

Abstract
The signal transducers and activators of transcription (STAT) proteins are a group of transcriptional factors. Among them, STAT5 initiates a pro-survival signaling cascade. So far, little has been known about the role of STAT5 in cerebral ischemia and reperfusion. This study examines the phosphorylation status of STAT5 in hippocampal CA1 in the early stage after transient global cerebral ischemia in rats. Our data show that the phosphorylation of STAT5 was increased in hippocampal CA1 at 1 hour and 3 hours ischemia. Taking advantage of the neuroprotective effect of erythropoietin (EPO) in CA1, we further demonstrated that the administration of EPO enhanced the phosphorylation of STAT5, with SATA5a being phosphorylated earlier. The enhanced phosphorylation of STAT5 in the EPO-treated group was accompanied by the upregulation of STAT5 downstream gene products, Bcl-xL and XIAP. Consequently, ischemic CA1 neuronal damage was attenuated by the administration of EPO. Both the enhancement of STAT5 phosphorylation and the neuroprotection rendered by EPO were blocked by Tyrphostin, a selective inhibitor for Janus kinase 2, which is an upstream kinase of STAT5. These findings suggest an association between the activation of STAT5 and CA1 neuronal survival after cerebral ischemia.
Keywords: STAT5, CA1, ischemia, EPO
Introduction
The signal transducers and activators of transcription (STAT) proteins are a group of transcriptional factors. So far, seven STAT members have been described: STAT1, 2, 3, 4, 5a, 5b and 6. Each STAT protein has three major structures: a DNA binding domain, a src-homology (SH2) domain for dimerization, and a conserved tyrosine residue which can be phosphorylated by Janus protein kinases (Battle and Frank, 2002, Paukku and Silvennoinen, 2004). This group of transcription factors responds rapidly to stimuli, because once activated through phosporylation, they dimerize and translocate into the nucleus without the involvement of second messengers (Aaronson and Horvath, 2002). The classical function of STATs is to mediate the signaling pathways of cytokines and growth factors. Some of the STAT members also are reported to be able to regulate the processes of apoptosis (Battle and Frank, 2002). While other STATs can mediate pro-apoptotic or anti-apoptotic signals depending on the conditions of cellular stimulation, STAT5 only demonstrates a pro-survival signal (Debierre-Grockiego, 2004).
STAT5 is phosphorylated at tyrosine 694 by Janus kinase 2 (JAK2) and thus gains the ability of translocation to the nucleus and the activity of binding to specific DNA sequences in the promoters of responsive genes to enhance gene transcription (Battle and Frank, 2002, Paukku and Silvennoinen, 2004). STAT5 was originally recognized as a transcription factor that regulates the β-casein gene in response to prolactin, but STAT5 is also activated by several other cytokines and growth factors, such as erythropoietin (EPO), growth hormone, and interleukins (Gobert et al., 1996, Battle and Frank, 2002, Debierre-Grockiego, 2004, Paukku and Silvennoinen, 2004). STAT5 is reported to play an important anti-apoptotic role in heart induced by cardiac ischemia (Yamaura et al., 2003), in endothelial cells induced by hypoxia (Dudley et al., 2005), and in the survival of hematopoietic cells (Battle and Frank, 2002, Debierre-Grockiego, 2004) and neuroblastoma SH-SY5Y cells (Um and Lodish, 2006). Downstream products of STA5 related to the regulation of apoptosis include Bcl-xL (Socolovsky et al., 1999) and the X-linked inhibitor of apoptosis protein (XIAP) (Mohapatra et al., 2003). Both Bcl-xL and XIAP demonstrate anti-apoptotic effects in ischemic brain (Zhang et al., 2004).
Erythropoietin is the major hormone that stimulates and regulates the production of red blood cells; in addition to its hematopoietic role, recent studies have established EPO as a potent neuroprotective peptide against ischemic brain injury (Sakanaka et al., 1998, Calapai et al., 2000, Catania et al., 2002, Chong et al., 2002, Wen et al., 2002). EPO is also demonstrated to be able to promote the phosphorylation and activation of STAT5 through JAK2 (Damen et al., 1995, Gobert et al., 1996, Parganas et al., 1998, Siren et al., 2001, Sola et al., 2005). Following cerebral ischemia, apoptosis is an important cell death mechanism, especially in the injury of hippocampal CA1 neurons induced by global ischemia (Zhang et al., 2004). Although STAT5 protein has been demonstrated to be expressed in the hippocampus and cortex of both embryonic and adult rat brain (De-Fraja et al., 1998, De-Fraja et al., 2000), it remains unclear whether STAT5 plays a role in the regulation of cell death following cerebral ischemia.
The purpose of this study is to determine the status of STAT5 phosphorylation in hippocampal CA1 in the early phase following global cerebral ischemia in rats, induced by an ischemic model of four vessel-occlusion (4VO). This model mimics cardiac arrest and resuscitation, and is characterized by the selective and delayed cell death of hippocampal CA1 neurons. In addition, we also determine whether the administration of EPO enhances the phosphorylation of STAT5 in CA1; if so, whether the enhanced phosphorylation of STAT5 in CA1 mediates the neuroprotective effect of EPO; and the potential mechanism of STAT5’s anti-apoptotic role in ischemic CA1.
Material and Methods
The rat model of global ischemia and EPO infusion
All animal experiments were approved by and carried out in accordance with the NIH Guide for the Care and Use of Laboratory Animals using male Sprague-Dawley rats (300 to 330 g, Hilltop Lab Animals, Scottdale, PA). Anesthesia was induced with 5% isoflurane and maintained with 2% isoflurane in a mixture of 30% O2 and 70% N2O through intubation and mechanical ventilation. Transient global ischemia was induced for 12 min followed by reperfusion using a previously described four-vessel occlusion method (Chen et al., 1998, Cao et al., 2002). Blood pressure, blood gases, and blood glucose concentration were monitored and remained in the normal range throughout the experiments through left femoral artery catheterization. Rectal temperature was continually monitored and kept at 37–37.5°C using a heating pad and a temperature-regulated heating lamp. Brain temperature was monitored using a 29-Ga thermocouple implanted in the left caudate-putamen and kept at 35.8°C ± 0.2°C during ischemia. Electroencephalograms were monitored in all rats to ensure isoelectricity within 10 seconds after the induction of ischemia (PowerLab, ADInstruments, Colorado Springs, CO). Sham operations were performed in additional animals using identical anesthetic and surgical procedures, except that the common carotid arteries were not occluded.
Recombinant human EPO (rhEPO, R&D Systems, Minneapolis, MN) was prepared in 0.1% BSA in PBS at a concentration of 5 U/μl and infused using a 10-μl Hamilton syringe into the right intracerebral ventricle (ICV) using the following coordinates from bregma: anteroposterior, −0.8 mm; lateral, 1.5 mm; depth, 3.5 mm. In selected experiments, rats were subjected to ICV infusion of both EPO and JAK2 inhibitor Tyrphostin AG490 (Sigma). Infusions of agents were performed 20 min after ischemia using procedures described previously (Chen et al., 1998).
Histology
For histological analysis with hematoxylin stain, animals were grouped randomly with 8–10 rats in each group. Three days after 12 min of global ischemia, the animals were sacrificed, and the brains removed and frozen in isopentane cooled by dry ice. In the EPO-treated group, 50 U of rhEPO was injected into the right ICV of rats 20 min after ischemia; the same volume of vehicle was infused in control group. Coronal sections of 20 μm in thickness were cut from fresh-frozen brains at the level of dorsal hippocampus and stained with hematoxylin. The total numbers of healthy neurons in the CA1 regions were counted microscopically by two investigators blind to the experimental conditions.
Detection of DNA fragmentation by nick-translation
The DNA polymerase I-mediated biotin-dATP nick-translation (PANT) assay (Nagayama et al., 2000) was performed on fresh-frozen brain sections to detect single-strand DNA breaks. The sections were prepared as described previously. Then sections were air-dried, fixed with 10% formalin for 10 min, and washed three times in PBS. The sections were then fixed in ethanol/acetic acid (2:1, vol/vol) for 5 min and washed three times in PBS. After the sections were permeabilized with 1% Triton X-100 for 20 min and quenched with 2% H2O2 for 15 min, they were washed three times in PBS. Then, sections were incubated in a moist-air chamber at 37°C for 90 min with a PANT reaction mixture containing 5 mM MgCl2, 10 mM 2-mercaptoethanol, 20 μg/ml bovine serum albumin, dGTP, dCTP, and dTTP at 30 μM each, 29 μM biotinylated dATP, 1 μM dATP, and 40 U/ml Escherichia coli DNA polymerase I (Sigma, Woodlands, TX) in PBS (pH 7.4). The reaction was terminated by two PBS washes. After washing for 5 min in PBS containing bovine serum albumin (0.5 mg/ml), the slides were incubated with streptavidin-horseradish peroxidase (Vectastain Elite ABC, Vector Laboratories, Burlingame, CA) in PBS containing bovine serum albumin for 90 min at room temperature. Detection of the biotin-streptavidin-peroxidase complex was carried out by incubating the sections with 0.5 mg/ml 3, 3′-diaminobenzidine in PBS (pH 7.4) and 0.05% H2O2. To determine nonspecific labeling, selected sections were incubated in the reaction buffer without DNA polymerase I. The total numbers of ISEL-positive neurons in the CA1 regions were counted microscopically by two investigators blind to the experimental conditions.
Western blot analysis
Western blotting was performed using the standard method previously described (Cao et al., 2003). Rats were sacrificed at indicated time points after 12 min global ischemia, or 24 hr after sham operation (n=4 per experimental condition, unless otherwise indicated). In EPO-treated groups, 50 U of rhEPO was injected into the right ICV 20 min after the ischemia. The whole hippocampus from each side of the brain was dissected out and placed on a smooth surface, then the CA1 region was carefully separated from the rest of hippocampal residual as described previously (Elmer et al., 1998). The CA1 region of the hippocampus was then homogenized in cell lysis buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, and 1 μg/ml leupeptin. The protein samples were then sonicated, and the total protein extracts were subjected to Western blot analysis using standard methods (Cao et al., 2003). Blots were probed with antibodies recognizing STAT5, phospho-STAT5 (p-STAT5) at tyr694; Bcl-xL; caspase-9; XIAP (Cell Signaling Technology, Beverly, MA); and EPO receptor (Santa Cruz Biotechnology, Santa Cruz, CA). After incubation with the appropriate secondary antibodies conjugated with horseradish peroxidase (Santa Cruz Biotechnology), positive bands were visualized through incubating membranes with LumiGLO chemiluminescent substrate (Kirkegaard & Perry Laboratories, Inc., Gaithersburg, MD), and then the membranes were exposed to X-ray films. Gel analysis was accomplished with the assistance of a computerized scanning program (MCID; Imaging Research Inc., St. Catharines, Ontario, Canada). To determine the nuclear translocation of p-STAT5, the hippocampi were isolated, and the CA1 regions were dissected. Nuclear protein was extracted as described previously (Cao et al., 2003), and analysized by Western blot.
Immunoprecipitation
Immunoprecipitation was performed to measure the expression of p-STAT5a and p-STAT5b separately after ischemia, using a procedure described previously (Cao et al., 2002) and followed by Western blots. Briefly, total protein was isolated from the CA1 sector of the hippocampi at 0.5, 1 and 3 hr after ischemia (n = 4) using the cell lysis buffer described previously. Equal amounts of protein (150 μg per sample) from each experimental condition were subjected to immunoprecipitation using the rabbit anti-STAT5a and -STAT5b antibodies (Zymed Laboratories, Inc., South San Francisco, CA). The resulting immunoprecipitates were then analyzed by immunoblotting with anti-phospho-STAT5 at tyr694 antibody (Cell Signaling, Danvers, MA). Gel analysis was processed with the assistance of MCID.
Statistical analysis
All data were presented as mean values ± standard error. Statistical significance among means was assessed by ANOVA followed by post hoc Scheffe’s tests, with p< 0.05 considered statistically significant.
Results
The phosphorylation of STAT5 was increased in CA1 following global cerebral ischemia
We first confirmed the expression of STAT5 in hippocampal CA1 regions of rats by Western blots. As shown in Figure 1A, STAT5 was detected in CA1 regions. To investigate the phosphorylation level of STAT5 in hippocampal CA1 in the early phase following cerebral ischemia, we employed transient global cerebral ischemia induced by 4-vessel occlusion in rats. Whole-cell protein extracts were prepared from the CA1 of hippocampi at various time points after 12 min of global ischemia or after sham operation. Immunoblot analysis was performed using antibodies against phosphorylated (p-) forms of STAT5 at tyr694 and nonphosphorylated STAT5. As shown in Figure 1A, the p-STAT5 level was detectable in CA1 in sham-operated rats; following ischemia, the level of p-STAT5 was increased at 1 and 3 hr, and then subsided to basal level after 6 hr of reperfusion. Densitometric analysis, shown in Figure 1B, demonstrated that the relative immunoreactivities for p-STAT5 were significantly increased at 1 hr after reperfusion, peaking at 3 hr and returning to sham control levels by 6 hr. There was no significant change in the level of total STAT5 in hippocampal CA1 following global cerebral ischemia.
Figure 1. Increased phosphorylation of STAT5 in rat hippocampal CA1 in the early stage following global cerebral ischemia.
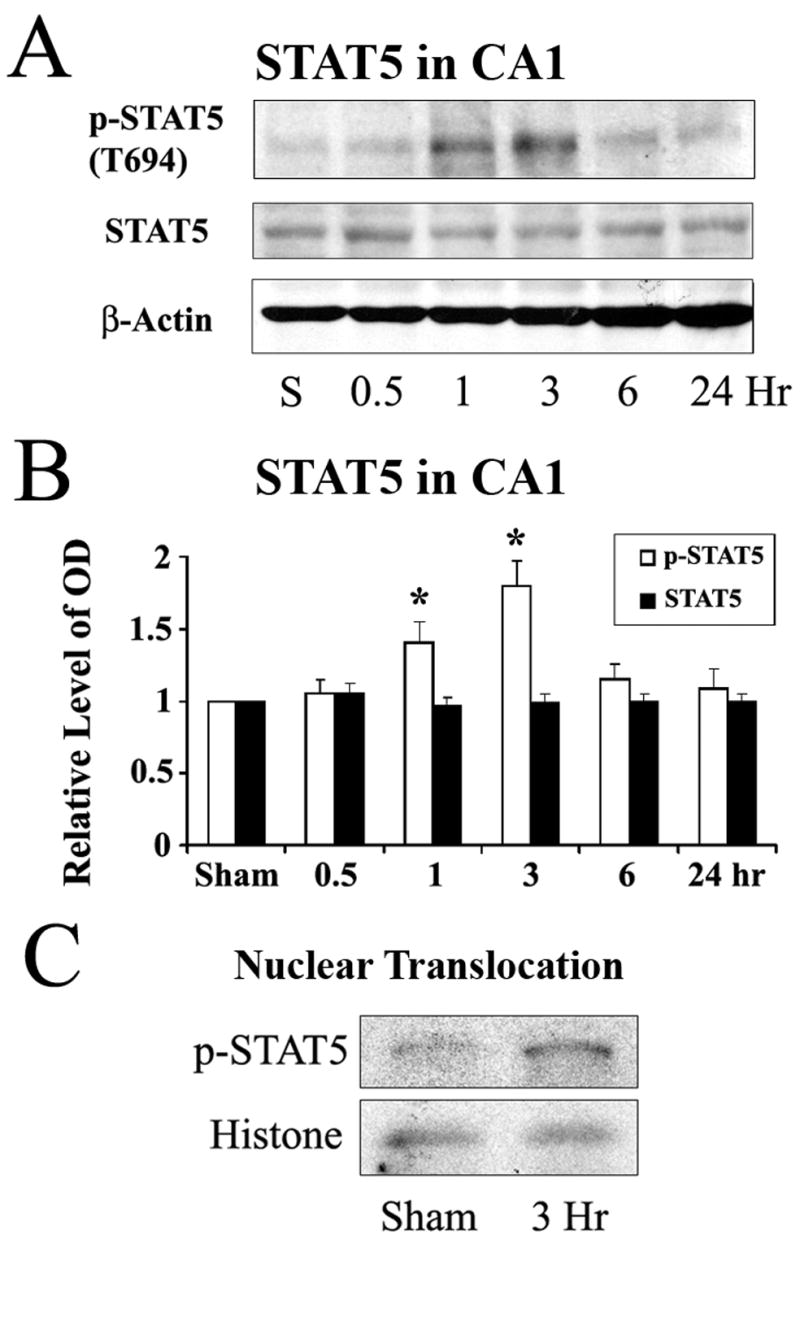
(A) Representative Western blots showing the levels of phosphorylated and total STAT5 in rat hippocampal CA1 at serial time points following ischemia. (B) The average levels of p-STAT5 in the CA1 region determined by Western blot were increased at 1 hr and 3 hr following ischemia compared to the sham-operated group. Data are mean ± SEM, assessed by ANOVA and post hoc Scheffe’s tests. *P<0.05 vs. the sham-operated. (C) Representative Western blot with nuclear preparation of hippocampal CA1 demonstrating increased nuclear translocation of p-STAT5 at 3 hr after ischemia.
As a transcriptional factor, p-STAT5 needs to be translocated into nucleus to bind a special sequence of DNA. We next detected whether the phosphorylated form of STAT5 was translocated into nuclei. The nuclear fraction of CA1 neurons was prepared from sham-operated rats and ischemic rats at 3 hr after global ischemia; the preparation was then analyzed by Western blot. As shown in Figure 1C, there was an increased nuclear distribution of p-STAT5 at 3 hr after ischemia in CA1, parallel with the change in p-STAT5 following ischmia, indicating that the phosphorylated form of STAT5 was translocated into nuclei.
The administration of EPO further enhanced the phosporylation of STAT5 in CA1 following global cerebral ischemia
Based on the concept that EPO has a neuroprotective effect against ischemic injury, EPO activates STAT5, and STAT5 upregulates anti-aopototic downstream gene products, we hypothesized that p-STAT5 levels in CA1 may be further enhanced by the administration of exogenous EPO, and that the enhanced phosphorylation of STAT5 may contribute to the neuroprotective effects of EPO.
To address our hypotheses, we first determined whether the EPO receptor was expressed in CA1 neurons and if the level of EPO receptor was changed following ischemia. As shown in Figure 2A, EPO receptor was expressed in CA1 regions at a readily detectable level, and there was no significant change in the level of EPO receptor within the first 24 hr after ischemia. We next determined if the administration of EPO enhances the phosphorylation of STAT5 in CA1 following global cerebral ischemia. Fifty Units of rhEPO in 10 μl of vehicle was infused into the ICV of rat brains 20 min after ischemia; then the CA1 sectors were isolated at 1 hr and 3 hr after ischemia. In another set of rats, only 10 μl of vehicle was infused into the ICV 20 min after ischemia, and the CA1 sectors were also isolated at the same time points as in EPO-treated animals. Whole-cell protein was extracted from the CA1, and immunoblot analysis was performed. As shown in Figures 1B and 1C, the increases in p-STAT5 levels at 1 and 3 hr following ischemia were further enhanced by the administration of 50 U of EPO on the basis of already existing increases in p-STAT5 levels in vehicle-treated animals. The differences between EPO- and vehicle-treated groups were significant at both 1 hr and 3 hr after ischemia (Figures 2B and 2C). No significant difference in the level of total STAT5 was detected between the EPO group and vehicle group. These findings suggest that the level of p-STAT5 in hippocampal CA1 was increased at 1 hr and peaked at 3 hr following global cerebral ischemia in rats, and that the administration of EPO further enhanced the phosphorylation of STAT5 and nuclear translocation of p-STAT5 in CA1.
Figure 2. Erythropoietin further enhances the phosphorylation of STAT5 in hippocampal CA1 during the early stage after global cerebral ischemia in rats.
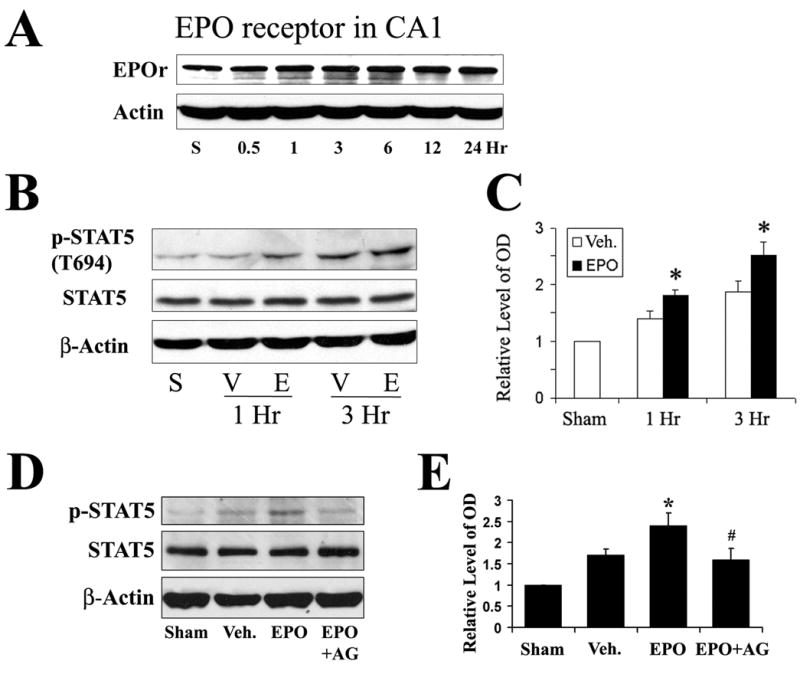
(A) Representative Western blots showing the expression of EPO receptor in rat CA1 regions. (B) Representative Western blots showing the protein levels of phosphorylated and total STAT5 in the CA1 at early time points following ischemia from S (sham-operated), V (vehicle-treated ischemia) and E (EPO-treated 20 min after ischemia) groups. (C) The average levels of phospho-STAT5 in the CA1 region determined by Western blot were increased in the EPO-treated ischemia group at 1 and 3 hr following global ischemia compared to the vehicle-treated ischemia group. Data are mean ± SEM, assessed by ANOVA and post hoc Scheffe’s tests. *P<0.05 vs. vehicle-treated ischemia group at the same time point. (D) Representative Western blots showing protein levels of phosphorylated and total STAT5 in the CA1 region at 3 hr following ischemia. Sham, sham-operated animal; Veh., vehicle-treated ischemia; EPO, EPO treatment (50 Units, 20 min after ischemia); EPO+AG, EPO and AG490-treated ischemia (50 Units of EPO and 2.5 μg of AG490, 20 min after ischemia). (E) Western blots of phospho-STAT5 in CA1 at 3 hr after global ischemia were quantified, showing that AG490 blocked the EPO-enhanced phosphorylation of STAT5. Data are mean ± SEM, assessed by ANOVA and post hoc Scheffe’s tests. *P<0.05 vs. vehicle group, # P<0.05 vs. EPO group, >0.05 vs. vehicle group.
EPO-induced activation of STAT5 is mediated by JAK2, through phosphorylating tyrosine 343 on the EPO receptor, and then recruiting and phosphorylating STAT5 (Damen et al., 1995, Parganas et al., 1998, Digicaylioglu and Lipton, 2001). To further determine the role of STAT5, we inhibited the activation of JAK2, the upstream pathway of STAT5, using a selective JAK2 inhibitor Tyrphostin AG490. AG490 (2.5 μg in 5 μL DMSO/PBS) was infused into the ICV immediately following the infusion of EPO. The animals were sacrificed at 3 hr after ischemia, and protein extracts from the CA1 region were analyzed using Western blot. While the level of p-STAT5 increased following ischemia alone, EPO further increased the amount of p-STAT5 to an even higher level. Inhibition of JAK2 using AG490 returned the EPO-enhanced p-STAT5 levels to those of the ischemic animals (Figures 2D-2E).
Different phosphorylation levels of STAT5a and STAT5b were found in CA1 following global cerebral ischemia
STAT5 proteins are encoded by two distinct but closely related genes, stat5a and stat5b (Liu et al., 1995, Debierre-Grockiego, 2004). STAT5a and STAT5b proteins share 96% identity, with the difference in their C-terminals, and both of them can be phosphorylated at tyrosine 694 (Ehret et al., 2001, Battle and Frank, 2002, Debierre-Grockiego, 2004, Paukku and Silvennoinen, 2004). There is, however, a difference between the functions of STAT5 and STAT5b, as demonstrated by the single and double knockout studies of STAT5 (Teglund et al., 1998, Park et al., 1999, Zhang et al., 2000, Debierre-Grockiego, 2004), and it appears that only STAT5a is required for ischemic tolerance in heart (Yamaura et al., 2003).
To detect the phosphorylation levels of STAT5a and STAT5b in CA1 separately following global ischemia and treatment with EPO, we performed immunoprecipitation with STAT5a and STAT5b antibodies, followed by Western blots with anti-phospho-STAT5 antibody. The antibody against p-STAT5 recognizes the phosphorylated forms of both STAT5a and STAT5b. There was no significant alteration in phosphorylation status of STAT5a in CA1 following ischemia in vehicle-treated groups. In the EPO-treated groups, however, STAT5a phosphorylation was increased at 1 hr compared with sham-operated group and vehicle-treated group, and then subsided 3 hr later (Figures 3A and 3B). Similar to p-STAT5a, p-STAT5b was not significantly increased in the CA1 in vehicle-treated groups; compared with sham-operated group, the treatment with EPO resulted in an increased phosphorylation of p-STAT5b 3 hr after ischemia (Figures 3C and 3D). Therefore, both pSTAT5a and 5b might contribute to the increases in pSTAT5, with 5a being the earlier contributor and 5b the later one.
Figure 3. Alteration of phosphorylation levels of STAT5a and STAT5b in rat CA1 following global ischemia.
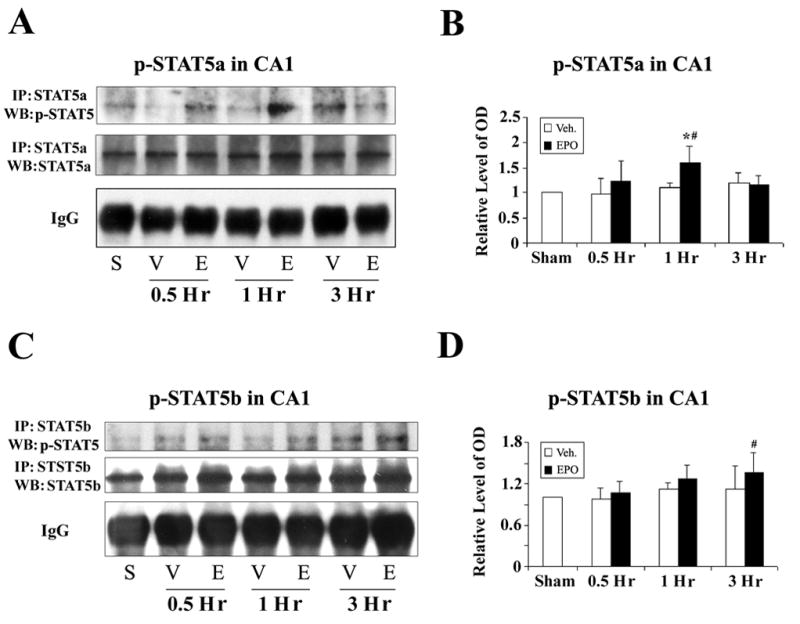
(A) Immunoprecipitation (IP) was performed using anti-STAT5a antibody, and the resulting immunoprecipitates were analyzed with Western blots (WB) using anti-phospho-STAT5 antibody. Representative blots show phospho-STAT5a levels in the CA1 region at early time points following global ischemia from S (sham-operated), V (vehicle-treated ischemia) and E (EPO-treated 20 min after ischemia) groups. (B) The average levels of phospho-STAT5a in the CA1 region from IP/Western blots were increased in the EPO-treated ischemia group at 1 hr following global ischemia, compared with the vehicle-treated ischemia group. *P<0.05 vs. vehicle-treated group at the same time point; #P<0.05 vs sham-operated group. (C) IP was performed with anti-STAT5b antibody, and then the resulting immunoprecipitates were analyzed with WB using anti-phospho-STAT5 antibody. Representative blots showing the levels of phosphorylated STAT5b in CA1 following global ischemia from S (sham-operated), V (vehicle-treated ischemia) and E (EPO-treated 20 min after ischemia) groups. (D) The average levels of phospho-STAT5b in CA1 on IP/Western blots were calculated and presented. #P<0.05 vs sham-operated group. No significant difference between vehicle- and EPO-treated ischemia groups was detected.
The expression of Bcl-xL and XIAP was upregulated by the administration of EPO in CA1 following global cerebral ischemia
Target genes of STAT5 include those suppressing the mitochondrial apoptotic pathway, such as Bcl-xL (Socolovsky et al., 1999, Kirito et al., 2002), and XIAP (Mohapatra et al., 2003). Since EPO was shown to be able to enhance the phosphorylation of STAT5 in CA1 following ischemia, we next investigated whether the administration of EPO can upregulate downstream genes of STAT5. As shown in Figures 4A and 4B, the expression of Bcl-xL was upregulated at 6 hr following ischemia in the EPO-treated group compared with the vehicle-treated group, and was maintained at relatively high levels through 24 hr after ischemia. XIAP is considered an effective apoptosis inhibitor by its suppression of caspase-9 (Takahashi et al., 1998, Fan et al., 2006), and the caspase-9-dependent intrinsic pathway could be the primary mechanism responsible for the activation of caspase-3 and cell death in ischemic hippocampal CA1 (Cao et al., 2002). Therefore, we next investigated whether there was a change in the expression of XIAP in CA1 following ischemia. Consistent with the previous report (Siegelin et al., 2005), we detected a slight increased level of XIAP at 24 hr post-ischemia in hippocampi over vehicle-treated rats, and EPO further enhanced the expression of XIAP at 24 hr after ischemia as shown in Figures 4C and 4D. Consequently, the cleavage of caspase-9 was attenuated in the EPO-treated group compared with the vehicle-treated group, as shown in Figures 4E and 4F. These findings suggest that the mitochondrial apoptotic pathway in ischemic CA1 in EPO-treated animals was attenuated through the upregulation of Bcl-xL and XIAP as well as the reduction of caspase-9 cleavage.
Figure 4. EPO treatment upregulates the expression of Bcl-xL and XIAP in CA1 following global cerebral ischemia in rat.
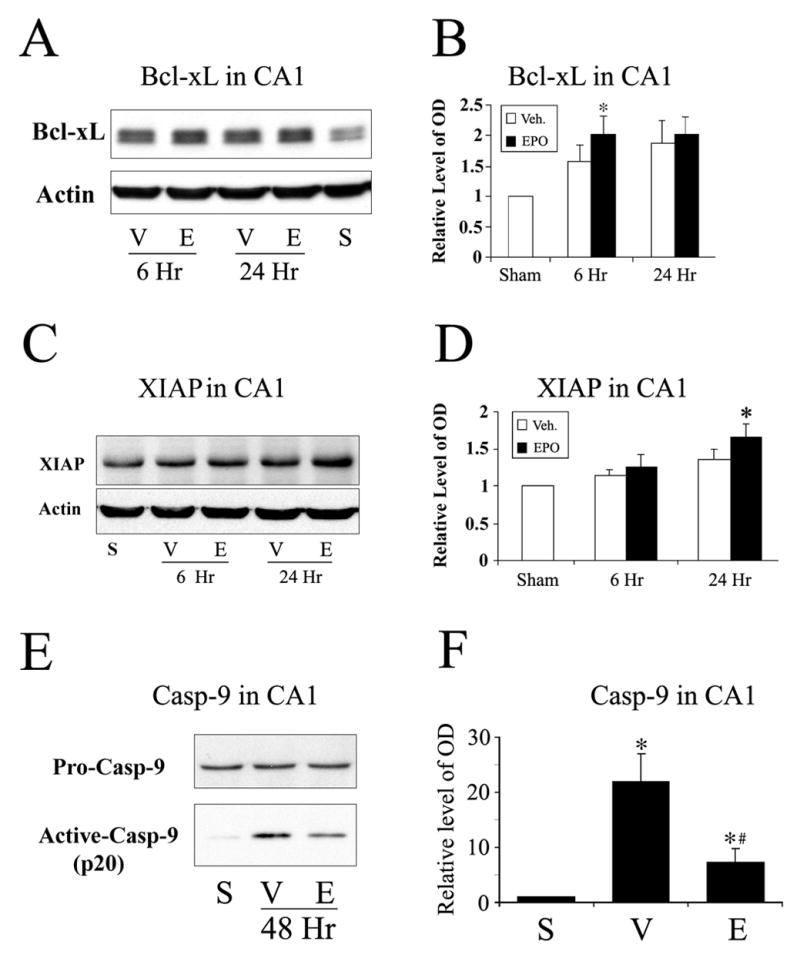
(A) Representative Western blots showing the expression of Bcl-xL in the CA1 region at time points indicated in S (sham-operated), V (vehicle-treated ischemia) and E (EPO-treated 20 min after ischemia) groups. (B) The average levels of Bcl-xL in the CA1 from Western blots were increased in EPO-treated rats at 6 hr after ischemia. Data are mean ± SEM, assessed by ANOVA and post hoc Scheffe’s tests. *P<0.05 vs. EPO-treated group, n=5. (C) Representative Western blots showing the expression of XIAP in the CA1 region at time points indicated in S (sham-operated), V (vehicle-treated ischemia) and E (EPO-treated 20 min after ischemia) groups. (D) The average level of XIAP in the CA1 from Western blots was increased in EPO-treated rats at 24 hr after ischemia. Data are mean ± SEM, assessed by ANOVA and post hoc Scheffe’s tests. *P<0.05 vs. EPO-treated group, n=5. (E) Representative Western blots showing the levels of full-length and cleaved forms of caspase-9 in the CA1 at 48 hr after ischemia. S, sham-operated; V, vehicle-treated ischemia; and E, EPO treatment 20 min after ischemia. (F) The average levels of cleaved form of caspase-9 from Western blots were decreased in EPO-treated rats at 48 hr after ischemia. Data are mean ± SEM, assessed by ANOVA and post hoc Scheffe’s tests. *P<0.05 vs. sham-operated group; #P<0.05 vs. EPO-treated group. S, sham-operated; V, vehicle-treated ischemia; and E, EPO treatment 20 min after ischemia.
EPO protected CA1 neurons from ischemic damage
After finding that EPO enhanced the phosphorylation of STAT5 and upregulated the expression of Bcl-xL and XIAP in CA1, we hypothesized that EPO may protect CA1 neurons against ischemic death induced by global cerebral ischemia in rats via STAT5 signaling pathway. To determine if EPO eventually attenuates CA1 neuronal death induced by global ischemia, we subjected the rats to 12 min of global cerebral ischemia, followed by infusion of either vehicle or 50 U of rhEPO into ICV at 20 min after ischemia. Three days later, the brains were removed; fresh-frozen sections in the dorsal hippocampal level were stained with hematoxylin and PANT assay. As shown in the upper panel of Figures 5A and 5B, the number of healthy CA1 neurons in the EPO-treated group was higher than that in the vehicle-treated group. Consistently, the number of neurons with DNA fragmentation was lower in the EPO-treated group than that in the vehicle-treated group as demonstrated in the lower panel of Figure 5A and Figure 5C, indicating that EPO protected CA1 neurons from ischemic damage induced by global cerebral ischemia. Consistent with the inhibition of STAT5 phosphorylation by AG490 shown previously in Figures 2E and 2F, the ICV infusion of AG490 (2.5 μg in 5 μL DMSO/PBS, which was a safe dose for rats) largely blocked the neuroprotective effects of EPO against CA1 neuronal death (Figures 5B and 5C), suggesting an important role of STAT5 in EPO-mediated neuroprotection.
Figure 5. EPO protects hippocampal CA1 neurons against ischemic injury in rat, which can be blocked by AG490.
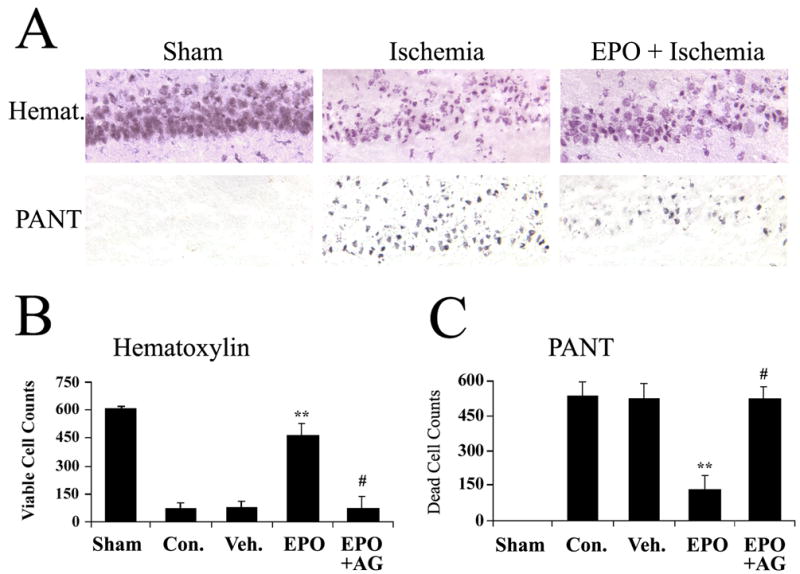
(A) Representative photomicrographs of hippocampal CA1 regions at 72 hr after global ischemia in rat. Upper panel, hematoxylin stain; lower panel, PANT stain showing dead cells with DNA damage. 20X. (B) Live CA1 neurons were counted and the mean number was plotted for each group. Data are mean ± SEM, assessed by ANOVA and post hoc Scheffe’s tests. **P<0.01 vs. control (ischemia only) and vehicle-treated ischemia groups. #<0.05 vs. EPO-treated ischemia group (50U, 20 min after ischemia) and P>0.05 vs. control and vehicle-treated groups. Sham, sham-operated group; Con., ischemia only group; Veh., vehicle-treated ischemia group; EPO, EPO-treated group (50 Units, 20 min after ischemia); EPO+AG, EPO and AG490-treated ischemia group (50 Units of EPO and 2.5 μg of AG490, 20 min after ischemia). (C) Dead (PANT-positive) CA1 neurons were counted and the mean number was plotted for each group. Data are mean ± SEM, assessed by ANOVA and post hoc Scheffe’s tests. **P<0.01 vs. control (ischemia only) and vehicle-treated groups; #<0.05 vs. EPO-treated group (50U, 20 min after ischemia) and P>0.05 vs. control (ischemia only) and vehicle-treated groups. Sham, sham-operated group; Con., ischemia only group; Veh., vehicle-treated ischemia group; EPO, EPO-treated group (50 Units, 20 min after ischemia); EPO+AG, EPO and AG490-treated group (50 Units of EPO and 2.5 μg of AG490, 20 min after ischemia).
Discussion
Our data show that the phosphorylation of STAT5 was increased in hippocampal CA1 at 1 and 3 hr following global cerebral ischemia in rats, accompanied by increased nuclear translocation of p-STAT5; and that the administration of EPO further enhanced the phosphorylation of STAT5, with SATA5a being phosphorylated earlier. The enhanced phosphorylation of STAT5 in the EPO-treated group was accompanied by the upregulation of downstream gene products Bcl-xL and XIAP, and was associated with reduced CA1 neuronal death. The JAK2 inhibitor Tyrphostin AG490 blocked the EPO-mediated enhancement of STAT5 phosphorylation and the EPO-mediated neuroprotection against CA1 neuronal death after global cerebral ischemia.
The activation and role of STAT5 in CA1 after global cerebral ischemia have not been investigated previously. We detected an increased phosphorylation of STAT5 in CA1 as early as 1 to 3 hr after ischemia. The mechanisms for this phosphorylation of STAT5 remain unclear. In addition to EPO, several other factors, including IL-2, IL-6, growth hormone, prolactin, and granulocyte macrophage colony-stimulating factor, can also activate STAT5 via JAK signaling pathway (Battle and Frank, 2002, Debierre-Grockiego, 2004). After cerebral ischemia and reperfusion, one or more of these factors may play a role in the activation of STAT5.
The role of STAT5 in the EPO-mediated neuroprotection against cerebral ischemia has been controversial. Previous in vitro study has shown that the DNA binding activity of STAT5 was not increased following EPO stimulation (Ruscher et al., 2002). However, it needs to be pointed out that this result was seen under a condition without ischemic challenge, and that the phosphorylation of STAT5 was not investigated. In contrast, another study showed that the level of p-STAT5 in cultured hippocampal neurons was maintained at a relatively higher level by EPO at 15 hr of hypoxia, but was lower than that in the normoxia condition (Siren et al., 2001). At 1 day and 3 days after focal cerebral ischemia in neonatal rats, the level of p-STAT5 was higher in EPO groups than that in ischemic groups, similar to the in vitro study discussed above (Sola et al., 2005). However, 15 hr after hypoxia in neuronal culture and day 1 and day 3 after focal cerebral ischemia in animal models represented the late stages of ischemic insult, at which time robust cell death have already occurred. What these results likely reflect is the possibility that there were more viable cells in the EPO-treated sample than in the untreated sample, emphasizing the neuroprotective effect of EPO. Whether STAT5 plays a role in EPO-mediated neuroprotection before the point of no return, i.e., in the early stage after ischemia in vivo, has not been reported previously. STAT5 recently was reported to be necessary to EPO-mediated neuroprotection against apoptosis in differentiated neuroblastoma cells (Um and Lodish, 2006). Our results shown that EPO further enhanced the phosphorylation of STAT5, and the administration of JAK2 inhibitor blocked the enhancement of STAT5 phosphorylation and neuroprotection rendered by EPO, suggesting STAT5 may play an important role in EPO-mediated neuroprotection.
The effect of EPO on the expression of Bcl-xL has been controversial. Ruscher and co-workers found no upregulation of Bcl-xL following EPO treatment in cultured neurons without OGD challenge (Ruscher et al., 2002), whereas Wen and co-workers reported an increased expression of Bcl-xL in both in vivo and in vitro studies following ischemic insults (Wen et al., 2002). Our results support the notion that the expression of Bcl-xL is upregulated following ischemia and EPO treatment. In addition, XIAP, another gene product of STAT5, was also found upregulated following ischemia and EPO treatment in our study, indicating that the action of STAT5 in EPO-mediated neuroprotection may be carried out by Bcl-xL and XIAP, suppressing caspase-9 activation and the mitochondrial apoptotic pathway.
STAT5a protein (96 kDa) and STAT5b protein (94 kDa) share 96% identity, with the difference in their C-terminals (Debierre-Grockiego, 2004). Both STAT5a and STAT5b proteins can be phosphorylated at tyrosine 694, and therefore gain their activity of translocation to nucleus and binding to specific DNA sequences (TTCN2–4GAA) in the promoters of responsive genes (Ehret et al., 2001, Battle and Frank, 2002, Debierre-Grockiego, 2004, Paukku and Silvennoinen, 2004). STAT5a has been found to play an important role against myocardial ischemia-reperfusion injury, demonstrated by the loss of ability to achieve ischemic tolerance of heart in STAT5a knockout mice (Yamaura et al., 2003). Our data show that the phosphorylation of STAT5a occurred earlier than that of STAT5b in ischemic CA1 in the EPO-treated group, suggesting a more important role for STAT5a.
STAT5 has also demonstrated biological activities other than transcriptional regulations. Recently, it was reported that there appeared to be a cross-talk between the PI3K and STAT5 pathways (Gesbert and Griffin, 2000, Rosa Santos et al., 2000, Kirito et al., 2002, Nyga et al., 2005). Activated STAT5 interacted with the regulatory subunit of PI3K, which was required for the activation of PI3K (Rosa Santos et al., 2000, Nyga et al., 2005). On the other hand, the PI3K-AKT pathway could synergize with STAT5 to enhance the expression of Bcl-xL, possibly by enhancing STAT5 nuclear translocation and decreasing its degradation (Gesbert and Griffin, 2000, Debierre-Grockiego, 2004). In addition, STAT5 has also been demonstrated to interact with cAMP-response element binding protein (CREB). STAT5 transcriptional activity was enhanced by the phosphorylation of CREB, which was induced by EPO and stem cell factor (Boer et al., 2002, Boer et al., 2003, Gewinner et al., 2004). CREB is a transcriptional factor whose downstream gene products, such as brain-derived neurotrophic factor (BDNF), have neuroprotective effects (Finkbeiner et al., 1997). Recently, EPO also was shown to be able to increase the phosphorylation of CREB and upregulate BDNF expression in neurons (Viviani et al., 2005). Taken together, the activity of STAT5 in CA1 in the EPO-treated group may be augmented through its interaction with AKT and CREB.
In summary, this study demonstrates that the phosphorylation and activation of STAT5 may be involved in the function of EPO in reducing experimental ischemic injury induced by global cerebral ischemia. Together with previous findings (Sakanaka et al., 1998, Calapai et al., 2000, Catania et al., 2002, Wen et al., 2002, Zhang et al., 2006), our results suggest that EPO may be useful in treating ischemic incidents in humans.
Acknowledgments
This work was supported by the NIH Grants NS43802, NS45048, NS36736 and NS38560 to J.C. from the NINDS. J.C. is a recipient of the Established Investigator Award (240135N) from the American Heart Association. J.C. was also supported in part by the Geriatric Research, Education and Clinical Center, Veterans Affairs Pittsburgh Health Care System. We thank Carol Culver for editorial assistance, and Pat Strickler for secretarial support.
Abbreviations used
- EPO
erythropoietin
- ICV
intracerebral ventricle
- JAK2
Janus kinase 2
- STAT5
signal transducer and activator of transcription 5
- XIAP
X-linked inhibitor of apoptosis protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aaronson DS, Horvath CM. A road map for those who don't know JAK-STAT. Science. 2002;296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- Battle TE, Frank DA. The role of STATs in apoptosis. Curr Mol Med. 2002;2:381–392. doi: 10.2174/1566524023362456. [DOI] [PubMed] [Google Scholar]
- Boer AK, Drayer AL, Rui H, Vellenga E. Prostaglandin-E2 enhances EPO-mediated STAT5 transcriptional activity by serine phosphorylation of CREB. Blood. 2002;100:467–473. doi: 10.1182/blood.v100.2.467. [DOI] [PubMed] [Google Scholar]
- Boer AK, Drayer AL, Vellenga E. Stem cell factor enhances erythropoietin-mediated transactivation of signal transducer and activator of transcription 5 (STAT5) via the PKA/CREB pathway. Exp Hematol. 2003;31:512–520. doi: 10.1016/s0301-472x(03)00075-4. [DOI] [PubMed] [Google Scholar]
- Calapai G, Marciano MC, Corica F, Allegra A, Parisi A, Frisina N, Caputi AP, Buemi M. Erythropoietin protects against brain ischemic injury by inhibition of nitric oxide formation. Eur J Pharmacol. 2000;401:349–356. doi: 10.1016/s0014-2999(00)00466-0. [DOI] [PubMed] [Google Scholar]
- Cao G, Clark RS, Pei W, Yin W, Zhang F, Sun FY, Graham SH, Chen J. Translocation of apoptosis-inducing factor in vulnerable neurons after transient cerebral ischemia and in neuronal cultures after oxygen-glucose deprivation. J Cereb Blood Flow Metab. 2003;23:1137–1150. doi: 10.1097/01.WCB.0000087090.01171.E7. [DOI] [PubMed] [Google Scholar]
- Cao G, Luo Y, Nagayama T, Pei W, Stetler RA, Graham SH, Chen J. Cloning and characterization of rat caspase-9: implications for a role in mediating caspase-3 activation and hippocampal cell death after transient cerebral ischemia. J Cereb Blood Flow Metab. 2002;22:534–546. doi: 10.1097/00004647-200205000-00005. [DOI] [PubMed] [Google Scholar]
- Catania MA, Marciano MC, Parisi A, Sturiale A, Buemi M, Grasso G, Squadrito F, Caputi AP, Calapai G. Erythropoietin prevents cognition impairment induced by transient brain ischemia in gerbils. Eur J Pharmacol. 2002;437:147–150. doi: 10.1016/s0014-2999(02)01292-x. [DOI] [PubMed] [Google Scholar]
- Chen J, Nagayama T, Jin K, Stetler RA, Zhu RL, Graham SH, Simon RP. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci. 1998;18:4914–4928. doi: 10.1523/JNEUROSCI.18-13-04914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Hematopoietic factor erythropoietin fosters neuroprotection through novel signal transduction cascades. J Cereb Blood Flow Metab. 2002;22:503–514. doi: 10.1097/00004647-200205000-00001. [DOI] [PubMed] [Google Scholar]
- Damen JE, Wakao H, Miyajima A, Krosl J, Humphries RK, Cutler RL, Krystal G. Tyrosine 343 in the erythropoietin receptor positively regulates erythropoietin-induced cell proliferation and Stat5 activation. Embo J. 1995;14:5557–5568. doi: 10.1002/j.1460-2075.1995.tb00243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debierre-Grockiego F. Anti-apoptotic role of STAT5 in haematopoietic cells and in the pathogenesis of malignancies. Apoptosis. 2004;9:717–728. doi: 10.1023/B:APPT.0000045785.65546.a2. [DOI] [PubMed] [Google Scholar]
- De-Fraja C, Conti L, Govoni S, Battaini F, Cattaneo E. STAT signalling in the mature and aging brain. Int J Dev Neurosci. 2000;18:439–446. doi: 10.1016/s0736-5748(00)00007-1. [DOI] [PubMed] [Google Scholar]
- De-Fraja C, Conti L, Magrassi L, Govoni S, Cattaneo E. Members of the JAK/STAT proteins are expressed and regulated during development in the mammalian forebrain. J Neurosci Res. 1998;54:320–330. doi: 10.1002/(SICI)1097-4547(19981101)54:3<320::AID-JNR3>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Digicaylioglu M, Lipton SA. Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-kappaB signalling cascades. Nature. 2001;412:641–647. doi: 10.1038/35088074. [DOI] [PubMed] [Google Scholar]
- Dudley AC, Thomas D, Best J, Jenkins A. A VEGF/JAK2/STAT5 axis may partially mediate endothelial cell tolerance to hypoxia. Biochem J. 2005;390:427–436. doi: 10.1042/BJ20050351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehret GB, Reichenbach P, Schindler U, Horvath CM, Fritz S, Nabholz M, Bucher P. DNA binding specificity of different STAT proteins. Comparison of in vitro specificity with natural target sites. J Biol Chem. 2001;276:6675–6688. doi: 10.1074/jbc.M001748200. [DOI] [PubMed] [Google Scholar]
- Elmer E, Kokaia Z, Kokaia M, Carnahan J, Nawa H, Lindvall O. Dynamic changes of brain-derived neurotrophic factor protein levels in the rat forebrain after single and recurring kindling-induced seizures. Neuroscience. 1998;83:351–362. doi: 10.1016/s0306-4522(97)00387-4. [DOI] [PubMed] [Google Scholar]
- Fan Y-F, Lu C-Z, Xie J, Zhao Y-X, Yang G-Y. Apoptosis inhibition in ischemic brain by intraperitoneal PTD-BIR3-RING (XIAP) Neurochemistry International. 2006;48:50–59. doi: 10.1016/j.neuint.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME. CREB: a major mediator of neuronal neurotrophin responses. Neuron. 1997;19:1031–1047. doi: 10.1016/s0896-6273(00)80395-5. [DOI] [PubMed] [Google Scholar]
- Gesbert F, Griffin JD. Bcr/Abl activates transcription of the Bcl-X gene through STAT5. Blood. 2000;96:2269–2276. [PubMed] [Google Scholar]
- Gewinner C, Hart G, Zachara N, Cole R, Beisenherz-Huss C, Groner B. The coactivator of transcription CREB-binding protein interacts preferentially with the glycosylated form of Stat5. J Biol Chem. 2004;279:3563–3572. doi: 10.1074/jbc.M306449200. [DOI] [PubMed] [Google Scholar]
- Gobert S, Chretien S, Gouilleux F, Muller O, Pallard C, Dusanter-Fourt I, Groner B, Lacombe C, Gisselbrecht S, Mayeux P. Identification of tyrosine residues within the intracellular domain of the erythropoietin receptor crucial for STAT5 activation. Embo J. 1996;15:2434–2441. [PMC free article] [PubMed] [Google Scholar]
- Kirito K, Watanabe T, Sawada K, Endo H, Ozawa K, Komatsu N. Thrombopoietin regulates Bcl-xL gene expression through Stat5 and phosphatidylinositol 3-kinase activation pathways. J Biol Chem. 2002;277:8329–8337. doi: 10.1074/jbc.M109824200. [DOI] [PubMed] [Google Scholar]
- Liu X, Robinson GW, Gouilleux F, Groner B, Hennighausen L. Cloning and expression of Stat5 and an additional homologue (Stat5b) involved in prolactin signal transduction in mouse mammary tissue. Proc Natl Acad Sci U S A. 1995;92:8831–8835. doi: 10.1073/pnas.92.19.8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra S, Chu B, Wei S, Djeu J, Epling-Burnette PK, Loughran T, Jove R, Pledger WJ. Roscovitine inhibits STAT5 activity and induces apoptosis in the human leukemia virus type 1-transformed cell line MT-2. Cancer Res. 2003;63:8523–8530. [PubMed] [Google Scholar]
- Nagayama T, Lan J, Henshall DC, Chen D, O'Horo C, Simon RP, Chen J. Induction of oxidative DNA damage in the peri-infarct region after permanent focal cerebral ischemia. J Neurochem. 2000;75:1716–1728. doi: 10.1046/j.1471-4159.2000.0751716.x. [DOI] [PubMed] [Google Scholar]
- Nyga R, Pecquet C, Harir N, Gu H, Dhennin-Duthille I, Regnier A, Gouilleux-Gruart V, Lassoued K, Gouilleux F. Activated STAT5 proteins induce activation of the PI 3-kinase/Akt and Ras/MAPK pathways via the Gab2 scaffolding adapter. Biochem J. 2005;390:359–366. doi: 10.1042/BJ20041523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parganas E, Wang D, Stravopodis D, Topham DJ, Marine J-C, Teglund S, Vanin EF, Bodner S, Colamonici OR, van Deursen JM. Jak2 Is Essential for Signaling through a Variety of Cytokine Receptors. Cell. 1998;93:385–395. doi: 10.1016/s0092-8674(00)81167-8. [DOI] [PubMed] [Google Scholar]
- Park SH, Liu X, Hennighausen L, Davey HW, Waxman DJ. Distinctive roles of STAT5a and STAT5b in sexual dimorphism of hepatic P450 gene expression. Impact of STAT5a gene disruption. J Biol Chem. 1999;274:7421–7430. doi: 10.1074/jbc.274.11.7421. [DOI] [PubMed] [Google Scholar]
- Paukku K, Silvennoinen O. STATs as critical mediators of signal transduction and transcription: lessons learned from STAT5. Cytokine Growth Factor Rev. 2004;15:435–455. doi: 10.1016/j.cytogfr.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Rosa Santos SC, Dumon S, Mayeux P, Gisselbrecht S, Gouilleux F. Cooperation between STAT5 and phosphatidylinositol 3-kinase in the IL-3-dependent survival of a bone marrow derived cell line. Oncogene. 2000;19:1164–1172. doi: 10.1038/sj.onc.1203418. [DOI] [PubMed] [Google Scholar]
- Ruscher K, Freyer D, Karsch M, Isaev N, Megow D, Sawitzki B, Priller J, Dirnagl U, Meisel A. Erythropoietin is a paracrine mediator of ischemic tolerance in the brain: evidence from an in vitro model. J Neurosci. 2002;22:10291–10301. doi: 10.1523/JNEUROSCI.22-23-10291.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakanaka M, Wen TC, Matsuda S, Masuda S, Morishita E, Nagao M, Sasaki R. In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc Natl Acad Sci U S A. 1998;95:4635–4640. doi: 10.1073/pnas.95.8.4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegelin MD, Kossatz LS, Winckler J, Rami A. Regulation of XIAP and Smac/DIABLO in the rat hippocampus following transient forebrain ischemia. Neurochem Int. 2005;46:41–51. doi: 10.1016/j.neuint.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Siren AL, Fratelli M, Brines M, Goemans C, Casagrande S, Lewczuk P, Keenan S, Gleiter C, Pasquali C, Capobianco A, Mennini T, Heumann R, Cerami A, Ehrenreich H, Ghezzi P. Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc Natl Acad Sci U S A. 2001;98:4044–4049. doi: 10.1073/pnas.051606598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socolovsky M, Fallon AE, Wang S, Brugnara C, Lodish HF. Fetal anemia and apoptosis of red cell progenitors in Stat5a−/−5b−/− mice: a direct role for Stat5 in Bcl-X(L) induction. Cell. 1999;98:181–191. doi: 10.1016/s0092-8674(00)81013-2. [DOI] [PubMed] [Google Scholar]
- Sola A, Rogido M, Lee BH, Genetta T, Wen TC. Erythropoietin after focal cerebral ischemia activates the Janus kinase-signal transducer and activator of transcription signaling pathway and improves brain injury in postnatal day 7 rats. Pediatr Res. 2005;57:481–487. doi: 10.1203/01.PDR.0000155760.88664.06. [DOI] [PubMed] [Google Scholar]
- Takahashi R, Deveraux Q, Tamm I, Welsh K, Assa-Munt N, Salvesen GS, Reed JC. A single BIR domain of XIAP sufficient for inhibiting caspases. J Biol Chem. 1998;273:7787–7790. doi: 10.1074/jbc.273.14.7787. [DOI] [PubMed] [Google Scholar]
- Teglund S, McKay C, Schuetz E, van Deursen JM, Stravopodis D, Wang D, Brown M, Bodner S, Grosveld G, Ihle JN. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998;93:841–850. doi: 10.1016/s0092-8674(00)81444-0. [DOI] [PubMed] [Google Scholar]
- Um M, Lodish HF. Antiapoptotic effects of erythropoietin in differentiated neuroblastoma SH-SY5Y cells require activation of both the STAT5 and AKT signaling pathways. J Biol Chem. 2006;281:5648–5656. doi: 10.1074/jbc.M510943200. [DOI] [PubMed] [Google Scholar]
- Viviani B, Bartesaghi S, Corsini E, Villa P, Ghezzi P, Garau A, Galli CL, Marinovich M. Erythropoietin protects primary hippocampal neurons increasing the expression of brain-derived neurotrophic factor. J Neurochem. 2005;93:412–421. doi: 10.1111/j.1471-4159.2005.03033.x. [DOI] [PubMed] [Google Scholar]
- Wen TC, Sadamoto Y, Tanaka J, Zhu PX, Nakata K, Ma YJ, Hata R, Sakanaka M. Erythropoietin protects neurons against chemical hypoxia and cerebral ischemic injury by up-regulating Bcl-xL expression. J Neurosci Res. 2002;67:795–803. doi: 10.1002/jnr.10166. [DOI] [PubMed] [Google Scholar]
- Yamaura G, Turoczi T, Yamamoto F, Siddqui MA, Maulik N, Das DK. STAT signaling in ischemic heart: a role of STAT5A in ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285:H476–482. doi: 10.1152/ajpheart.00079.2003. [DOI] [PubMed] [Google Scholar]
- Zhang F, Signore AP, Zhou Z, Wang S, Cao G, Chen J. Erythropoietin protects CA1 neurons against global cerebral ischemia in rat: Potential signaling mechanisms. J Neurosci Res. 2006;83:1241–1251. doi: 10.1002/jnr.20816. [DOI] [PubMed] [Google Scholar]
- Zhang F, Yin W, Chen J. Apoptosis in cerebral ischemia: executional and regulatory signaling mechanisms. Neurol Res. 2004;26:835–845. doi: 10.1179/016164104X3824. [DOI] [PubMed] [Google Scholar]
- Zhang S, Fukuda S, Lee Y, Hangoc G, Cooper S, Spolski R, Leonard WJ, Broxmeyer HE. Essential role of signal transducer and activator of transcription (Stat)5a but not Stat5b for Flt3-dependent signaling. J Exp Med. 2000;192:719–728. doi: 10.1084/jem.192.5.719. [DOI] [PMC free article] [PubMed] [Google Scholar]