mM glutamate (S+G) or with 3 mM succinate and 1μM rotenone (Rot, S+R) as indicated. In addition, where indicated the incubation medium was supplemented with 1μM cyclosporin A (CsA) and 100μM ADP (plus 1μM oligomycin) (panel a,e) or with 1 mM ATP (panel c). In e, numbers show the time of incubation with BAXoligo, CsA, ADP and oligomycin in minutes. Panels b, d, and f show statistical analyses of cytochrome c release in different conditions. In all cases, cytochrome c release with alamethicin was taken as 100%. In b, *p>0.05 between the BAXoligo-induced cytochrome c release in the experiments with succinate plus glutamate versus succinate plus rotenone, N=5; **p>0.01 between the BAXoligo-induced cytochrome c release with and without CsA, ADP, and oligomycin. In d, *p<0.05 between BAXoligo-induced cytochrome c release with and without ATP, N=5. In f, *p<0.05, **p<0.01 between the effect of vehicle and BAXoligo, N=3. In g, mitochondria were incubated under very gentle shaking for 30 minutes at 37°C with 10.8μg/ml BAXoligo. Then, BAXoligo insertion was evaluated as described in Materials and Methods. Where indicated, mitochondria were incubated with 1μM cyclosporin A (CsA) and 100μM ADP (plus 1μM oligomycin). COX IV immunoblots were used as a control for equal loading.
In addition to cytochrome c release, BAXoligo resulted in a large amplitude swelling of brain mitochondria as judged by the decrease in light scattering of mitochondrial suspension measured at 90° to the incident beam (Fig. 6). Alamethicin produced a maximal decrease in light scattering corresponding to the maximal extent of mitochondrial swelling [27]. To compare different light scattering experiments, we assumed the maximal swelling produced by alamethicin as 100% and estimated the extent of swelling produced by BAXoligo under different conditions as a percentage from the maximal swelling (X%) (Fig. 6b). An aliquot of the dialysis buffer used for oligomerization of BAXoligo and containing 1% octyl glucoside failed to produce significant change in light scattering (Fig. 6a). BAXoligo caused the largest decrease in light scattering when mitochondria were fueled with succinate plus glutamate (Fig. 6b), while with succinate plus rotenone the decrease in light scattering was smaller (Fig. 6c). Importantly, a combination of cyclosporin A and ADP suppressed the mitochondrial swelling induced by BAXoligo (Fig. 6d). Similar effect was obtained with 1mM ATP (Fig. 6e). Figure 6f summarizes the results of the light scattering measurements. Thus, BAXoligo induced a large amplitude mitochondrial swelling sensitive to the mPT inhibitors, suggesting mPT involvement.
Figure 6. In brain mitochondria BAXoligo induced large amplitude mitochondrial swelling sensitive to the inhibitors of the mPT.
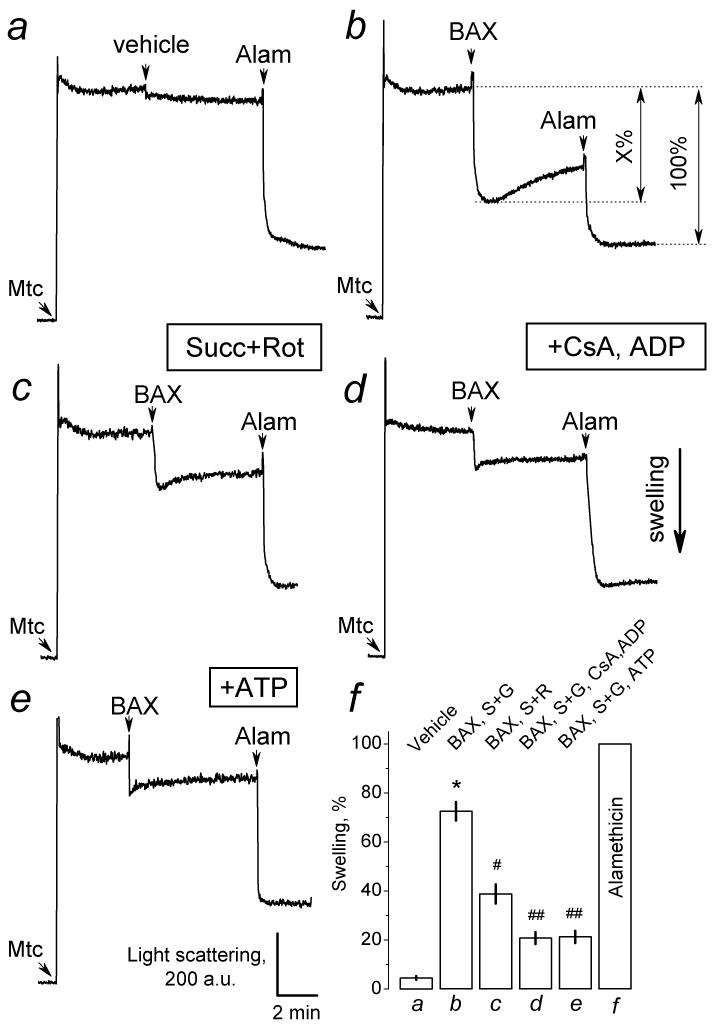
In a, b, d, and e, the standard incubation medium was supplemented with 3 mM succinate plus 3 mM glutamate; in c - with 3 mM succinate plus 1μM rotenone (Rot). In a, 8μl of the dialysis buffer for BAXoligo solubilization (vehicle) were added as indicated into a cuvette with a total volume of 400μl of the incubation medium. In b-e, 10.8μg/ml BAXoligo was added as indicated. In all experiments, 30μg/ml alamethicin was added at the end of recordings to obtain maximal swelling. In d, the incubation medium was supplemented with 1μM cyclosporin A (CsA), 100μM ADP, and 1μM oligomycin. In e, the incubation medium was supplemented with 1 mM ATP. In f, a graph summarizing assessments of mitochondrial swelling is presented. The maximal amplitude of a decrease in light scattering was assumed to reflect the maximal mitochondrial swelling taken as 100% (see in Panel b). The extent of mitochondrial swelling induced by BAXoligo was calculated as a percentage of the maximal swelling. *p>0.01 between the effect of vehicle and BAXoligo in the experiments with succinate plus glutamate; #p>0.05 between the effect of BAXoligo with succinate plus glutamate and with succinate plus rotenone; ##p>0.01 for the effect of BAXoligo with or without CsA, ADP, and oligomycin, and for the effect of BAXoligo with or without ATP (N=3).
To further examine mitochondrial morphological changes, we performed transmission electron microscopy (TEM) with isolated brain mitochondria treated with BAXoligo (Fig. 7). All mitochondria were divided in three morphological classes including condensed (C), swollen (S), and mitochondria with tubular cristae (T) shown in Figure 7a, b, and c respectively. The results of morphometric analysis performed in a blind manner are shown in Figure 7g. A vast majority of organelles treated with the vehicle appeared to be in the condensed state with a significant vacuolization of matrices (Fig. 7d) typical for the isolated brain mitochondria [13,27,38]. Treatment of mitochondria with BAXoligo caused swelling of organelles (Fig. 7e). A few mitochondria had particular matrix structures, which we defined as tubular cristae (Fig. 7c). Pretreatment of mitochondria with mPT inhibitors prevented mitochondrial swelling (Fig. 7f). However, mitochondria did not retain their initial morphology. With mPT inhibitors, the tubular configuration of cristae appeared to be prevalent (Fig. 7f). Thus, BAXoligo caused a dramatic mitochondrial remodeling, which was sensitive to mPT inhibitors and, therefore, might involve the mPT.
Figure 7. The changes in mitochondrial morphology induced by BAXoligo visualized by transmission electron microscopy with isolated brain mitochondria. The effect of cyclosporin A and ADP.
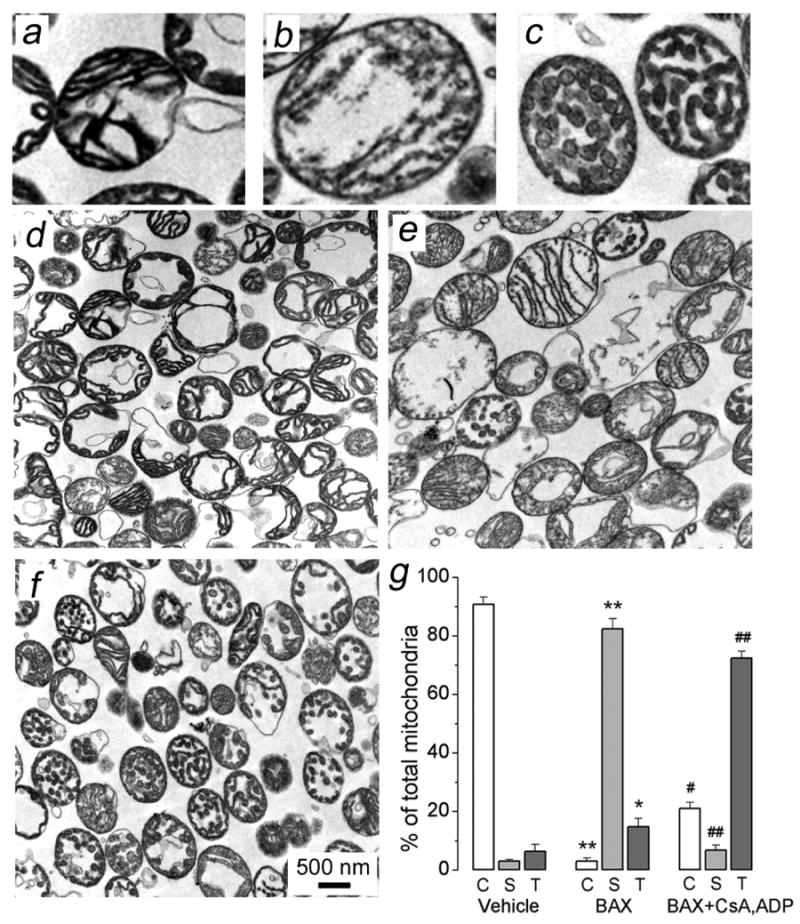
Three distinct types of mitochondrial morphology were identified: condensed (C, representative mitochondria are shown in Panel a), swollen (S, representative mitochondria are shown in Panel b), and mitochondria with tubular cristae (T, representative mitochondria are shown in Panel c). In d, mitochondria were treated with a vehicle (6μl of the dialysis buffer for BAXoligo in 300μl of incubation medium) under very gentle shaking for 30 minutes at 37°C. In e, mitochondria were treated with 10.8μg/ml BAXoligo. In f, mitochondria were pre-treated with 1μM cyclosporin A (CsA) and 100μM ADP plus 1μM oligomycin for 3 minutes prior to the addition of 10.8μg/ml BAXoligo, to which mitochondria were incubated for the next 30 minutes. In g, the results of morphometric analysis are expressed as a percentage from the total number of analyzed mitochondria. Mitochondria were counted in a blind manner, the total number of mitochondria per condition was 550-600. *p>0.05, **p>0.01 for the corresponding types of mitochondria after treatment with vehicle versus BAXoligo; #p>0.05, ##p>0.01 for the corresponding types of mitochondria after treatment with BAXoligo in the presence or absence of CsA, ADP, and oligomycin.
The release of cytochrome c (Fig. 2d-f) occurred much longer after the onset of the mPT induced by BAXoligo (Fig. 6b). To examine whether cytochrome c release correlated with the time course of tubular cristae formation, we performed additional electron microscopy evaluation of mitochondrial morphology over time following BAXoligo addition (Fig. 8). We found that tubular cristae were formed already after 2 minutes of incubation with BAXoligo (Fig. 8c). Then, over time the number of mitochondria with tubular cristae declined and number of swollen mitochondria increased. Thus, BAXoligo-induced cytochrome c release did not correlate with the time course of tubular cristae formation and rather paralleled mitochondrial swelling. However, this does not rule out an important role of tubular cristae formation as a step in structural rearrangement of mitochondria leading to complete cytochrome c release.
Figure 8. Time-dependence of BAXoligo-induced morphological changes in brain mitochondria.
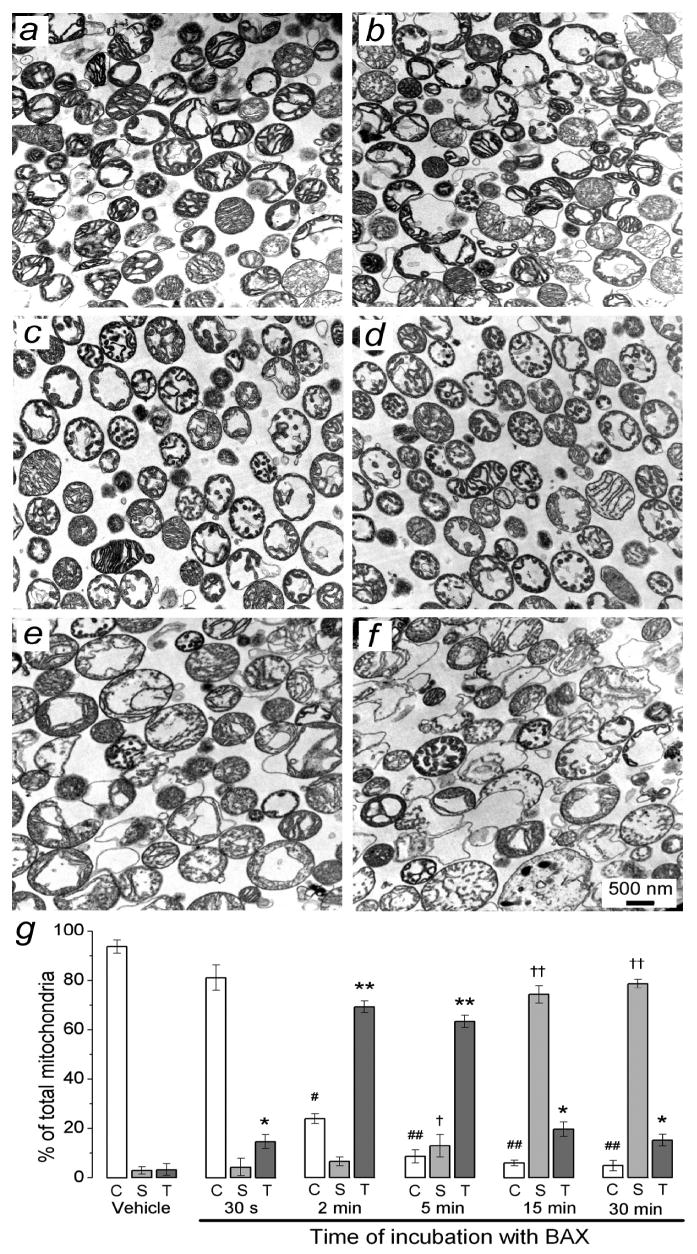
In a, mitochondria were treated with a vehicle (6μl of the dialysis buffer for BAXoligo in 300μl of incubation medium) under very gentle shaking for 30 minutes at 37°C. In b-f, brain mitochondria were fixed after 30 seconds, 2, 5, 15 and 30 minutes of incubation with 10.8μg/ml BAXoligo, respectively. In g, the results of morphometric analysis are expressed as a percentage from the total number of analyzed mitochondria. C, condensed mitochondria; S, swollen mitochondria; T, mitochondria with tubular cristae. Mitochondria were counted similar as described in the legend to Fig. 7. *p>0.05 and **p>0.01, #p>0.05 and ##p>0.01, †p>0.05 and ††p>0.01 for the corresponding types of mitochondria after treatment with vehicle versus BAXoligo.
In addition to the release of cytochrome c and large amplitude swelling, BAXoligo resulted in mitochondrial depolarization in the concentration-dependent manner (Fig. 9a,d,g). In contrast to depolarization induced by a combination of tBID and monomeric BAX [13,14], depolarizations induced by BAXoligo were abrupt and profound. At the end of the experiments, mitochondria were treated with Ca2+ to induce the Ca2+-dependent mPT and completely depolarize organelles. Pretreatment of mitochondria with CsA and ADP (Fig. 6b,e,h) or with ATP suppressed depolarizations induced by BAXoligo (Fig. 6c,f,i). The mPT inhibitors also protected against Ca2+-induced sustained depolarization, but only in the experiments in which Ca2+ was added after low or moderate BAXoligo (3.6 or 7.2μg/ml). With a high BAXoligo (10.8μg/ml), the inhibitors of the mPT failed to preclude sustained depolarization induced by Ca2+ (Fig. 6e,f), probably due to massive loss of cytochrome c (Fig. 2) and impaired capacity of the respiratory chain. Thus, in addition to the cytochrome c release and mitochondrial swelling, brain mitochondria responded to BAXoligo by depolarization, which appeared to be sensitive to mPT inhibitors and, therefore, associated with the induction of the mPT.
Figure 9. In isolated brain mitochondria BAXoligo caused mitochondrial depolarization sensitive to the inhibitors of the mPT.
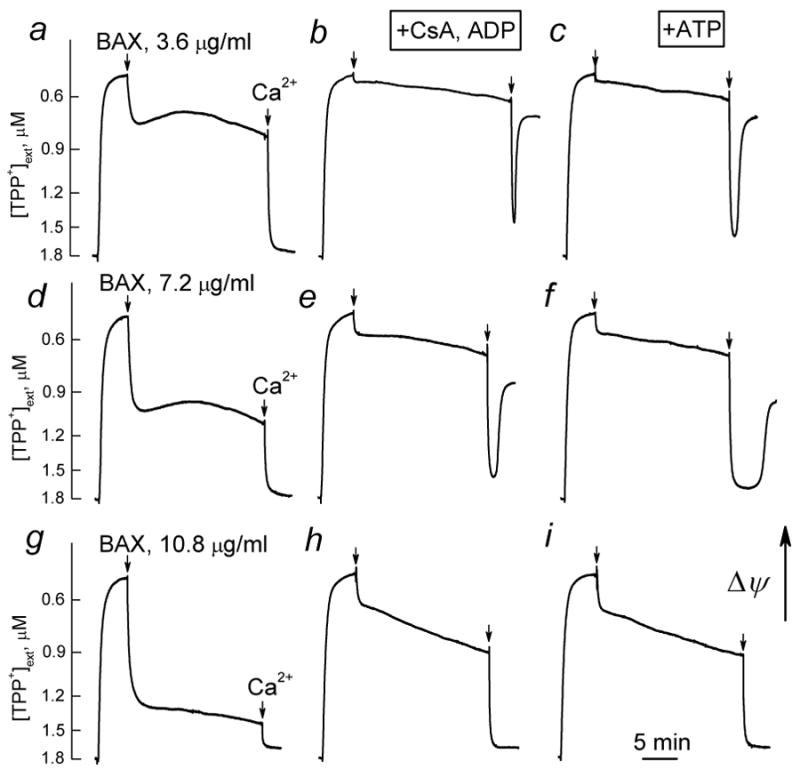
Different concentrations of BAXoligo were added as indicated. At the end of the experiments, 267μM Ca2+ was added to completely depolarize mitochondria. In b, e, and h, the incubation medium was supplemented with 1μM cyclosporin A (CsA), 100μM ADP, and 1μM oligomycin. In c, f, and i, the incubation medium was supplemented with 1 mM ATP. In the control experiments, an aliquot of the dialysis buffer for BAXoligo with 1% octyl glucoside (6μl added into a 300μl of the incubation medium) failed to affect mitochondrial membrane potential (not shown).
The large amplitude swelling of isolated brain mitochondria produced by BAXoligo might lead to the rupture of the OMM, which subsequently would result in a cytochrome c escape from the intermembrane space [27,39]. Alternatively, BAXoligo could specifically permeabilize the OMM. In order to evaluate the role of mitochondrial swelling in the OMM permeabilization, we compared mitochondrial swelling and the release of cytochrome c induced by BAXoligo or a bolus of Ca2+. Previously, we have shown that in the standard 125 mM KCl-based incubation medium, isolated brain mitochondria undergo large amplitude swelling without substantial release of cytochrome c [27]. Similar observation has been made by other investigators with mitochondria isolated from Xenopus eggs [16]. It was concluded that under these circumstances the extent of swelling appeared to be insufficient to rupture the OMM and release cytochrome c [16,27]. We confirmed these findings in the present study. Indeed, with all tested oxidative substrates, Ca2+ produced a significant decrease in light scattering of mitochondrial suspension, indicative of mitochondrial swelling, which was comparable with a decrease in light scattering produced by BAXoligo (compare Figs. 6b,c and 10a-c). This suggested similar swelling of organelles treated with BAXoligo or Ca2+. Indeed, TEM confirmed that a significant fraction of mitochondria treated with Ca2+ appeared to be swollen similar to mitochondria treated with BAXoligo (compare Figs. 7e and 10f). However, Ca2+, in contrast to BAXoligo, did not produce a detectable cytochrome c release (Fig. 10h) while BAXoligo caused a massive release of cytochrome c (Fig. 2). Thus, it seems likely that in addition to mitochondrial swelling and possible rupture of the OMM, which we cannot rule out, BAXoligo causes dramatic permeabilization of the OMM by another as yet unidentified mechanism.
Figure 10. In the standard 125 mM KCl-based incubation medium, Ca2+ induced mitochondrial swelling but not the release of cytochrome c.

In a-d, the results of light scattering assays are shown. Mitochondria were incubated in the media with different oxidative substrates as indicated. 267μM Ca2+ and 30μg/ml alamethicin were added as indicated. The extent of mitochondrial swelling was assessed as described in the legend to Figure 3. In d, *p>0.05 for the effect of Ca2+ in the experiments with succinate plus glutamate and with succinate plus rotenone, N=3. In e-g, the results of transmission electron microscopy are shown. In these experiments, mitochondria were incubated in the standard incubation medium supplemented with 3 mM succinate and 3 mM glutamate. In e and f, mitochondria were incubated under very gentle shaking for 30 minutes at 37°C. In f, mitochondria were treated with 267μM Ca2+. In g, a graph summarizing estimations of mitochondrial swelling similar to those described in the legend to Figure 7. *p>0.01 for the corresponding types of mitochondria prior to and after treatment with Ca2+. In h, western blot analysis of cytochrome c release from mitochondria treated with 267μM Ca2+ or 30μg/ml alamethicin. In i, the statistical analysis of cytochrome c release induced by Ca2+. Cytochrome c release with alamethicin was taken as 100%.
The results presented so far indicate that in isolated brain mitochondria BAXoligo induces cytochrome c release that parallels an induction of the mPT. Ca2+ is the most prominent inducer of the mPT [21]. Without additional testing, we could not rule out that calcium might contaminate BAXoligo preparations used in our experiments. To address this issue we assessed calcium contamination in our BAXoligo preparation using the Ca2+-selective electrode (Fig. 11a). These experiments revealed that BAXoligo preparations used in our experiments did not contain appreciable amounts of Ca2+. Nevertheless, we examined the cytochrome c release induced by BAXoligo in the presence of 1 mM EGTA and did not find any difference with experiments where we used 10μM EGTA (Fig. 11b). Thus, all data obtained with recombinant BAXoligo could be attributed to the action of this protein and to Ca2+ contamination.
Figure 11. Preparation of BAXoligo did not contain appreciable amounts of Ca2+. BAXoligo induced similar cytochrome c release in the eincubation media supplemented with 10μM or 1 mM EGTA.
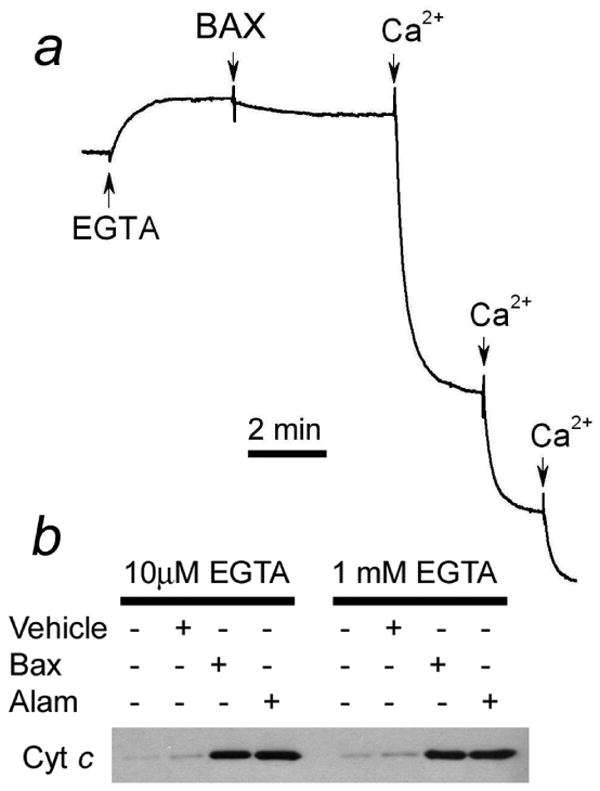
In a, a representative trace obtained with a Ca2+-sensitive electrode. Where indicated, 10μM EGTA, 10.8μg/ml BAXoligo or 25μM Ca2+ were added. In b, cytochrome c release from brain mitochondria induced by 10.8μg/ml BAXoligo or 30μg/ml alamethicin in the incubation media supplemented either with 10μM or with 1 mM EGTA. Respiratory substrates: 3 mM succinate plus 3 mM glutamate.
Earlier, it was proposed that oxidative stress and lipid peroxidation could contribute to BAXoligo-induced cytochrome c release from isolated liver mitochondria [22]. In the following experiments, we addressed the question of whether the intensity of oxidative stress, judged as the rate of ROS generation by mitochondria, correlated with the release of cytochrome c induced by BAXoligo or alamethicin. In mitochondria, superoxide radical O2−·, a primary reactive oxygen species, is converted by Mn-superoxide dismutase into H2O2 which can be easily followed with Amplex Red assay [40]. With succinate as a substrate, mitochondrial generation of ROS is linked to the reverse electron flow from Complex II to Complex I of the respiratory chain and can be effectively inhibited by mild mitochondrial depolarization [41,42]. In our experiments, BAXoligo decreased the rate of ROS generation in a concentration-dependent manner (Fig. 12), according to its ability to depolarize mitochondria (Fig. 9). FCCP and alamethicin produced even stronger suppression of ROS generation. CsA and ADP attenuated inhibition of ROS generation by BAXoligo (Fig. 12), but not by FCCP or alamethicin (not shown). A combination of CsA and ADP attenuated the inhibition of ROS generation by BAXoligo presumably due to protection of Δψ and, therefore, preservation of the reverse electron flow in the respiratory chain. In the presence of mPT inhibitors, ROS generation was high (Fig. 12), but the release of cytochrome c was significantly diminished (Fig. 5). On the other hand, mPT inhibitors failed to affect the inhibition of ROS generation induced by alamethicin (not shown). Thus, in our experiments with isolated brain mitochondria the intensity of oxidative stress and the release of cytochrome c induced by BAXoligo or alamethicin had an inverse correlation. Therefore, it seems unlikely that lipid peroxidation associated with the oxidative stress contributed to the release of cytochrome c from isolated brain mitochondria.
Figure 12. Recombinant BAXoligo, FCCP or alamethicin inhibited ROS generation by isolated brain mitochondria. Cyclosporin A and ADP attenuated the BAXoligo-induced inhibition of ROS generation.
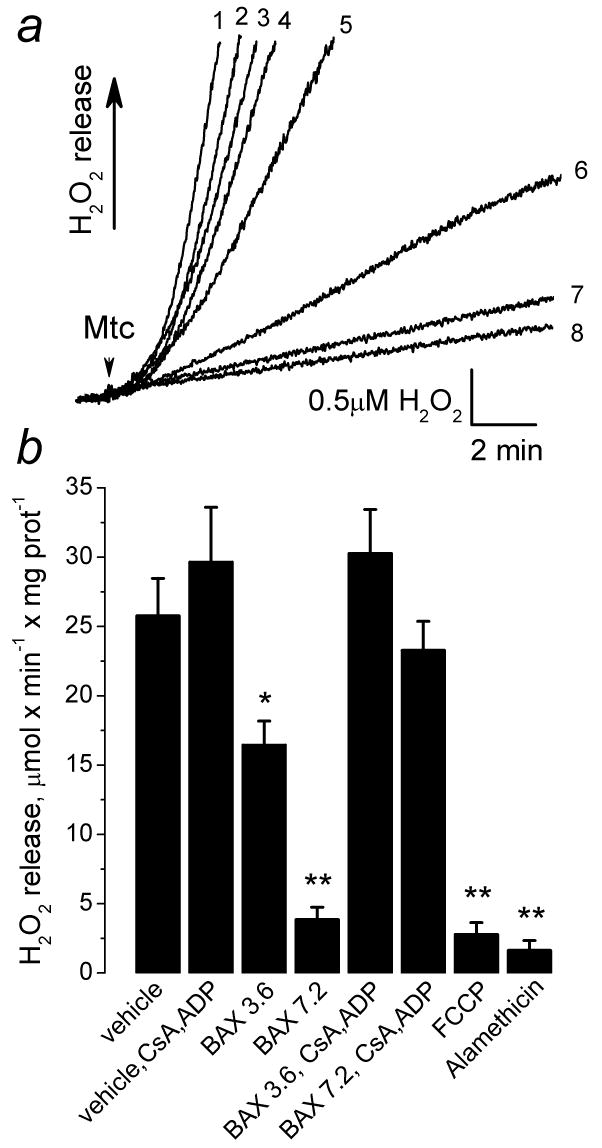
In a, the representative traces obtained under different experimental conditions are shown as: 1 – vehicle plus 1μM cyclosporin A (CsA), 100μM ADP, and 1 μM oligomycin (Oligo); 2 – 3.6μg/ml BAXoligo, CsA, ADP, and Oligo; 3 – vehicle; 4 – 7.2μg/ml BAXoligo, CsA, ADP, Oligo; 5 – 3.6μg/ml BAXoligo; 6 – 7.2μg/ml BAXoligo; 7 – 1μM FCCP; 8 – 30μg/ml alamethicin. All additions were made prior to the mitochondria. 5.5μl of the dialysis buffer for BAXoligo solubilization was used as a vehicle. In b, a graph summarizes the data obtained with the Amplex Red assay under different experimental conditions. The concentration of BAXoligo is given in μg/ml. *p>0.05, **p>0.01 for H2O2 generation after treatment with vehicle versus treatment with BAXoligo or alamethicin, N=3.
Discussion
The release of mitochondrial intermembrane proteins plays a key role in execution of the apoptotic program [43]. The cell-free experimental model of isolated mitochondria in conjunction with the use of recombinant pro-apoptotic proteins proved to be a very helpful tool in the elucidation of these mechanisms. However, most studies have been performed with liver mitochondria [1,9,12,22,36,44] whereas the interaction of pro-apoptotic proteins with isolated brain mitochondria has been less studied [13,14,19,45]. As with any model system, results obtained with isolated mitochondria have some limitations and cannot be applied directly to situation in the cell. However, the experiments with whole cells often suffer from ambiguity based on less than ideal experimental approaches, difficulty to control intracellular environment, and enormous complexity of cell organization at structural and biochemical levels. Therefore, we strongly believe that preparations of isolated purified mitochondria represent an excellent model system, which allows precise control over experimental conditions and, thus, significantly facilitates detailed dissection of molecular mechanisms of mitochondrial involvement into apoptotic program.
The main finding of our study with isolated brain mitochondria is that recombinant BAXoligo induces massive cytochrome c release by the mechanism associated with the mPT but without involvement of oxidative stress. Several different lines of experimental evidence support this conclusion. First, BAXoligo produced cytochrome c release, which was suppressed by inhibitors of the mPT. Second, BAXoligo produced large amplitude mitochondrial swelling, which was also inhibited by mPT inhibitors. Third, BAXoligo caused abrupt and profound mitochondrial depolarization that was antagonized by mPT inhibitors. Fourth, BAXoligo led to the inhibition of ROS generation and mPT inhibitors attenuated inhibition of ROS generation by BAXoligo. Finally, cytochrome c release induced by BAXoligo had an inverse correlation with the rate of ROS generation. Thus, it appeared that in the experiments with brain mitochondria BAXoligo resulted in both the mPT and complete cytochrome c release, which was accompanied by dramatic mitochondrial remodeling and did not require oxidative stress.
In early studies with isolated liver mitochondria, BAXoligo was found to be able to cause cytochrome c release associated with the mPT [9,11,36,46]. Correspondingly, it was hypothesized that cytochrome c release occurred as a result of mitochondrial swelling and the rupture of the OMM. Some investigators proposed that the mPT played a regulatory role in facilitating BAX translocation into the membrane [47] or in promoting a protein-induced efflux of cytochrome c into the intermembrane space by remodeling mitochondrial cristae [12]. On the other hand, a large group of investigators failed to demonstrate a link between the protein-induced cytochrome c release and the mPT [1,13,14,16,19,44,48]. For instance, in rat liver mitochondria, BAXoligo neither induced the mPT nor preventing the mPT with EGTA attenuated cytochrome c release induced by BAXoligo [44]. Later, another group of investigators showed independence of BAXoligo-induced cytochrome c release from the mPT using isolated liver mitochondria from cyclophilin D-knockout mice [49]. Thus, in isolated liver mitochondria, BAXoligo did not induce the mPT, and BAXoligo-induced cytochrome c release from liver mitochondria appeared to be mPT-independent.
The reasons for discrepancies in results from different groups are not clear but variations in the properties of mitochondrial preparations, in the experimental protocols and in the activity of recombinant BAXoligo might contribute to this controversy. The elusive nature of CsA inhibition of the mPT [50] as well as our limited knowledge about molecular composition and the mechanisms of induction of the mPT [51,52] obviously also contribute to this problem. To identify mPT induction in our experiments, we used ATP or a combination of CsA and ADP, which were previously found to be the most effective ways to inhibit the mPT [25,27]. With the use of these inhibitors, we clearly demonstrated that BAXoligo induces mPT in isolated brain mitochondria and that mPT is involved in cytochrome c release induced by BAXoligo.
The mPT is considered a process of induction/activation of the huge proteinaceous pore that increases the permeability of the IMM up to 1.5kDa [37]. Ca2+ is the most prominent inducer of the pore. Although it is known that Ca2+ has to enter mitochondria in order to induce the mPT, the precise Ca2+ targets and the mechanisms leading to the mPT is still not quite clear [21,52]. Ca2+, however, is not the only inducer of the mPT. There are number of agents and factors that can also lead to induction of the mPT in the absence of Ca2+ [10,12,46,53]. In our experiments, BAXoligo produced mitochondrial depolarization and large amplitude swelling of organelles, both typical features of the mPT [54]. These effects of BAXoligo were sensitive to mPT inhibitors. Based on these observations, we concluded that BAXoligo induced mPT in isolated brain mitochondria. Since these experiments were performed in the absence of Ca2+, we concluded that BAXoligo-induced mPT was Ca2+-independent.
An inhibition of the mPT strongly attenuated cytochrome c release induced by BAXoligo in our experiments. Thus, induction of the mPT and mitochondrial remodeling appeared to be important for BAXoligo-induced cytochrome c release. The most straightforward model posits the mPT-associated mitochondrial swelling as a mechanism of rupture of the OMM, leading to escape of cytochrome c from the intermembrane space [39,55,56]. In our experiments, Ca2+ also induced swelling of brain mitochondria, but failed to release cytochrome c. It is conceivable that under these experimental conditions Ca2+-induced swelling was insufficient to rupture the OMM. Contrariwise, BAXoligo produced mitochondrial swelling similar to Ca2+, but resulted in a complete cytochrome c release. This suggested either a larger amplitude of swelling or an additional, more specific mechanism of OMM permeabilization, independent from swelling. Since TEM images of BAXoligo- and Ca2+-treated mitochondria look strikingly similar, the latter explanation seems more likely.
If BAXoligo can permeabilize the OMM independently from swelling, then, the next question is how could an inhibition of the mPT and suppression of swelling diminish the release of cytochrome c? One plausible explanation consists in the assumption that BAXoligo induces mPT-dependent remodeling of mitochondria, manifested in unfolding of mitochondrial cristae, providing opening of the closed spaces limited by cristae and, thus, facilitating escape of cytochrome c. This could be better understood by keeping in mind that intra-cristae regions may contain up to 85% of the total cytochrome c, whereas only about 15% is contained in the intermembrane space [57]. Thus, by wrapping matrix regions, cristae could restrict free diffusion of cytochrome c. This hypothesis was proposed earlier for interaction of tBID with isolated liver mitochondria [12]. In this study, tBID caused distinct mitochondrial remodeling, which could be attenuated by CsA and therefore linked to the mPT [12]. Interestingly, tBID applied to mouse liver mitochondria led to a prevalent appearance of mitochondria with tubular cristae similar to those observed in our experiments with BAXoligo and mPT inhibitors. In our experiments, most of the brain mitochondria treated with BAXoligo in the absence of mPT inhibitors appeared to be swollen and only a few had tubular cristae. It is conceivable that in our experiments an inhibition of the mPT stopped mitochondrial remodeling at the intermediate stage characterized by tubular cristae. Thus, our results argue in favor of the essential role of mitochondrial remodeling in cytochrome c release induced by BAXoligo. Therefore, it seems likely that different factors, which promote the mPT (e.g. Ca2+ and oxidative stress) and hence favor mitochondrial remodeling, could facilitate BAXoligo-induced cytochrome c release while factors, which inhibit the mPT could impede the release of cytochrome c.
Previously, it was hypothesized that cytochrome c bound to the outer surface of the IMM forms two distinct pools [22]. The “loosely bound” cytochrome c appeared to be electrostatically attached to the IMM via interaction with anionic lipids, mainly cardiolipin [58]. In addition, it has been proposed that some cytochrome c molecules are anchored to the lipid membrane due to hydrophobic interactions and, thus, form a pool of “tightly bound” cytochrome c, which represents only about 10% of the total cytochrome c [32]. Peroxidation of cardiolipin might disrupt the interaction between cytochrome c and cardiolipin, increasing the fraction of “loosely bound” cytochrome c [59,60]. It was proposed that “loosening” of cytochrome c might serve as a first step in the process of cytochrome c release from isolated liver mitochondria [22]. Hence, oxidative stress could amplify the release of cytochrome c by increasing its detachment from the membrane. However, oxidative stress could also promote the mPT [37], which we found to be associated with the cytochrome c release induced by BAXoligo in brain mitochondria. The release of cytochrome c induced in our experiments either by BAXoligo or by alamethicin was not accompanied by the increased generation of ROS. On the contrary, the generation of ROS, which could potentially cause lipid peroxidation, was drastically diminished. Nevertheless, alamethicin as well as BAXoligo resulted in a complete cytochrome c release. Since this release proceeded without activation of ROS generation, oxidative stress appeared to play a dispensable role in the BAXoligo-induced release of cytochrome c from brain mitochondria. The experiments with replacement of the standard KCl-based medium for the low ionic strength mannitol-sucrose medium indicated that the attachment of cytochrome c to the IMM is governed primarily by weak electrostatic interactions which could be easily interrupted in high ionic strength KCl-based medium [31,32]. Consequently, it seems likely that the massive release of cytochrome c induced by BAXoligo proceeds by a mechanism involving permeabilization of the OMM accompanied by mPT-dependent mitochondrial remodeling without necessity for oxidative stress-dependent loosening of cytochrome c attachment to the IMM.
Acknowledgments
We gratefully acknowledge Mrs. Caroline Miller and Dr. Vincent Gattone (Electron Microscopy Center, Indiana University School of Medicine) for their help with electron microscopy. This work was supported by the NIH/NINDS R01 NS 050131 to NB.
Abbreviations
- mPT
mitochondrial permeability transition
- OMM
outer mitochondrial membrane
- IMM
inner mitochondrial membrane
- Δψ
mitochondrial membrane potential
- TPP+
tetraphenylphosphonium cation
- CsA
cyclosporin A
- VDAC
voltage-dependent anion channel
- COX IV
cytochrome oxidase subunit IV
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Eskes R, Antonsson B, Osen-Sand A, Montessuit S, Richter C, Sadoul R, Mazzei G, Nichols A, Martinou JC. Bax-induced cytochrome C release from mitochondria is independent of the permeability transition pore but highly dependent on Mg2+ ions. J Cell Biol. 1998;143:217–224. doi: 10.1083/jcb.143.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 3.Eskes R, Desagher S, Antonsson B, Martinou JC. Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol Cell Biol. 2000;20:929–935. doi: 10.1128/mcb.20.3.929-935.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, Thompson CB, Korsmeyer SJ. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000;14:2060–2071. [PMC free article] [PubMed] [Google Scholar]
- 5.Korsmeyer SJ, Wei MC, Saito M, Weiler S, Oh KJ, Schlesinger PH. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000;7:1166–1173. doi: 10.1038/sj.cdd.4400783. [DOI] [PubMed] [Google Scholar]
- 6.Montessuit S, Mazzei G, Magnenat E, Antonsson B. Expression and purification of full-length human Bax alpha. Protein Expression & Purification. 1999;15:202–206. doi: 10.1006/prep.1998.1010. [DOI] [PubMed] [Google Scholar]
- 7.Antonsson B, Montessuit S, Lauper S, Eskes R, Martinou JC. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem J. 2000;345(Pt 2):271–278. [PMC free article] [PubMed] [Google Scholar]
- 8.Pastorino JG, Simbula G, Yamamoto K, Glascott PA, Jr, Rothman RJ, Farber JL. The cytotoxicity of tumor necrosis factor depends on induction of the mitochondrial permeability transition. J Biol Chem. 1996;271:29792–29798. doi: 10.1074/jbc.271.47.29792. [DOI] [PubMed] [Google Scholar]
- 9.Narita M, Shimizu S, Ito T, Chittenden T, Lutz RJ, Matsuda H, Tsujimoto Y. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc Natl Acad Sci U S A. 1998;95:14681–14686. doi: 10.1073/pnas.95.25.14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zamzami N, El Hamel C, Maisse C, Brenner C, Munoz-Pinedo C, Belzacq AS, Costantini P, Vieira H, Loeffler M, Molle G, Kroemer G. Bid acts on the permeability transition pore complex to induce apoptosis. Oncogene. 2000;19:6342–6350. doi: 10.1038/sj.onc.1204030. [DOI] [PubMed] [Google Scholar]
- 11.Tafani M, Karpinich NO, Hurster KA, Pastorino JG, Schneider T, Russo MA, Farber JL. Cytochrome c release upon Fas receptor activation depends on translocation of full-length bid and the induction of the mitochondrial permeability transition. J Biol Chem. 2002;277:10073–10082. doi: 10.1074/jbc.M111350200. [DOI] [PubMed] [Google Scholar]
- 12.Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, Korsmeyer SJ. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell. 2002;2:55–67. doi: 10.1016/s1534-5807(01)00116-2. [DOI] [PubMed] [Google Scholar]
- 13.Brustovetsky N, Dubinsky JM, Antonsson B, Jemmerson R. Two pathways for tBID-induced cytochrome c release from rat brain mitochondria: BAK-versus BAX-dependence. J Neurochem. 2003;84:196–207. doi: 10.1046/j.1471-4159.2003.01545.x. [DOI] [PubMed] [Google Scholar]
- 14.Brustovetsky T, Antonsson B, Jemmerson R, Dubinsky JM, Brustovetsky N. Activation of calcium-independent phospholipase A (iPLA) in brain mitochondria and release of apoptogenic factors by BAX and truncated BID. J Neurochem. 2005;94:980–994. doi: 10.1111/j.1471-4159.2005.03248.x. [DOI] [PubMed] [Google Scholar]
- 15.Kim TH, Zhao Y, Barber MJ, Kuharsky DK, Yin XM. Bid-induced cytochrome c release is mediated by a pathway independent of mitochondrial permeability transition pore and Bax. J Biol Chem. 2000;275:39474–39481. doi: 10.1074/jbc.M003370200. [DOI] [PubMed] [Google Scholar]
- 16.von Ahsen O, Renken C, Perkins G, Kluck RM, Bossy-Wetzel E, Newmeyer DD. Preservation of mitochondrial structure and function after Bid- or Bax-mediated cytochrome c release. J Cell Biol. 2000;150:1027–1036. doi: 10.1083/jcb.150.5.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wigdal SS, Kirkland RA, Franklin JL, Haak-Frendscho M. Cytochrome c release precedes mitochondrial membrane potential loss in cerebellar granule neuron apoptosis: lack of mitochondrial swelling. J Neurochem. 2002;82:1029–1038. doi: 10.1046/j.1471-4159.2002.01049.x. [DOI] [PubMed] [Google Scholar]
- 18.Madesh M, Antonsson B, Srinivasula SM, Alnemri ES, Hajnoczky G. Rapid kinetics of tBid-induced cytochrome c and Smac/DIABLO release and mitochondrial depolarization. Journal of Biological Chemistry. 2002;277:5651–5659. doi: 10.1074/jbc.M108171200. [DOI] [PubMed] [Google Scholar]
- 19.Polster BM, Basanez G, Young M, Suzuki M, Fiskum G. Inhibition of Bax-induced cytochrome c release from neural cell and brain mitochondria by dibucaine and propranolol. J Neurosci. 2003;23:2735–2743. doi: 10.1523/JNEUROSCI.23-07-02735.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Priault M, Cartron PF, Camougrand N, Antonsson B, Vallette FM, Manon S. Investigation of the role of the C-terminus of Bax and of tc-Bid on Bax interaction with yeast mitochondria. Cell Death Differ. 2003;10:1068–1077. doi: 10.1038/sj.cdd.4401270. [DOI] [PubMed] [Google Scholar]
- 21.Rasola A, Bernardi P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis. 2007;12:815–833. doi: 10.1007/s10495-007-0723-y. [DOI] [PubMed] [Google Scholar]
- 22.Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci U S A. 2002;99:1259–1263. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldstein JC, Munoz-Pinedo C, Ricci JE, Adams SR, Kelekar A, Schuler M, Tsien RY, Green DR. Cytochrome c is released in a single step during apoptosis. Cell Death Differ. 2005;12:453–462. doi: 10.1038/sj.cdd.4401596. [DOI] [PubMed] [Google Scholar]
- 24.Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch Biochem Biophys. 1979;195:453–459. doi: 10.1016/0003-9861(79)90371-0. [DOI] [PubMed] [Google Scholar]
- 25.Novgorodov SA, Gudz TI, Milgrom YM, Brierley GP. The permeability transition in heart mitochondria is regulated synergistically by ADP and cyclosporin A. J Biol Chem. 1992;267:16274–16282. [PubMed] [Google Scholar]
- 26.Andreyev AY, Fahy B, Fiskum G. Cytochrome c release from brain mitochondria is independent of the mitochondrial permeability transition. FEBS Lett. 1998;439:373–376. doi: 10.1016/s0014-5793(98)01394-5. [DOI] [PubMed] [Google Scholar]
- 27.Brustovetsky N, Brustovetsky T, Jemmerson R, Dubinsky JM. Calcium-induced cytochrome c release from CNS mitochondria is associated with the permeability transition and rupture of the outer membrane. J Neurochem. 2002;80:207–218. doi: 10.1046/j.0022-3042.2001.00671.x. [DOI] [PubMed] [Google Scholar]
- 28.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 29.Kamo N, Muratsugu M, Hongoh R, Kobatake Y. Membrane potential of mitochondria measured with an electrode sensitive to tetraphenyl phosphonium and relationship between proton electrochemical potential and phosphorylation potential in steady state. J Membr Biol. 1979;49:105–121. doi: 10.1007/BF01868720. [DOI] [PubMed] [Google Scholar]
- 30.Shalbuyeva N, Brustovetsky T, Bolshakov A, Brustovetsky N. Calcium-dependent spontaneously reversible remodeling of brain mitochondria. J Biol Chem. 2006;281:37547–37558. doi: 10.1074/jbc.M607263200. [DOI] [PubMed] [Google Scholar]
- 31.Jacobs EE, Sanadi DR. The reversible removal of cytochrome c from mitochondria. J Biol Chem. 1960;235:531–534. [PubMed] [Google Scholar]
- 32.Cortese JD, Voglino AL, Hackenbrock CR. Multiple conformations of physiological membrane-bound cytochrome c. Biochemistry. 1998;37:6402–6409. doi: 10.1021/bi9730543. [DOI] [PubMed] [Google Scholar]
- 33.Catisti R, Vercesi AE. The participation of pyridine nucleotides redox state and reactive oxygen in the fatty acid-induced permeability transition in rat liver mitochondria. FEBS Lett. 1999;464:97–101. doi: 10.1016/s0014-5793(99)01677-4. [DOI] [PubMed] [Google Scholar]
- 34.Cassarino DS, Parks JK, Parker WD, Jr, Bennett JP., Jr The parkinsonian neurotoxin MPP+ opens the mitochondrial permeability transition pore and releases cytochrome c in isolated mitochondria via an oxidative mechanism. Biochim Biophys Acta. 1999;1453:49–62. doi: 10.1016/s0925-4439(98)00083-0. [DOI] [PubMed] [Google Scholar]
- 35.Chauvin C, Oliveira FDe, Ronot X, Mousseau M, Leverve X, Fontaine E. Rotenone inhibits the mitochondrial permeability transition-induced cell death in U937 and KB cells. J Biol Chem. 2001;276:41394–41398. doi: 10.1074/jbc.M106417200. [DOI] [PubMed] [Google Scholar]
- 36.Pastorino JG, Tafani M, Rothman RJ, Marcinkeviciute A, Hoek JB, Farber JL, Marcineviciute A. Functional consequences of the sustained or transient activation by Bax of the mitochondrial permeability transition pore. J Biol Chem. 1999;274:31734–31739. doi: 10.1074/jbc.274.44.31734. [DOI] [PubMed] [Google Scholar]
- 37.Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
- 38.Hansson MJ, Mansson R, Mattiasson G, Ohlsson J, Karlsson J, Keep MF, Elmer E. Brain-derived respiring mitochondria exhibit homogeneous, complete and cyclosporin-sensitive permeability transition. J Neurochem. 2004;89:715–729. doi: 10.1111/j.1471-4159.2004.02400.x. [DOI] [PubMed] [Google Scholar]
- 39.Petit PX, Goubern M, Diolez P, Susin SA, Zamzami N, Kroemer G. Disruption of the outer mitochondrial membrane as a result of large amplitude swelling: the impact of irreversible permeability transition. FEBS Lett. 1998;426:111–116. doi: 10.1016/s0014-5793(98)00318-4. [DOI] [PubMed] [Google Scholar]
- 40.Adam-Vizi V, Chinopoulos C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol Sci. 2006;27:639–645. doi: 10.1016/j.tips.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 41.Korshunov SS, Korkina OV, Ruuge EK, Skulachev VP, Starkov AA. Fatty acids as natural uncouplers preventing generation of O2·- and H2O2 by mitochondria in the resting state. FEBS Lett. 1998;435:215–218. doi: 10.1016/s0014-5793(98)01073-4. [DOI] [PubMed] [Google Scholar]
- 42.Votyakova TV, Reynolds IJ. DeltaPsi(m)-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. J Neurochem. 2001;79:266–277. doi: 10.1046/j.1471-4159.2001.00548.x. [DOI] [PubMed] [Google Scholar]
- 43.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 44.Gogvadze V, Robertson JD, Zhivotovsky B, Orrenius S. Cytochrome c release occurs via Ca2+-dependent and Ca2+-independent mechanisms that are regulated by Bax. J Biol Chem. 2001;276:19066–19071. doi: 10.1074/jbc.M100614200. [DOI] [PubMed] [Google Scholar]
- 45.Starkov AA, Polster BM, Fiskum G. Regulation of hydrogen peroxide production by brain mitochondria by calcium and Bax. J Neurochem. 2002;83:220–228. doi: 10.1046/j.1471-4159.2002.01153.x. [DOI] [PubMed] [Google Scholar]
- 46.Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HL, Prevost MC, Xie Z, Matsuyama S, Reed JC, Kroemer G. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- 47.De Giorgi F, Lartigue L, Bauer MK, Schubert A, Grimm S, Hanson GT, Remington SJ, Youle RJ, Ichas F. The permeability transition pore signals apoptosis by directing Bax translocation and multimerization. FASEB J. 2002;16:607–609. doi: 10.1096/fj.01-0269fje. [DOI] [PubMed] [Google Scholar]
- 48.Jurgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC. Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci U S A. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 50.Brustovetsky N, Dubinsky JM. Limitations of cyclosporin A inhibition of the permeability transition in CNS mitochondria. J Neurosci. 2000;20:8229–8237. doi: 10.1523/JNEUROSCI.20-22-08229.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Forte M, Bernardi P. Genetic dissection of the permeability transition pore. J Bioenerg Biomembr. 2005;37:121–128. doi: 10.1007/s10863-005-6565-9. [DOI] [PubMed] [Google Scholar]
- 52.Bernardi P, Krauskopf A, Basso E, Petronilli V, Blalchy-Dyson E, Di Lisa F, Forte MA. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006;273:2077–2099. doi: 10.1111/j.1742-4658.2006.05213.x. [DOI] [PubMed] [Google Scholar]
- 53.Zoratti M, Szabo I, De Marchi U. Mitochondrial permeability transitions: how many doors to the house? Biochim Biophys Acta. 2005;1706:40–52. doi: 10.1016/j.bbabio.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 54.Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- 55.Skulachev VP. Why are mitochondria involved in apoptosis? Permeability transition pores and apoptosis as selective mechanisms to eliminate superoxide-producing mitochondria and cell. FEBS Lett. 1996;397:7–10. doi: 10.1016/0014-5793(96)00989-1. [DOI] [PubMed] [Google Scholar]
- 56.Scarlett JL, Murphy MP. Release of apoptogenic proteins from the mitochondrial intermembrane space during the mitochondrial permeability transition. FEBS Lett. 1997;418:282–286. doi: 10.1016/s0014-5793(97)01391-4. [DOI] [PubMed] [Google Scholar]
- 57.Bernardi P, Azzone GF. Cytochrome c as an electron shuttle between the outer and inner mitochondrial membranes. J Biol Chem. 1981;256:7187–7192. [PubMed] [Google Scholar]
- 58.Demel RA, Jordi W, Lambrechts H, van Damme H, Hovius R, de Kruijff B. Differential interactions of apo- and holocytochrome c with acidic membrane lipids in model systems and the implications for their import into mitochondria. J Biol Chem. 1989;264:3988–3997. [PubMed] [Google Scholar]
- 59.Nomura K, Imai H, Koumura T, Kobayashi T, Nakagawa Y. Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. Biochem J. 2000;351:183–193. doi: 10.1042/0264-6021:3510183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Petrosillo G, Ruggiero FM, Pistolese M, Paradies G. Reactive oxygen species generated from the mitochondrial electron transport chain induce cytochrome c dissociation from beef-heart submitochondrial particles via cardiolipin peroxidation. Possible role in the apoptosis. FEBS Lett. 2001;509:435–438. doi: 10.1016/s0014-5793(01)03206-9. [DOI] [PubMed] [Google Scholar]