

Abstract
Background
Essential tremor (ET) is the most common movement disorder and its pathophysiology is unknown. We hypothesize that increased membrane excitability in motor circuits has a key role in the pathogenesis of ET. Specifically, we propose that neural circuits controlling ballistic movements are inherently unstable due to their underlying reciprocal innervation. Such instability is enhanced by increased neural membrane excitability and the circuit begins to oscillate. These oscillations manifest as tremor.
Methods
Postural limb tremor was recorded in 22 ET patients and then the phenotype was simulated with a conductance-based neuromimetic model of ballistic movements. The model neuron was Hodgkin-Huxley type with added hyperpolarization activated cation current (Ih), low threshold calcium current (IT), and GABA and glycine mediated chloride currents. The neurons also featured the neurophysiological property of rebound excitation after release from sustained inhibition (post-inhibitory rebound). The model featured a reciprocally innervated circuit of neurons that project to agonist and antagonist muscle pairs.
Results
Neural excitability was modulated by changing Ih and/or IT. Increasing Ih and/or IT further depolarized the membrane and thus increased excitability. The characteristics of the tremor from all ET patients were simulated when Ih was increased to ~10× the range of physiological values. In contrast, increasing other membrane conductances, while keeping Ih at a physiological value, did not simulate the tremor. Increases in Ih and IT determined the frequency and amplitude of the simulated oscillations.
Conclusion
These simulations support the hypothesis that increased membrane excitability in potentially unstable, reciprocally innervated circuits can produce oscillations that resemble ET. Neural excitability could be increased in a number of ways. In this study membrane excitability was increased by up-regulating Ih and IT. This approach suggests new experimental and clinical ways to understand and treat common tremor disorders.
Introduction
Essential tremor (ET) is a common neurological disorder characterized by postural tremor that worsens with movement. The pathophysiological mechanism of ET is unclear. However, a number of drugs that reduce neuronal membrane excitability are beneficial in ET. For example, propranolol – a commonly used beta-blocker that reduces membrane excitability [1] – is an effective drug for treating ET [2]. GABA-mimetic inhibitory agents such as gabapentin can reduce ET [3]. Ethanol, which enhances GABA and glycine mediated currents, and inhibits the excitatory glutamatergic transmission (ultimately decreasing neural excitability), ameliorates ET [4-6]. Improvement in ET was also reported with topiramate [7]. Topiramate has a GABA-mimetic effect and also inhibits glutamatergic (excitatory) transmission [8].
Therefore we hypothesize that increased membrane excitability in pre-motor neurons has a key role in pathogenesis of ET. First, we present two fundamental concepts that support this idea. Based upon these concepts, we then test our hypothesis with a conductance based computational model that simulates tremor.
Concept 1 – Sherrington's 'burden': reciprocal inhibition and rebound depolarization
Excitation of an agonist muscle and simultaneous inhibition of its antagonist is necessary for efficient force generation during movement. This is Sherrington's principle for reciprocal innervation. For example, when we flex an elbow, the flexor group of muscles receives excitatory impulses from the corresponding neurons. The same neurons also inhibit neurons innervating the antagonist muscle group, the extensors. Figure 1A schematically illustrates this phenomenon. The green lines are excitatory neural projections and red lines are inhibitory. Reciprocally inhibitory neural circuits are present in many central areas responsible for limb movement [9]. Furthermore, some limb movement related neurons also exhibit a rebound increase in their firing rate when inhibition is removed – post-inhibitory rebound (PIR). For example, PIR is observed in neurons of premotor areas for limb movements, such as the thalamus and inferior olive [10].
Figure 1.
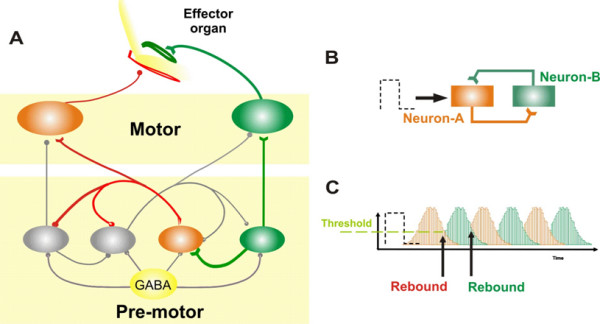
(A) The circuit of reciprocally innervated neurons for controlling ballistic movements. The excitatory premotor neurons send excitatory projections to the motor neurons innervating the agonist muscle group. At the same time this neuron also sends an excitatory projection to the inhibitory neuron innervating the motor neuron for the antagonist muscle group. In addition, mutual inhibitory connections exist between premotor neurons. These mutually inhibitory connections predispose the neural circuit to instability and oscillations. (B, C) Demonstration of oscillations in a two-neuron circuit. Neuron-A inhibits neuron-B and vice-versa. A small pulse to neuron-A increases its discharge and thus inhibits neuron-B. Once the discharge of the neuron drops inhibition from neuron-B is removed. This results in a rebound increase in the neuron-B firing rate. Since neuron-B also inhibits neuron-A, the same phenomenon of post-inhibitory rebound repeats for neuron-A. In the panel 'C' response of neuron A is schematized with red bars, while the green trace is response from neuron B.
Under physiological circumstances, with normal levels of external inhibition, PIR provides sufficient force to generate prompt, high-speed ballistic movements. Reciprocal innervation and PIR also make neural circuits inherently unstable and prone to oscillations [11,12]. Figure 1, panels B and C, illustrate why a circuit of reciprocally innervated neurons with PIR is prone to oscillations. A neural circuit is formed by two mutually inhibitory neurons (neuron-A and -B). A small pulse of neural activity, either from spontaneous fluctuations in neural activity or a small spontaneous movement, activates neuron-A. The discharge from neuron-A inhibits neuron-B. Following the termination of the input to neuron-A, its discharge drops and inhibition upon neuron-B is removed. Neuron-B in turn shows PIR and starts to fire. Since neuron-B also inhibits neuron-A, when firing in neuron-B stops, PIR occurs in neuron-A. Thus the reciprocally-innervated neural circuit begins to oscillate (Figure 1C).
Nature's solution to this inherent instability in a reciprocally innervated neural circuit is to add enough external inhibition to keep the excitability of the constituent neurons under control. For example, we have proposed that oscillations in an analogous circuit controlling ballistic eye movements (saccades) are normally prevented by tonic external glycinergic inhibition [12]. Similarly, abolishing GABAergic inhibition in GABA mutant mice is known to cause tremor [13]. However, oscillations in reciprocally innervating circuits can be seen in the presence of normal external inhibition. For example, in patients with ET GABA mutation was not found [14]. In the following section we introduce a concept explaining the basis of oscillations in reciprocally innervated circuits when external inhibition is intact.
Concept 2 – Increased excitability can make reciprocally innervated neurons unstable
Oscillations in reciprocally innervated circuits could emerge if the relative effect of intact external inhibition is reduced by an increased excitability within the reciprocally-innervated neurons themselves. Therefore it is possible that increased neural excitability can overcome the effects of normal external inhibition. There could be a number of causes of increased excitability including an increase in either the hyperpolarization activated cation current (Ih) or the low threshold calcium current (IT) [15,16] or alterations in the intracellular levels of second messengers and the regulators that influence the activation kinetics of these ion channels [16-18].
Testing the hypothesis
We recorded postural limb tremor in 22 patients with ET, and used a neuromimetic model to simulate their tremor. We tested our hypothesis by simulating a Hodgkin-Huxley type, conductance-based, neuromimetic model of pre-motor burst neurons responsible for ballistic limb movements. This model has the following features: (1) A circuit consisting of reciprocally innervating excitatory and inhibitory neurons. (2) Physiologically-realistic membrane kinetics of the premotor neurons determined by specific and physiologically plausible subsets of membrane ion channels. The latter also determines the excitability of the membrane. (3) These model neurons had a property of rebound firing after sustained inhibition - post-inhibitory rebound (PIR). By increasing specific membrane conductances that are known to increase PIR and neural excitability, such as Ih and IT, we could simulate the range of frequencies of tremor recorded from ET patients.
Methods
Patient selection and tremor recordings
We studied 22 ET patients, who gave written, informed consent before enrolling in the study.
Inclusion and exclusion criteria
Patients were recruited from the movement disorders clinic. Patients had bilateral postural tremor of their hands. We excluded patients with dystonia, drug-induced tremor, psychogenic tremor, and orthostatic tremor. Subjects with enhanced physiological tremor, which often resembles ET, were excluded. The frequency of the tremor and the effects of loading (putting weight on the outstretched limb) during recording of postural tremor condition were used to exclude patients with enhanced physiological tremor. Loading reduces the frequency of enhanced physiological tremor.
Limb tremor was recorded with a three-axis accelerometer attached with a piece of surgical tape to the top of the middle phalanx of the index finger (FlexAble Systems, Fountain Hills, AZ). Patients held their arms outstretched against gravity with palms toward the floor (postural tremor). Typical tremor frequency in ET does not exceed 15 Hz, thus we sampled at 100 Hz (more than three times the minimum Nyquist-Shannon sampling rate). The raw acceleration signal recorded by the three-axis accelerometer contains high-frequency noise, inherent in all acceleration sensing systems, and low-frequency noise due to changes in the attitude of the limb relative to gravity (sway). These artifacts were removed by de-trending and digital filtering that involved three-point averaging. The 1 G (9.8 m/s2) gravity vector, which is much larger than the tremor accelerations, was determined from the 3-D calibration and removed off-line [19].
Data from each axis of the accelerometer and the magnitude of the acceleration vector (square root of the sum of the acceleration squared on all three axes) were processed separately. Cycle-by-cycle analysis was performed. A cycle was defined as follows: first we removed any bias from the de-trended data (i.e., normalized amplitude = actual amplitude – mean amplitude). This kept the peaks of the cycles positive and the troughs negative. The time when the data trace changed from a negative value to a positive value (i.e., the positively moving zero-crossing) was recorded. The time of the first positively moving zero-crossing marks the beginning and the next positively moving zero-crossing marks the end of the given cycle. The inverse of the cycle period yields the cycle frequency; the difference between the peak and trough gives the peak-to-peak amplitude.
Computational simulations
Here we summarize the key features of our computational model. Readers are referred to additional files 1, 2 and 3 for more details on methodology of computational simulations. The Hodgkin-Huxley equations were implemented to simulate action potentials. GABA and glycine mediated inhibitory chloride conductance was implemented for inhibition, and non-NMDA and NMDA glutamatergic channels for excitation. Kinematics of CaV3 channels (carrying IT) and four subtypes of HCN channel (HCN1-HCN4; carrying Ih) were included to simulate PIR and to modulate neuronal excitability. The activation kinetics of each of the subtypes of ion channels carrying Ih are different, HCN-1 being the fastest and HCN-4 the slowest, with the others having intermediate activation time constants [20].
The following equation describes the time evolution of the membrane potential of the brain stem neurons:
| C*dV/dt = -IL - IT - n1Ih1- n2Ih2- n3Ih3- n4Ih4- INa - IK - ICl - INMDA - InonNMDA | (1) |
where V is the membrane potential of the neuron, C is the membrane capacitance (1 μF/cm2) and n1-n4 is a rate scaling factor determining the ion channel expression profile in the neuron. IL, IT, Ih1-4, INa, and IK, denote the leak current, low-threshold calcium current, hyperpolarization activated current (carried by HCN1-4), fast sodium current and delayed rectifier potassium current, respectively. ICl is the synaptic current mediated by glycinergic and GABAergic neurotransmitters. INMDA and InonNMDA are synaptic currents mediated by NMDA and non-NMDA sensitive glutamate receptors. The parameters n1-n4 represent the relative expression strength of Ih channels. The model simulated a set of four burst neurons – inhibitory agonist, inhibitory antagonist, excitatory agonist, excitatory antagonist. We assumed a small, yet physiologically plausible, variability in the ion channel expression profiles of the simulated burst neurons, which in turn, accounted for minor (physiological) variability in their membrane properties. Supplementary figure 1 (additional file 2 ) schematizes the model organization, synaptic weights, and mathematical functions implemented in the model. Additional details of the model are in additional file 1 as well as in Miura and Optican (2006) and Shaikh, et al., (2007).
Results
Model simulations
We simulated membrane properties of reciprocally-innervated burst neurons within a local feedback loop model for ballistic movements. Each neuron was a conductance-based single-compartment model. The major conductances simulating the membrane properties of the model neurons are schematized in Figure 2A. The details of the model organization and the mathematical equations driving these ion currents are explained in methods and additional file 1, 2 and 3. The model is compatible with the known anatomical organization of neural circuits for limb movements. The membrane properties of the model burst neurons are also consistent with the known profiles of limb-movement sensitive pre-motor neurons. The model simulated normal ballistic limb movements when the ion currents and excitability of the simulated burst neuron membranes were within physiological limits (Ih = 0.1 mSeimens; IT = 2.0 mSeimens; and resting membrane potential = -68 mV). Increases in Ih and IT further depolarized the resting membrane potential producing increased neural excitability (Figure 2B). Oscillations resembling ET were simulated when Ih and IT in the model neurons were increased. The increase in these currents resulted in alternating bursts of action potentials in the neurons innervating the sets of agonist and antagonist muscles (Figure 2C). In Figure 2C the membrane potential is plotted along the y-axis and time along the x-axis. The alternating bursts of discharge reflect the circuit oscillations and produce the limb tremor. The simulated oscillations (the model output) are illustrated as the grey trace in Figure 2D. The common time scale in Figure 2C and 2D facilitates the comparison of the simulated oscillations (model output) with the alternating bursts of discharge in the pairs of neurons innervating agonist and antagonist muscles. The black trace in Figure 2D illustrates a representative postural limb tremor recorded from one ET patient. The simulated tremor superimposes fairly well on the postural limb tremor from the ET patient. The frequency of the simulated tremor (5.9 Hz) is close to the actual tremor frequency (5.7 Hz). The correlation coefficient between the two waveforms was 0.9.
Figure 2.

(A) A traditional Hodgkin-Huxley model of cell membranes with multiple ion channels was used to generate the action potential. In order to simulate physiologically realistic neural behavior, ion channels such as hyperpolarization activated cation currents (Ih) and low threshold calcium current (IT) were also included. NMDA and non-NMDA excitatory glutamatergic channels as well as GABA sensitive inhibitory channels were also included. The grey box schematizes the burst neuron, while its grey outline schematizes the cell membrane. The ion channels span the membrane thickness. dV is the rate of change in the membrane potential over period 'dt'. C is the membrane capacitance (1 μF/cm2) and n1-n4 is a rate scaling factor determining the ion channel expression profile in the neuron. IL, IT, Ih1-4, INa, and IK, denote the leak current, low-threshold calcium current, hyperpolarization activated current (carried by HCN1-4), fast sodium current and delayed rectifier potassium current, respectively. ICl is the synaptic current mediated by glycinergic and GABAergic neurotransmitters. INMDA and InonNMDA are synaptic currents mediated by NMDA and non-NMDA sensitive glutamate receptors. (B) The effects of changing Ih (x-axis) and IT (y-axis) on the resting membrane potential (color coded) in the simulated neuron. As expected, increases in Ih and IT further depolarize the neuron. A depolarizing shift in the resting membrane potential reflects increased neural excitability. (C) Illustration of bursts of action potential spikes from the agonist and antagonist burst neurons. The alternate spiking behavior of these neurons is evident when they are plotted along the same time scale (x-axis). (D) Simulation (grey trace) of essential tremor (black trace) is shown. The tremor amplitude (y-axis) is plotted against time (x-axis). The time scale for simulated essential tremor is the same as the time scale for the traces representing the spiking behavior of the burst neurons. The frequency of tremor recorded from the patient is 5.7 Hz, which is closely simulated by the neuromimetic model (5.9 Hz). The amplitude of the simulated tremor also resembles the one recorded from the ET patient.
Postural tremor in ET has a relatively wide range of frequencies [21]. Therefore, we asked if the conductance-based membrane model could simulate the inter-subject variability in the tremor frequency. The range of frequencies and corresponding amplitude of the postural tremor from 22 ET patients are illustrated in Figure 3A. The tremor frequency in these ET patients ranged from 3–11 Hz. We simulated this variability in frequency by changing the value of Ih and IT. How the amplitude and frequency of tremor depend on the value of Ih and IT in our model is shown in Figure 3B and 3C. In these figures the x- and y-axes represent IT and Ih, respectively. The oscillation frequency (Hz) and amplitude (degree) are color coded and plotted along the z-axes. Tremor occurred when conductance through Ih was larger than 1 mS/cm2 (approximately 10 times larger than its physiological value, 0.1 mS/cm2; S: seimens) [22,23]. The effects of increase in Ih on changes in frequency and amplitude, for a given constant value of IT, were investigated. Increasing Ih, while keeping IT at a constant value, increased the frequency of tremor (slope ± 95% confidence interval: 0.5 ± 0.05). However, when IT was kept constant, there was only a slight decrease in the tremor amplitude when Ih was increased (slope ± 95% confidence interval: -0.02 ± 0.10). Then we investigated the changes in the frequency and amplitude of tremor when IT was increased but Ih was kept at a constant value. With a constant Ih, the tremor frequency decreased when IT was increased (slope ± 95% confidence interval: -3.9 ± 0.1). In a similar analysis, the amplitude of tremor, however, increased with increasing IT (slope ± 95% confidence interval: 1.1 ± 0.01).
Figure 3.
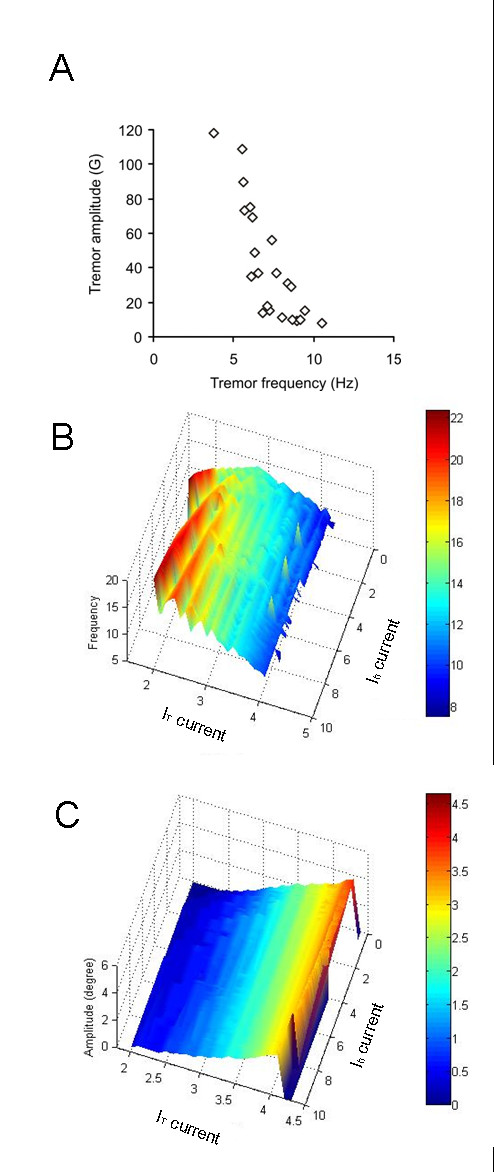
(A) Correlation of frequency and amplitude of tremor in 22 ET patients. Each data point represents one patient. A negative correlation was noted between the frequency and amplitude in 22 ET patients. Also note the frequency of ET ranges from 3–11 Hz in the patients. G = 0.0098 m/s2. (B) The frequency of the oscillations is determined primarily by Ih and IT, which in turn depend upon the distribution of ion channel subtypes (upper panels). The frequency of oscillation is predominantly determined by Ih, while its amplitude is predominantly determined by IT. Notice in panel 'B' that as we increase IT the red color appears sooner for higher values of Ih but is not present when Ih is relatively low (~2 ms). (C) The amplitude of tremor is predominantly determined by the value of IT.
If Ih was increased above the physiological values, ET was simulated for any IT that was 2.75 mS/cm2 or larger (normal IT: 2.0 mS/cm2, reference 26). In contrast, if Ih was near its physiological range, increasing IT alone did not simulate oscillations.
We tuned the model to match the tremor frequency measured in each of the 22 ET patients. Then we plotted the observed versus simulated frequency for each patient as colored diamonds, indicating the model values of Ih and IT required for that patient (Figure 4). There was a nearly perfect correlation between the frequency of simulated tremor and the corresponding postural tremor in the patients (all the data points fall along the black dashed equality line). The data points in Figure 4 are color-coded according to the value IT or Ih. The higher values of Ih correspond to ET patients with higher tremor frequencies (Figure 4A). Consistent with the results in Figure 3, the range of Ih to simulate ET was 10 – 80 times larger than its normal physiological value. The corresponding range of IT to simulate the characteristics of ET was only 1.3 – 3 times higher than its physiological value. Our results suggest that the variability of tremor frequency among ET patients is determined by both Ih and IT. Tremor frequency increases with increasing Ih and decreases with increasing IT.
Figure 4.
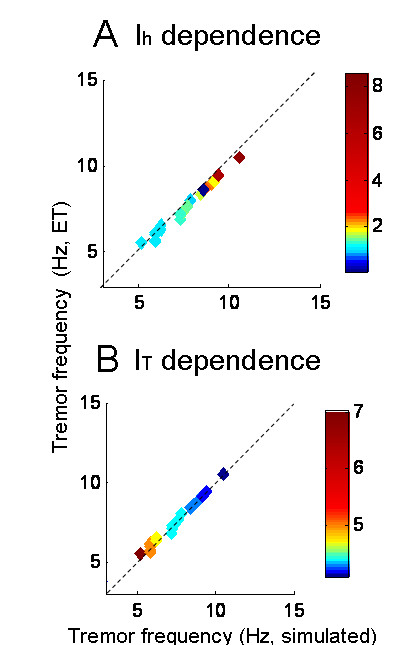
Strong correlation between the frequency of simulated tremor and corresponding postural tremor frequency in the ET patients. Note that all the data points fall along the equality line suggesting a strong correlation. The color coding illustrates the value of IT or Ih in the model that was required to simulate tremor from each ET patient. (A) The higher values of Ih correspond to higher amplitude as well as frequency of tremor. The range of modeled Ih conductances to simulate the ET is 10 – 80 times larger than its physiological value. (B) The higher values of IT in the model corresponds to the lower frequency. In contrast to Ih, the range of IT to simulate ET is only 1.3 – 3 times higher than its physiological value. These results imply that tremor frequency amongst ET is predominantly determined by the Ih (as compared with IT).
In summary, simulations support our hypothesis that an increase in premotor neural excitability, caused by increasing Ih and IT, results in oscillations of a reciprocally innervated neural circuit. These oscillations resemble ET. The frequency of tremor is determined by both IT and Ih.
Discussion
Essential tremor (ET) is a common but poorly understood neurological disorder [25,26]. Patients with ET, however, are reasonably well treated with drugs that reduce membrane excitability. Therefore, we asked whether or not increased membrane excitability could play a critical role in the pathogenesis of ET.
How increased membrane excitability affects motor control?
Increases in membrane excitability could affect motor control in many ways. One mechanism is by destabilizing the circuits comprised of reciprocally innervated neurons. Such circuits exist at many levels in the central nervous system. This pattern of reciprocal innervation between pre-motor neurons projecting to a pair of agonist-antagonist muscles is fundamental for efficient force generation during ballistic movements [9]. The stability in these circuits requires adequate external inhibition. Either removal of external inhibition [12] or increasing the excitability of the constituent neurons (as proposed here) could lead to oscillations that produce tremor.
Can hyperexcitability cause oscillations in motor circuits that lead to tremor?
We used a conductance-based model of burst neurons with physiologically realistic membrane properties and anatomically realistic neural connections to test this hypothesis. The cardinal features of this model were threefold: 1) increased neural excitability secondary to increase in Ih and/or IT, 2) post-inhibitory rebound (PIR), and 3) inherent circuit instability resulting from reciprocal innervation between the neurons projecting to agonist and antagonist muscle pairs. The model simulated oscillations resembling ET when Ih was increased (with or without an increase in IT). While suggesting an overall conceptual framework underlying oscillatory behavior, our model can not pinpoint the specific anatomical neural networks that produce ET. Nevertheless, we can reasonably suggest that the following anatomical regions might be involved.
Neural circuits that might oscillate
Circuits in the thalamus, inferior olive, cerebrum and cerebellum are involved in the generation of limb movements. One mechanism for tremor is that a group of neurons within a single nucleus develops an abnormal oscillatory mode. In this mode, a neural discharge is followed by a prolonged hyperpolarization that terminates in rebound spikes. Thus, each neuron oscillates independently. Synchronization of such independently oscillating neurons could result in rhythmic activity that becomes strong enough to cause gross motor oscillations. Electrotonic coupling through connexin gap junctions can facilitate synchronization in premotor nuclei such as the inferior olive [27]. Cells of the inferior olive also express ion channels that carry Ih and IT. Moreover, Ih is thought to influence the synchronization of oscillations in the inferior olive [28]. It is possible that increased intracellular levels of cAMP in the inferior olive increases Ih conductance to facilitate these oscillations. Octanol, which reduces synchronized oscillations in the inferior olive, also reduces ET [29,30].
Reciprocal innervation occurs in many neural circuits controlling limb movements [9,12]. For example, thalamo-cortical relay (TC) neurons send glutamatergic excitatory projections to thalamic reticular (TR) neurons. TR neurons send GABA mediated inhibitory projections to TC neurons [31-33]. TR neurons mutually inhibit each other via inhibitory collaterals [31-33]. Thus interactions between TC and TR neurons make reciprocal loops with positive feedback from the TC to TR neurons, negative feedback from TR to TC neurons, and mutually inhibitory TR neurons [34]. The globus pallidus internus (GPi) sends inhibitory projections to TC and TR neurons in the motor thalamus [35,36]. Furthermore, thalamic and GPi neurons carry Ih and IT, and exhibit post-inhibitory rebound [1,15,37,38]. Hence, the reciprocally innervated neural network formed by the TC and TR neurons might be inherently unstable [12]. Physiologically, this network is under control by external inhibition from the GPi.
Bursting behavior in neurons, which is fundamental for making a circuit prone to oscillations, is also seen in the subthalamic nucleus [39]. Reciprocal innervation – a key feature for oscillating neural circuits – is also apparent in the spinal cord [9]. Thus there are a number of areas related to motor control within the central nervous system that are comprised of neurons and circuits that are prone to oscillations.
Increasing the membrane excitability in our lumped model
In our model we increased the Ih to increase membrane excitability and produce oscillations. As this is a lumped model, we can not differentiate among increasing the maximal conductance of an individual channel, increasing the number of channels, or increasing the probability that a channel is open. Any combination of these changes would increase Ih. Likewise, there are a number of intracellular modulators regulating Ih including intracellular levels of calcium, cAMP, and pH [15,16] that could affect membrane excitability. Again, our simulated lumped model cannot tell us which of these factors might be the cause. We had to increase Ih by ten-fold to simulate tremor. Although the range of pathological changes in channel currents is not known, a ten-fold increase does not seem unreasonable, as experimental studies have shown that conductances can be changed several fold [17]. Thus, the conceptual underpinning of our lumped model seem plausible.
Naturally, we do not expect that all patients with ET have the same cause for their increased excitability and consequent tremor. But, with the wide range of ion channels that participate in the molecular cascade that accounts for PIR, many types of alterations could change membrane excitability and in turn lead to the phenotype of ET. Similarly, the effects of loss of inhibition from the cerebellar Purkinje neurons (which may be abnormal in some patients with ET, see below) might also change the membrane threshold and increase the excitability of the premotor neurons, and lead to the appearance of tremor in some ET patients [25,26]. Indeed, our model underscores the importance of external inhibition in preventing oscillations in a reciprocally innervated circuit. For example, removal of external inhibition in an analogous model accounted for limb tremor in a syndrome called micro-saccadic oscillation and limb tremor – microSOLT [12].
Some ET patients have the gly9 susceptibility variant of the DRD3 gene [40,41]. DRD3 receptor is expressed in the thalamus and substantia nigra where limb-movement related neurons are present [42]. Two of the possible effects of this mutation could be directly related to the regulation of membrane excitability. There is a prolonged intracellular action of mitogen-activated protein kinase (MAPK) in the gly9 variant of DRD3 [40]. The latter can cause increased intracellular levels of cAMP via excessive inhibition of phosphodiesterase E4 [43-45]. Conversely, it was also shown that gly9 variant in DRD3 gene is associated with reduced forskolin induced formation of cAMP [40]. Although it is not known what the intracellular levels of cAMP in ET are, it is possible that they are altered. If they are higher, Ih could be increased and thus lead to an increase in membrane excitability.
Co-existing changes in central nervous system to change oscillation kinematics
Our simulations suggested that increasing Ih, and thus increasing excitability, increases the tremor frequency. It was also reported that the frequency of ET decreases and the amplitude increases with age [46]. Aging might alter the profile of membrane ion channels; our model explains the effects of maximal ion conductance on the tremor frequency. Furthermore, our simulations suggest that a parallel increase in IT, when Ih is already increased, reduces the frequency of the tremor (Figure 3B) and increases its amplitude (Figure 3C). This explanation does not exclude the possibility that aging could also change other membrane and circuit properties including the latency of the long feedback loop around the burst neurons.
Caveats and future directions
Although our hypotheses remain to be proven experimentally, they suggest new approaches to understanding common tremor disorders. The conceptual scheme that we present here could also be used to analyze tremor disorders other than ET. However, the unique aspect of this model is that it can simulate oscillations by changing intrinsic membrane properties of the burst neurons and does not require any 'structural' changes in the anatomical organization or connectivity of the constituent neurons. The latter is relevant to ET, since there are no gross structural changes except in some cases in which there is a decrease in cerebellar Purkinje neurons [25,26]. Loss of external inhibition to reciprocally innervating circuits could be an important underlying pathophysiological mechanism component in other tremor disorders including Parkinson's disease, cerebellar tremor, and micro-saccadic oscillations and limb tremor [12,34].
Finally, treatment with pharmacological blockers targeted towards ion channels may offer therapeutic benefits. For example, although counterintuitive, interfering with the function of a normal ion channel to decrease membrane excitability in the face of impaired external inhibition might reduce oscillatory behavior. Such an approach is similar to that for some forms of inherited epilepsy in which seizures are presumably caused by an abnormal ion channel. Treatment then can target another, presumably intact channel [47].
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
AS formulated the hypothesis and experimental design, carried out the experimental studies, participated in the model development, analyzed data, and wrote the manuscript. KM developed the model. LO participated in the model development and manuscript writing. SR participated in model development. RT collected the experimental data and prepared the experimental data analysis software. DZ formulated the hypothesis, experimental and modeling design, and wrote the manuscript. All authors read, edited, and approved the final manuscript.
Supplementary Material
Supplementary material
Membrane based model for essential based tremor
Acknowledgments
Acknowledgements
The work was supported by grants from NIH EY01849, Intramural Division of the National Eye Institute (NIH, DHHS), Gustavus and Louise Pfeiffer Foundation, and Ataxia-telangiectasia Children's Project. The authors thank Mr. Dale Roberts and Mr. Adrian Lasker for comments and support.
Contributor Information
Aasef G Shaikh, Email: ashaikh@dizzy.med.jhu.edu.
Kenichiro Miura, Email: kmiura@brain.med.kyoto-u.ac.jp.
Lance M Optican, Email: lanceoptican@nih.gov.
Stefano Ramat, Email: steram@bioing.unipv.it.
Robert M Tripp, Email: bob@flexable.com.
David S Zee, Email: dzee@dizzy.med.jhu.edu.
References
- McCormick DA. Pape HC Noradrenergic and serotonergic modulation of a hyperpolarization-activated cation current in thalamic relay neurones. J Physiol. 1990;431:319–42. doi: 10.1113/jphysiol.1990.sp018332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilligan B. Propranolol in essential tremor. Lancet. 1972;2:980. doi: 10.1016/s0140-6736(72)92520-2. [DOI] [PubMed] [Google Scholar]
- Ondo W, Hunter C, Vuong KD, Schwartz K, Jankovic J. Gabapentin for essential tremor: a multiple-dose, double-blind, placebo-controlled trial. Mov Disord. 2000;15:678–82. doi: 10.1002/1531-8257(200007)15:4<678::aid-mds1012>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Mihic SJ, Ye Q, Wick MJ, Koltchine VV, Krasowski MD, Finn SE, Mascia MP, Valenzuela CF, Hanson KK, Greenblatt EP, Harris RA, Harrison NL. Sites of alcohol and volatile anaesthetic action on GABA(A) and glycine receptors. Nature. 1997;389:385–9. doi: 10.1038/38738. [DOI] [PubMed] [Google Scholar]
- Manto M, Laute MA. A possible mechanism for the beneficial effect of ethanol in essential tremor. Eur J Neurol. 2008;15:697–705. doi: 10.1111/j.1468-1331.2008.02150.x. [DOI] [PubMed] [Google Scholar]
- Eggers ED, Berger AJ. Mechanisms for the modulation of native glycine receptor channels by ethanol. J Neurophysiol. 2004;91:2685–95. doi: 10.1152/jn.00907.2003. [DOI] [PubMed] [Google Scholar]
- Connor GS. A double-blind placebo-controlled trial of topiramate treatment for essential tremor. Neurology. 2002;59:132–4. doi: 10.1212/wnl.59.1.132. [DOI] [PubMed] [Google Scholar]
- White HS. Comparative anticonvulsant and mechanistic profile of the established and newer antiepileptic drugs. Epilepsia. 1999;40:S2–S10. doi: 10.1111/j.1528-1157.1999.tb00913.x. [DOI] [PubMed] [Google Scholar]
- Sherrington CS. On reciprocal innervation of antagonistic muscles – tenth note. Roy Soc Proc. 1907;52:337–349. [Google Scholar]
- Llinás RR. The intrinsic electrophysiological properties of mammalian neurons: insights into central nervous system function. Science. 1988;242:1654–1664. doi: 10.1126/science.3059497. [DOI] [PubMed] [Google Scholar]
- Ramat S, Leigh RJ, Zee DS, Optican LM. Ocular oscillations generated by coupling of brainstem excitatory and inhibitory saccadic burst neurons. Exp Brain Res. 2005;160:89–106. doi: 10.1007/s00221-004-1989-8. [DOI] [PubMed] [Google Scholar]
- Shaikh AG, Miura K, Optican LM, Ramat S, Leigh RJ, Zee DS. A new familial disease of saccadic oscillations and limb tremor provides clues to mechanisms of common tremor disorders. Brain. 2007;130:3020–31. doi: 10.1093/brain/awm240. [DOI] [PubMed] [Google Scholar]
- Kralic JE, Criswell HE, Osterman JL, O'Buckley TK, Wilkie ME, Matthews DB, Hamre K, Breese GR, Homanics GE, Morrow AL. Genetic essential tremor in gamma-aminobutyric acidA receptor alpha1 subunit knockout mice. J Clin Invest. 2005;115:774–9. doi: 10.1172/JCI23625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Xie WJ, Le WD, Huang MS, Jankovic J. Genetic analysis of the GABRA1 gene in patients with essential tremor. Neurosci Lett. 2006;401:16–19. doi: 10.1016/j.neulet.2006.02.066. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Pape HC. Properties of a hyperpolarization-activated cation current and its role in rhythmic oscillation in thalamic relay neurones. J Physiol. 1990;431:291–318. doi: 10.1113/jphysiol.1990.sp018331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh AG, Finlayson PG. Excitability of auditory brainstem neurons, in vivo, is increased by cyclic-AMP. Hear Res. 2005;201:70–80. doi: 10.1016/j.heares.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Wainger BJ, DeGennaro M, Santoro B, Siegelbaum SA, Tibbs GR. Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature. 2001;411:805–10. doi: 10.1038/35081088. [DOI] [PubMed] [Google Scholar]
- Lüthi A, McCormick DA. Modulation of a pacemaker current through Ca(2+)-induced stimulation of cAMP production. Nat Neurosci. 1999;2:634–41. doi: 10.1038/10189. [DOI] [PubMed] [Google Scholar]
- Caligiuri MP, Tripp RM. A portable hand-held device for quantifying and standardizing tremor assessment. J Med Eng Technol. 2004;28:254–62. doi: 10.1080/03091900410001658111. [DOI] [PubMed] [Google Scholar]
- Moosmang S, Stielber J, Zong X, Biel M, Hofmann F, Ludwig A. Cellular expression and functional characterization of four hyperpolarization-activated pacemaker channels in cardiac and neuronal tissues. Eur J Biochem. 2001;268:1646–52. doi: 10.1046/j.1432-1327.2001.02036.x. [DOI] [PubMed] [Google Scholar]
- Deuschl G, Raethjen J, Lindemann M, Krack P. The pathophysiology of tremor. Muscle Nerve. 2001;24:716–735. doi: 10.1002/mus.1063. [DOI] [PubMed] [Google Scholar]
- Huguenard JR, McCormick DA. Simulation of the currents involved in rhythmic oscillations in thalamic relay neurons. J Neurophysiol. 1992;68:1373–83. doi: 10.1152/jn.1992.68.4.1373. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Huguenard JR. A model of the electrophysiological properties of thalamocortical relay neurons. J Neurophysiol. 1992;68:1384–400. doi: 10.1152/jn.1992.68.4.1384. [DOI] [PubMed] [Google Scholar]
- Destexhe A, Contreras D, Steriade M, Sejnowski TJ, Huguenard JR. In vivo, in vitro, and computational analysis of dendritic calcium currents in thalamic reticular neurons. J Neurosci. 1996;16:169–85. doi: 10.1523/JNEUROSCI.16-01-00169.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrad JE, Louis ED, Honig LS, Flores I, Ross GW, Pahwa R, Lyons KE, Faust PL, Vonsattel JP. Reduced purkinje cell number in essential tremor: a postmortem study. Arch Neurol. 2008;65:101–107. doi: 10.1001/archneurol.2007.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis ED, Vonsattel JP. The emerging neuropathology of essential tremor. Mov Disord. 2008;23:174–82. doi: 10.1002/mds.21731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotelo C, Llinas R, Baker R. Structural study of inferior olivary nucleus of the cat: morphological correlates of electrotonic coupling. J Neurophysiol. 1974;37:541–559. doi: 10.1152/jn.1974.37.3.541. [DOI] [PubMed] [Google Scholar]
- Bal T, McCormick DA. Synchronized oscillations in the inferior olive are controlled by the hyperpolarization-activated cation current Ih. J Neurophysiol. 1997;77:3145–56. doi: 10.1152/jn.1997.77.6.3145. [DOI] [PubMed] [Google Scholar]
- Shill HA, Bushara KO, Mari Z, Reich M, Hallett M. Open-label dose-escalation study of oral 1-octanol in patients with essential tremor. Neurology. 2004;62:2320–2. doi: 10.1212/wnl.62.12.2320. [DOI] [PubMed] [Google Scholar]
- Sinton CM, Krosser BI, Walton KD, Llinás RR. The effectiveness of different isomers of octanol as blockers of harmaline-induced tremor. Pflugers Arch. 1989;414:31–36. doi: 10.1007/BF00585623. [DOI] [PubMed] [Google Scholar]
- Sherman SM, Guillery RW. Exploring the Thalamus. Academic Press, San Diego; 2001. pp. 144–167. [Google Scholar]
- Pinault D. The thalamic reticular nucleus: Structure, function and concept. Brain Res Rev. 2004;46:1–31. doi: 10.1016/j.brainresrev.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Guillery RW, Harting JK. Structure and connections of the thalamic reticular nucleus: Advancing views over half a century. J Comp Neurol. 2003;463:360–371. doi: 10.1002/cne.10738. [DOI] [PubMed] [Google Scholar]
- Hadipour-Niktarash A. A computational model of how an interaction between the thalamocortical and thalamic reticular neurons transforms the low-frequency oscillations of the globus pallidus. J Comput Neurosci. 2006;20:299–320. doi: 10.1007/s10827-006-6673-5. [DOI] [PubMed] [Google Scholar]
- Parent A, Hazrati LN. Functional anatomy of the basal ganglia. Part I: The cortico-basal ganglia-thalamo-cortical loop. Brain Res Rev. 1995;20:91–127. doi: 10.1016/0165-0173(94)00007-c. [DOI] [PubMed] [Google Scholar]
- Takada M, Hattori T. Glycine: an alternative transmitter candidate of the pallidosubthalamic projection neurons in the rat. J Comp Neurol. 1987;262:465–72. doi: 10.1002/cne.902620402. [DOI] [PubMed] [Google Scholar]
- Nambu A, Llinas R. Electrophysiology of globus pallidus neurons in vitro. J Neurophysiol. 1994;72:1127–39. doi: 10.1152/jn.1994.72.3.1127. [DOI] [PubMed] [Google Scholar]
- Llinás R, Jahnsen H. Electrophysiology of mammalian thalamic neurones in vitro. Nature. 1982;297:406–408. doi: 10.1038/297406a0. [DOI] [PubMed] [Google Scholar]
- Beurrier C, Congar P, Bioulac B, Hammond C. Subthalamic nucleus neurons switch from single-spike activity to burst-firing mode. J Neurosci. 1999;19:599–609. doi: 10.1523/JNEUROSCI.19-02-00599.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanneteau F, Funalot B, Jankovic J, Deng H, Lagarde JP, Lucotte G, et al. A functional variant of the dopamine D3 receptor is associated with risk and age-at-onset of essential tremor. Proc Natl Acad Sci USA. 2006;103:10753–8. doi: 10.1073/pnas.0508189103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucotte G, Lagarde JP, Funalot B, Sokoloff P. Linkage with the Ser9Gly DRD3 polymorphism in essential tremor families. Clin Genet. 2006;69:437–40. doi: 10.1111/j.1399-0004.2006.00600.x. [DOI] [PubMed] [Google Scholar]
- Sóvágó J, Makkai B, Gulyás B, Hall H. Autoradiographic mapping of dopamine-D2/D3 receptor stimulated [35S]GTPgammaS binding in the human brain. Eur J Neurosci. 2005;22:65–71. doi: 10.1111/j.1460-9568.2005.04192.x. [DOI] [PubMed] [Google Scholar]
- Hoffmann R, Baillie GS, Mackenzie SJ, Yarwood SJ, Houslay MD. The MAP kinase ERK2 inhibits the cyclic AMP-specific phosphodiesterase, HSPDE4D3 by phosphorylating it at Ser579. EMBO J. 1999;18:893–903. doi: 10.1093/emboj/18.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houslay MD, Milligan G. Tailoring cAMP signaling responses through isoform multiplicity. Trends Biochem Sci. 1997;22:217–224. doi: 10.1016/s0968-0004(97)01050-5. [DOI] [PubMed] [Google Scholar]
- Houslay MD, Sullivan M, Bolger GB. The multi-enzyme PDE4 cyclic AMP specific phosphodiesterase family: intracellular targeting, regulation and selective inhibition by compounds exerting anti-inflammatory and anti-depressant actions. Adv Pharmacol. 1998;44:225–342. doi: 10.1016/s1054-3589(08)60128-3. [DOI] [PubMed] [Google Scholar]
- Elble RJ. Essential tremor frequency decreases with time. Neurology. 2000;55:1547–51. doi: 10.1212/wnl.55.10.1547. [DOI] [PubMed] [Google Scholar]
- Cannon SC. Pathomechanisms in Channelopathies of Skeletal Muscle and Brain. Annu Rev Neurosci. 2006;29:387–415. doi: 10.1146/annurev.neuro.29.051605.112815. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material
Membrane based model for essential based tremor