

Abstract
Physiological and pathological turnover of basement membranes liberates biologically active cryptic molecules. Several collagen-derived fragments possess anti-angiogenic activity. Arresten is the 26-kDa non-collagenous domain of type IV collagen α1 chain. It functions as an efficient inhibitor of angiogenesis and tumor growth in mouse models, but its anti-angiogenic mechanism is not completely known. Here we show that arresten significantly increases apoptosis of endothelial cells in vitro by decreasing the amount of anti-apoptotic molecules of the Bcl-family, Bcl-2 and Bcl-xL. Although the pro-apoptotic effect of arresten is endothelial cell specific in vitro, in mouse tumors arresten induced apoptosis both in endothelial and tumor cells. The tumor cell apoptosis is likely an indirect effect due to the inhibition of blood vessel growth into the tumor. The active site of arresten was localized by deletion mutagenesis within the C-terminal half of the molecule. We have previously shown that arresten binds toα1β1 integrin on human umbilical vein endothelial cells. However, the microvascular endothelial cells (MLECs) are more important in the context of tumor vasculature. We show here that arresten binds also to the microvascular endothelial cells via α1β1 integrin. Furthermore, it has no effect on Matrigel neovascularization or the viability of integrin α1 null MLECs. Tumors implanted on integrin α1 deficient mice show no integrin α1 expression in the host-derived vascular endothelium, and thus arresten does not inhibit the tumor growth. Collectively, this data sheds more light into the anti-angiogenic mechanism of arresten.
Keywords: angiogenesis, apoptosis, arresten, collagen IV, basement membrane, integrin
INTRODUCTION
Angiogenesis, the formation of new blood vessels, is a critical event in tumor growth and metastasis [1]. Solid tumors will remain smaller than a few millimeters in diameter, if they are not able to induce their own blood supply, and thus inhibition of tumor angiogenesis suppresses tumor growth [1, 2]. Capillary endothelial cells are supported by vascular basement membranes (VBM), which influence several aspects of endothelial cell (EC) behavior, such as cell proliferation [3, 4]. Type IV collagen network is the structural backbone of the VBM [5]. Several cryptic fragments from non-collagenous (NC1) domains of type IV and XVIII collagens, such as arresten, canstatin, endostatin and tumstatin, have been shown to possess anti-angiogenic activity [6, 7, 8, 9]. Although all these collagen-derived endogenous inhibitors of angiogenesis have similar molecular sizes, and all of them are proteolytic cleavage products from NC1 domains of VBM collagens, they bind to distinct cell surface receptors and affect different parts of the angiogenic process [10, 11].
Arresten is a 26 kDa anti-angiogenic fragment from the α1 chain of type IV collagen. This molecule inhibits endothelial cell proliferation, migration, tube formation and Matrigel neovascularization [9]. Furthermore, it inhibits the growth of human tumors in nude mice and the development of tumor metastasis. Arresten was shown to bind to α1β1 integrin and heparan sulphate proteoglycans (HSPG) [9]. Later, we showed that many of the anti-angiogenic properties of arresten are mediated through α1β1 integrin [11]. Integrin α1β1 is the major collagen and laminin receptor [12], and it is the only collagen receptor that is able to activate the Ras/Sch/mitogen activated protein kinase (MAPK) pathway, thus promoting cell proliferation [13]. Integrin α1β1 is abundantly expressed on microvascular endothelial cells, but the expression is lower or even absent on endothelial cells lining larger blood vessels [14]. Interestingly, when tumors were implanted into the α1 integrin deficient mice, the tumors showed decreased tumor vascularization and growth [13].
In the present study, we have further characterized the anti-angiogenic properties of arresten. We show that arresten significantly increases apoptosis of endothelial cells by down-regulating the amount of anti-apoptotic molecules Bcl-2 and Bcl-xL. The active anti-angiogenic site of arresten is located in the C-terminal part of the molecule. We confirm and further explore the role of α1β1 integrin as a functional receptor of arresten on microvascular vessels essential for tumor blood supply. Integrin α1 is required for the anti-survival effect of arresten. Furthermore, the tumors implanted on integrin α1 deficient mice show no integrin α1 positive vasculature, and consequently the growth of tumors and blood vessels in these mice is not inhibited by arresten.
MATERIALS AND METHODS
Production of recombinant arresten and arresten deletion mutants
Human recombinant arresten was cloned and produced in Escherichia coli as previously described [9]. Briefly, arresten was expressed in the pET22b (+) expression vector (Novagen, Madison, WI) in BL21 cells (Novagen). Protein expression was induced by isopropyl-1-thio-β-D-galactopyranoside (IPTG) to a final concentration of 1 mM. After a 3-hour induction, cells were harvested, lysed with a lysis buffer (6 M guanidine, 0.1 M NaH2PO4, 0.01 M Tris-HCl, pH 8.0), sonicated and centrifuged. The supernatant was passed through a Ni-nitriloacetic acid agarose column (Qiagen, Chatsworth, CA). Arresten was eluted with an increasing concentration of imidazole (10, 25, 40, 125 and 250 mM) in 8 M urea, 0.1 M NaH2PO4, 0.01 M Tris-HCl, pH 8.0, and refolded by dialyzing twice against PBS. The concentration and purity of the soluble part of arresten was assayed with a bicichoninic acid assay (Pierce, Rockford, IL, USA) and SDS-PAGE. The production of recombinant human his-tagged arresten resulted in a predominantly soluble 29 kDa protein, consisting of the 26 kDa native arresten and 3 kDa polylinker and 6-histidine tag sequences. In order to localize the active site of arresten, mutants of arresten were generated by deletion mutagenesis, and these were termed Arr-1 (the first 115 amino acids) and Arr-2 (the last 115 amino acids). Both proteins were produced in E. coli using the pET-28a vector expression system (Novagen), and purified by their his-tag sequence using a Ni-Agarose column as described above. Polymyxin B (5 μg/ml) (Sigma) was used in all assays to remove the possible endotoxin contamination of arresten purified from bacteria [15].
Production of a synthetic T7 peptide
The synthetic T7 peptide (TMPFLFCNVNDCNFASRNDYSYWL) derived from tumstatin [16] was synthesized at Tufts University Core Facility (Boston, MA), analyzed by mass-spectrophotometer analysis and purified by analytical HPLC.
Cell lines
Calf pulmonary aortic endothelial (C-PAE) cells, HT1080 fibrosarcoma cells, and renal cell carcinoma cells (786-0) were grown in DMEM containing 10% FBS 100 units/ml penicillin and 100 μg/ml streptomycin. Primary gingival fibroblasts (GF) were isolated and grown as previously [17]. Human prostate adenocarcinoma cells (PC-3) were grown in F12K containing 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin. Human tongue squamous cell carcinoma cells (HSC-3) were grown as described previously [18]. Human umbilical vein endothelial cells HUVEC (ATCC CRL-1730) and human primary microvascular endothelial (HMVEC, Lonza) cells were cultured in EGM-2-MV supplemented media (Clonetics/Lonza). CT26 colon carcinoma cells (CRL-2638 from ATCC) were cultured in DMEM supplemented with 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin. Primary mouse lung endothelial cell lines (MLEC) from wild type (Charles River) and α1 integrin deficient Balb/c mice [19, 20] were prepared as previously [11]. Briefly, the mice were sacrificed with cervical dislocation. The lungs were then perfused with ice-cold PBS-heparin (1U/ml), and the lung and heart were collected into cold Ham’s F-12 (Life Technologies). The lungs were digested with 0.1% collagenase for 1 hour at 37ºC, and plated to 0.1% gelatin coated flasks in MLEC media (40% Ham’s, 40% DMEM low glucose, 20% FBS, 1% penicillin-streptomycin, 2 mM L-glutamine, 100 μg/ml heparin (H3393, Sigma, St. Louis, MO) and 50 μg/ml endothelial mitogen (Biomedical Technologies)). Negative selection was performed with magnetic beads (Dynabeads M-450 Sheep anti-Rat IgG; Dynal, Oslo, Norway) conjugated with rat anti-mouse FcγII/III (Pharmingen, San Diego, CA), and positive selection was done twice with beads conjugated with rat anti-mouse ICAM-2 (Pharmingen).
Annexin V-FITC assay
C-PAE (0.5 × 105) cells were added to each well of a 6-well tissue culture plates in DMEM, 10% FBS for 12 hours. Fresh medium together with 175 nM of arresten, arresten deletion mutants (Arr-1 and Arr-2) or 40 ng/ml TNF-α was added at 2 and 4-hour time points. Control cells were treated with an equal volume of PBS. Subsequently, detached cells and adherent cells were pooled together and centrifuged at 1500 rpm. Cells were then washed with binding buffer (Clontech), and phosphatidyl-serine externalization (an early indicator of apoptosis) was measured by labeling with FITC-labeled annexin V (Clontech) according to the manufacturer’s instructions. Annexin-FITC labeled cells (1 × 104 cells/treatment) were counted using a Becton-Dickinson FACStar plus flow cytometer, and the data was further analyzed using standard Cell Quest software recording the shift in fluorescence peak corresponding to increased apoptosis. Duplicate assays were performed (two parallel samples each time), and the results are shown as a percentage of apoptotic cells ± SE. Additionally, the morphological changes in the C-PAE cells upon arresten treatment were analyzed microscopically.
The Caspase-3 assay
A suspension of 0.5×106 786-0, GF, HMVEC, HT1080, HSC-3 and PC-3 cells were plated on a 10 cm2 tissue culture plate in normal culture media, and the cells were allowed to attach for 24 hours, followed by serum-starvation in DMEM containing 2% FBS overnight. Next, the cells were treated in 2% serum with 3 ng/ml bFGF and arresten (350, 700 or 1400 nM), TNF α (40 ng/ml) or 0.01% BSA (negative control) for 16–18 hours. The cells and the media were then harvested and centrifuged for 10 minutes at 200×g. The ApoAlert CPP32/Caspase-3 Assay-kit (Clontech) was used to detect Caspase-3 enzyme activity in cells (4×107 cells/ml). DEVD-fmk, an inhibitor of caspase-3 was used to ensure specificity of the results.
In vitro TUNEL assay
C-PAE cells (5×105 cells/well) were grown in a 6-well fibronectin-coated plates (10μg/ml) in DMEM supplemented with 10% FBS and penicillin/streptomycin for 24 hours, followed by serum starvation (DMEM containing 2% FBS) overnight, and subsequently fresh media containing 3 ng/ml bFGF, 5 ng/ml VEGF and arresten (10–20 μg/ml) or TNF-α (40ng/ml) were added. Control samples received DMEM with 2% FBS, 3 ng/ml of bFGF and 5 ng/ml VEGF. After 24 hours, the floating and attached cells were collected, washed and resuspended in PBS at a concentration of 20×106 cells, and assayed by the Apoalert DNA fragmentation-kit (Clontech) according to the instructions of the manufacturer. The results were analyzed using a digital microscope (Nikon Microphot-SA) with the SPOT digital camera and software (Diagnostic Instrument Inc). Fifteen random fields were counted for each sample.
Immunofluorescence double staining for CD31 and TUNEL
Mice with subcutaneous CT26 tumors were treated with arresten or PBS, and sacrificed after 16 days. The tumors were removed, embedded in OCT and snap-frozen in liquid nitrogen. Double stainings were done from 8 μm cryosections of the tumors. The CD31 (endothelial cells, Pharmingen, clone MEC13.3) staining was done according to a previously published method [21] followed by TUNEL staining for apoptotic nuclei according to the manufacturers protocol (Roche) with a few modifications. Briefly, the TUNEL staining was done after the CD31 staining in acetone and acetone/chloroform fixed sections, and the permeabilization (0.1 % Triton X-100, 0.1 % sodium citrate in PBS) incubation time was increased to 5 min in RT. Finally the sections were mounted with Vectashield mounting media with DAPI (Vector Laboratories). Immunofluorescence microscopy was performed and analyzed using the Axioskop 2 fluorescent microscope, AxioCam HRC camera and the Axiovision 4.3 software (Zeiss) (magnification 200×). Apoptotic cells were counted and averaged from three tumors in control and arresten treated groups (three high power fields per tumor). The results are shown as the number of apoptotic cells ± SE. All mouse experiments were performed according to institutional animal care guidelines.
Western blotting of Bax, Bcl-2 and Bcl-xL
After a 24-h incubation with arresten (700 nM) or TNF-α (40 ng/ml) in low-serum media (2%), the HMVEC endothelial cells (90% confluent) were washed with cold PBS, lysed on ice in 1× lysis buffer (Cell Signaling) for 5 min, briefly sonicated and centrifuged for 10 min at 14000×g at 4°C. Protein concentration in the supernatants was measured with BCA Protein Assay Kit (Pierce) and 20 μg of proteins were separated using 12% SDS-PAGE followed by Coomassie Blue staining to verify equal loading in all lanes. The gels were destained and proteins were electrotransfered onto nitrocellulose membranes. Membranes were blocked with 5% non-fat milk for 1 h. Polyclonal anti-human Bax (Cell Signaling) and Bcl-xL (Cell Signaling) primary antibodies (dilution 1:1000) were incubated overnight at 4°C, and monoclonal anti-human Bcl-2 (Dako, clone 124) antibody (1:1000) was incubated overnight at RT followed by peroxidase-conjugated goat anti-rabbit or anti-mouse IgG (Dako, 1:200) for 1 h at RT. Immunoreactive proteins were visualized with ECL Western blotting detection reagents (Amersham). The treatments with arresten or TNF-α and the Western blotting were performed twice.
Cell attachment assay
The cell attachment assays were performed as previously described [22]. Briefly, 96-well plates were coated with 10 μg/ml arresten, 10 μg/ml fibronectin or 0.5 μg/ml vitronectin (BD Biosciences, San Jose, CA), followed by blocking with 30 mg/ml bovine serum albumin (Sigma) for an hour. HUVEC or primary MLECs from wild type or α1 integrin deficient mice (1×105 cells/ml) were incubated with a function blocking α1β1 integrin antibody (10 μg/ml) (a generous gift from Dr Humphrey Gardner, clone Ha31/8) [23], 1 or 5 μg/ml of cRGD (Arg-Gly-Asp) or RAD (Arg-Ala-Asp) peptides (Peptides International, Louisville, KY) for 15 minutes. Following this 100 μl of cell suspension/well was added onto plates and incubated for 45 min at 37ºC. After washing, cells were incubated 5 h - overnight in full media and the number of attached cells was determined with methylene blue staining [8]. Briefly, cells were washed with 1×PBS, fixed with 10% formalin in neutral buffered saline (Sigma) for 30 min at room temperature. After formalin removal, the cells were stained with 1% solution of methylene blue (Sigma) in 0.01 M borate buffer (pH 8.5) for 30 min at room temperature, followed by washing with borate buffer, and removal of methylene blue with 0.1 N HCl/ethanol (1:1). The results were analyzed on a microplate reader (Bio-Rad, Hercules, CA) at 655 nm.
Cell viability assay
Cell viability was measured with the MTT (3-(4,5-dimethylthiatzol-2-yl)-2,5-diphenyl tetrasolium bromide) (Chemicon, Temecula, CA, USA) assay. Wild type and α1 integrin deficient MLEC (0.4×104 cells/well) were plated in a 96-well plate and were allowed to attach overnight. The cells were then incubated with 0, 5, 50 and 500 nM of arresten for 24 h, after which MTT reagent was added to the cells for 6 hours. 80 ng/ml of TNF-α (Clontech, Palo Alto, CA) was used as a positive control for apoptosis. Viable cells produced black crystals in the presence of MTT, and the crystals were dissolved in isopropanol with 0.04 N HCl or DMSO. The resulting purple color was measured in a microplate reader (BioRad) at 570 nm.
Proliferation assay
Confluent C-PAE cells were trypsinized, and 0.4×105 cells/well were plated onto fibronectin-coated (10 μg/ml) 96-well plates, in 0.5% FBS DMEM. The following day, the media was changed to 20% FBS DMEM and stimulated with different concentrations of Arr-1 and Arr-2 (0.1, 0.5, 1, 5, 20 μg/ml; 7, 35, 70, 350 and 1400 nM respectively). Unstimulated cells were treated with 0.1% FBS in DMEM. Cells were allowed to proliferate for 24–48 hours, and the assay was stopped by formalin fixation before cells reached confluence, followed by methylene blue staining as previously described. For primary MLEC 0.7 × 105 cells/well, were plated to 96-well plates coated with 0.1% gelatin and 10 μg/ml fibronectin. After the cells had attached, they were serum starved (2% FBS) for 24 hours. The cells were then stimulated with media containing VEGF (10 ng/ml) with 0, 5, 50 or 500 nM of recombinant arresten or T7 peptide [16]. After 48 hours incubation, methylene blue staining was performed as described previously.
Matrigel plug assay
Matrigel (BD Biosciences) was thawed at 4°C, and mixtures containing 3% DMSO, 20 U/ml heparin (Pierce, Rockford, IL, USA), 100 ng/ml bFGF (R&D Systems, Minneapolis, MN) and 50 ng/ml VEGF (R&D Systems) with either PBS (control group, n=10 for wild type and n=9 for α1 integrin deficient mice), 200 μM T7 peptide (T7 group, n=9) or 200 μM recombinant human arresten (arresten group, n=10 for wild type and n=9 for α1 integrin deficient mice) were prepared and injected subcutaneously into the flank of the mice. Six days later the mice were sacrificed, the plugs removed and fixed in 10% formalin in neutral buffered saline (Sigma Diagnostics), embedded in paraffin and stained with hematoxylin and eosin (HE) or periodic acid-Schiff (PAS). The number of blood vessels per ten high power fields was counted and averaged (200×magnification).
In vivo tumor studies
CT26 mouse colon carcinoma cells that are tumorigenic in syngeneic wild type and α1 integrin deficient Balb/c mice [19, 20, 24] were used for the tumor studies. 0.5 × 106 viable cells were injected subcutaneously in the flank of 3-month old α1 integrin deficient and wild type littermates. Arresten, T7 peptide or PBS with 4% DMSO was injected intravenously daily at an equimolar dosage of 10 mg/kg (arresten) or 1 mg/kg (T7) for 16 days. Tumor volumes were measured using the standard formula, length ×width2× 0.52 [6]. The tumors were allowed to grow to approximately 500 mm3. Each group contained 6 or 7 mice, and two individual experiments were performed. To determine the amount of blood vessels in the tumors the CD31 immunohistochemical staining was performed as previously described [25]. Briefly, frozen sections were fixed in acetone (−20°C). Samples were incubated with a rat anti-mouse CD31 (Pharmingen, clone MEC13.3), hamster anti-rat/mouse integrin α1 (Pharmingen, clone Ha31/8) and hamster anti-mouse integrin β3 (Pharmingen, clone 2C9.G2) antibodies at room temperature for 2 h, washed three times in PBS, followed by incubation with a FITC-conjugated secondary antibody (Jackson ImmunoResearch Laboratories) at room temperature for 1 h. After washing in PBS, the slides were mounted with Vectashield (VectorLaboratories) anti-fade mounting medium and imaged. For controls, sections were directly incubated with secondary antibody. In each group, the numbers of CD31 positive blood vessels were counted in 20 to 30 fields at 400× magnification in a blinded fashion.
Statistical analysis
All values are expressed as mean ± SE. Student’s t test was used to identify significant differences in multiple comparisons. A level of p < 0.05 was considered statistically significant.
RESULTS
Arresten induces apoptosis of endothelial cells in vitro
The effect of arresten on endothelial cell apoptosis has not been studied in detail before. We show here that arresten treatment of endothelial C-PAE cells changed the cell morphology; the cells rounded up and detached indicating increased rate of apoptosis (Figure 1A and B). In order to study this induction of apoptosis in endothelial cells by arresten more closely, the annexin V-fluorescein isothiocyanate assay was used. In this assay a fluorescein isothiocyanate conjugate of annexin V that binds naturally to phosphatidylserine is used to detect the externalization of membrane phosphatidylserine, which occurs in the early stages of apoptosis. Arresten was shown to increase the amount of apoptotic cells 2.4-fold in 2 hours, and 3.9-fold in 4 hours (p < 0.05) (Figure 1C). TNF-α was used as a positive control for apoptosis and as expected it increased the amount of apoptotic cells 2.9-fold (2 hours) and 4.5-fold (4 hours) (Figure 1C). To analyze whether the apoptosis inducing property of arresten is endothelial cell specific, we performed a caspase-3 assay using various concentrations of arresten (350, 700 and 1400 nM) on HMVEC endothelial cells, HSC-3 oral carcinoma cells, PC-3 prostate adenocarcinoma cells, 789-0 renal cell carcinoma cells, HT1080 fibrosarcoma cells and primary gingival fibroblasts. TNF-α was used as positive control for apoptosis induction. Caspase-3 (CPP32) becomes activated during the early stages of apoptosis, initiating cellular breakdown by degrading structural and DNA repair proteins. We observed an increase in caspase-3 activity at arresten concentration of 700 nM in HMVEC. (Figure 1D). The increase was over 2-fold in HMVECs. Although the increase was not statistically significant, the p-value was less than 0.1 (Figure 1D). A specific inhibitor of caspase-3 (DEVD-fmk) decreased the protease activity to baseline, indicating that the increase in activity was specifically due to caspase-3. No increase in caspase-3 activity was observed in any of the non-endothelial cell lines (Figure 2A–E), indicating that the pro-apoptotic effect of arresten is endothelial cell specific in vitro.
Figure 1. Arresten induces endothelial cell apoptosis.
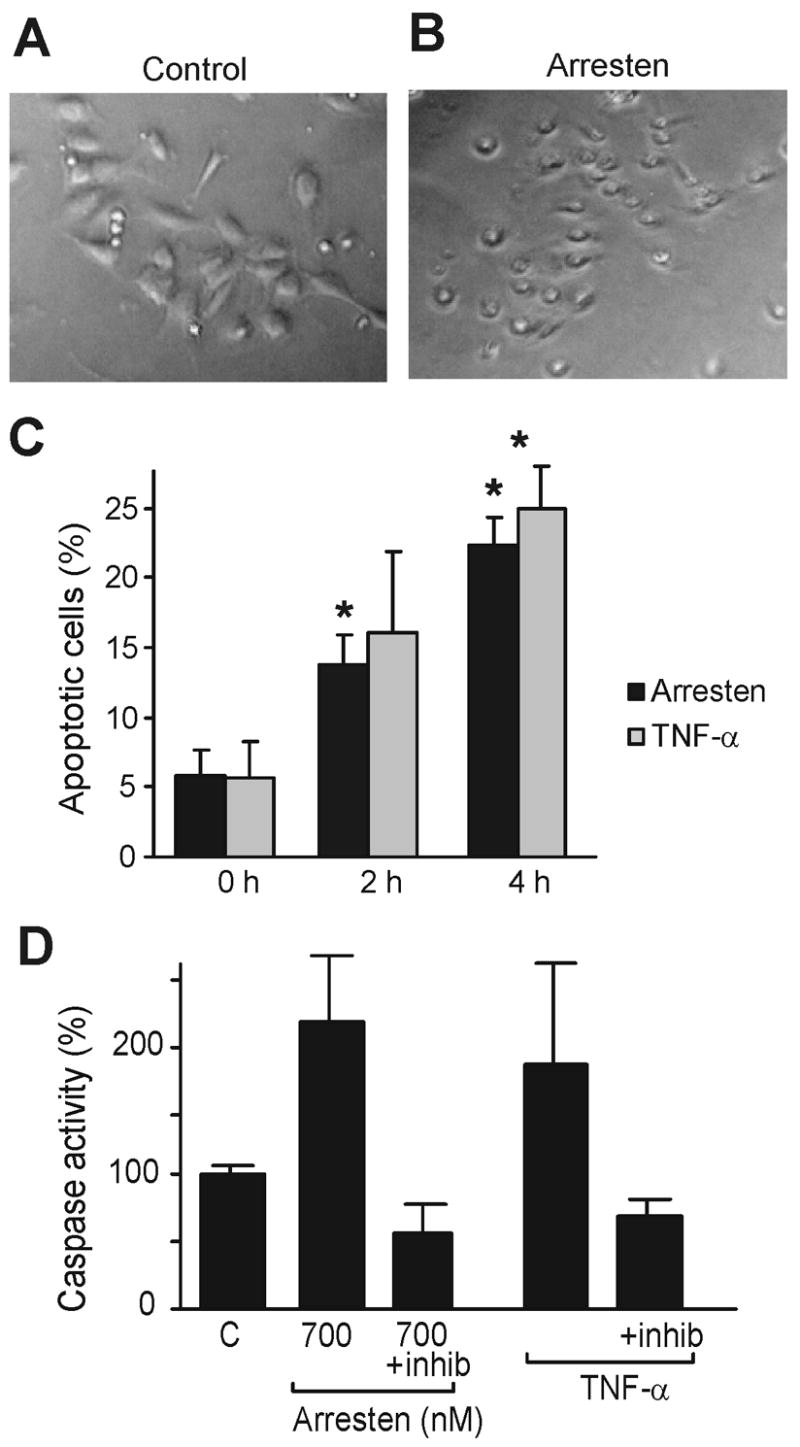
Annexin V-FITC staining was performed on C-PAE treated with 175 nM arresten for 0, 2 or 4 hours. Control cells received only PBS, and TNF- α (40 ng/ml) was used as a positive control for apoptosis. A–B. Arresten treatment induced morphological changes typical for apoptosis in the C-PAE cells (B) when compared to the control cells treated with PBS (A). C. The percentage of annexin V positive cells was plotted after different time points (0, 2 and 4 hours). Arresten induced apoptosis at a similar rate to TNF- α. This induction of apoptosis is statistically significant when compared to the cells treated with PBS (* p < 0.05). D. Arresten and TNF- α increased the activity of caspase-3 in HMVEC cells, when compared to cells treated with PBS alone (control) (p < 0.1). DEVD-fmk, a specific caspase-3 inhibitor (inhib), was used to ensure specificity of the assay. The caspase-3 assay was performed twice.
Figure 2. Arresten does not induce apoptosis in carcinoma, fibrosarcoma or fibroblast cell lines in vitro.

Arresten did not increase the activity of caspase-3 in HSC-3 oral squamous cell carcinoma cells (A), PC-3 prostate adenocarcinoma cells (B), 786-0 renal cell carcinoma cells (C), HT1080 fibrosarcoma cells (D) or primary gingival fibroblasts (E). Control cells received only PBS, and TNF- α (40 ng/ml) was used as a positive control for apoptosis. DEVD-fmk, a specific caspase-3 inhibitor (inhib), was used to ensure the specificity of the assay. The caspase-3 assay was performed twice (A, D, E) or once (B–C).
TUNEL assay to study apoptotic activity of arresten in vitro and in vivo
In the extensive DNA degradation that is characteristic for apoptosis, double-stranded, small DNA fragments as well as single strand breaks in the DNA are formed. These strand breaks can be detected by enzymatic labeling of the free 3′-OH-termini with modified nucleotides in the terminal deoxynucleotidyl transferase (TdT) reaction. We used the TUNEL (TdT-mediated X-dUTP nick-end-labeling) method to further analyze apoptosis induced by arresten. In the in vitro TUNEL assay we observed a 4-fold increase (p < 0.001) in the number of apoptotic C-PAE cells upon arresten treatment, when comparing to the untreated control (Figure 3A–E). TNF-α was used as a positive control and it induced apoptosis of the cells as expected (Figure 3E). In the in vivo TUNEL assay, tumor-bearing mice were sacrificed after 16 days of arresten or PBS treatment and cryosections of the tumors were stained by the TUNEL method. When the mice were treated with PBS only a very few apoptotic cells (green) were observed (Figure 3F, 3H). Arresten treatment (20 mg/kg) on the other hand significantly increased the number of apoptotic cells in the tumors of treated animals (Figure 3G, 3H), when compared to the control. Although the pro-apoptotic effect of arresten was endothelial cell specific in vitro, double staining of the tumors with TUNEL (green) and CD31 (endothelial cells, red) revealed that most of the apoptotic cells in arresten treated tumors were in fact tumor cells (Figure 3F-G). This is possibly an indirect effect of arresten; the inhibition of blood vessel growth by arresten starves the tumor cells and they undergo apoptosis. The quantification (mean ± SE) represents the total number of TUNEL positive cells (Figure 3H). Hot spot areas for TUNEL staining were selected for figures 3F–G.
Figure 3. Increased apoptosis after arresten treatment in vitro and in vivo in the TUNEL assay.

A-E. 0.5 × 106 C-PAE cells in 6-well plates were treated with arresten (350 and 700 nM), TNF- α (40 ng/ml) or PBS in media containing 3 ng/ml bFGF and 5 ng/ml VEGF. The cells were then collected and the TUNEL assay was performed. Apoptotic cells are shown in green (TdT(+) staining, A–B) and all cells are stained with red (propidium iodine, C–D). Arresten treatment (B) significantly increases the number of apoptotic cells, when compared to PBS treatment (A). E. The number of apoptotic cells per high power field was counted. Both arresten and TNF- α treatments significantly increase the number of apoptotic cells, when compared to cells treated with PBS (*** p < 0.001). F–H. Balb/c mice were implanted with subcutaneous CT26 tumors and treated with arresten or PBS injections for 16 days. Blood vessels in the tumor sections were detected by CD31 staining. To detect the DNA strand breaks characteristic to apoptotic cells, the TUNEL assay was performed. Double stainings of CD31 (red) and TUNEL (green) show that arresten significantly increases the total number of TUNEL positive apoptotic cells (G) compared to the control tumors (F) (200× magnification, hot spot areas for TUNEL staining selected for F-G). H. The number of apoptotic cells per high power field was counted (three tumors per experimental group, at least three fields were counted per tumor). Arresten treatment significantly increased the number of apoptotic cells in the tumors (mean ± SE, * p < 0.05). I. HMVEC cells treated with PBS (control, lane1), 700 nM arresten (lane 2) and 40 ng/ml TNF- α (lane 3) for 24 hours were lysed and 20 μg of protein per lane was separated and immunoblotted with primary antibodies against signaling molecules in the mitochondrial apoptosis pathway: no effect on Bax (pro-apoptotic) by arresten, down-regulation of Bcl-xL (anti-apoptotic) by arresten and down-regulation of Bcl-2 (anti-apoptotic) by arresten.
Induction of HMVEC apoptosis via down-regulation of Bcl-2 and Bcl-xL by arresten
To understand the pathways involved in the pro-apoptotic activity of arresten, we evaluated changes in the expression of Bcl-2, Bcl-xL and Bax. These Bcl-family members are significantly involved in the balance of pro-apoptotic and anti-apoptotic signals at the mitochondrial level. Bax is a pro-apoptotic molecule, whereas both Bcl-2 and Bcl-xL are anti-apoptotic. The balance of their expression determines whether cells undergo apoptosis [26]. Western blot analysis of their expression was done after 24-hour incubation of HMVEC cells. Arresten did not have any effect on the expression of Bax, but clearly decreased the expression of Bcl-xL and Bcl-2 (Figure 3I), thus shifting the balance and favoring the pro-apoptotic signaling. TNF-α was used as a positive control for apoptosis. Coomassie Blue staining of the SDS-PAGE gel was used to verify equal loading in all lanes (not shown).
Arresten deletion mutants have distinct effects on endothelial cells
Deletion mutants of arresten were generated in order to localize the active site of this molecule. By using deletion mutagenesis two arresten fragments were produced, and they were termed Arr-1 (the first 115 amino acids) and Arr-2 (the last 113 amino acids) (Figure 4A). Arr-2 was significantly more effective in inhibiting endothelial cell proliferation and inducing C-PAE endothelial cell apoptosis than Arr-1. The Arr-2 fragment was able to inhibit proliferation and return the number of cells to baseline at a concentration of 0.5 μg/ml (35 nM), while Arr-1 was not able to cause the same effect until concentrations of 20 μg/ml (1400 nM) (Figure 4B). Increased apoptosis can be detected in the annexin V assay by a shift in fluorescence peak. Arr-1 was unable to cause this shift in the fluorescence peak at a concentration of 0.5 μg/ml (35 nM) (Figure 4C), but at higher concentrations (5 μg/ml or 350 nM) apoptosis was also induced by Arr-1 (data not shown). On the other hand, Arr-2 caused a significant shift in fluorescence at both 0.5 μg/ml (35 nM) (Figure 4D) and 5 μg/ml (350 nM) concentrations (data not shown). These results strongly indicate that the active anti-angiogenic site is localized within the last 113 amino acids of the arresten molecule.
Figure 4. The localization of the site of the pro-apoptotic and anti-proliferative activity of arresten.
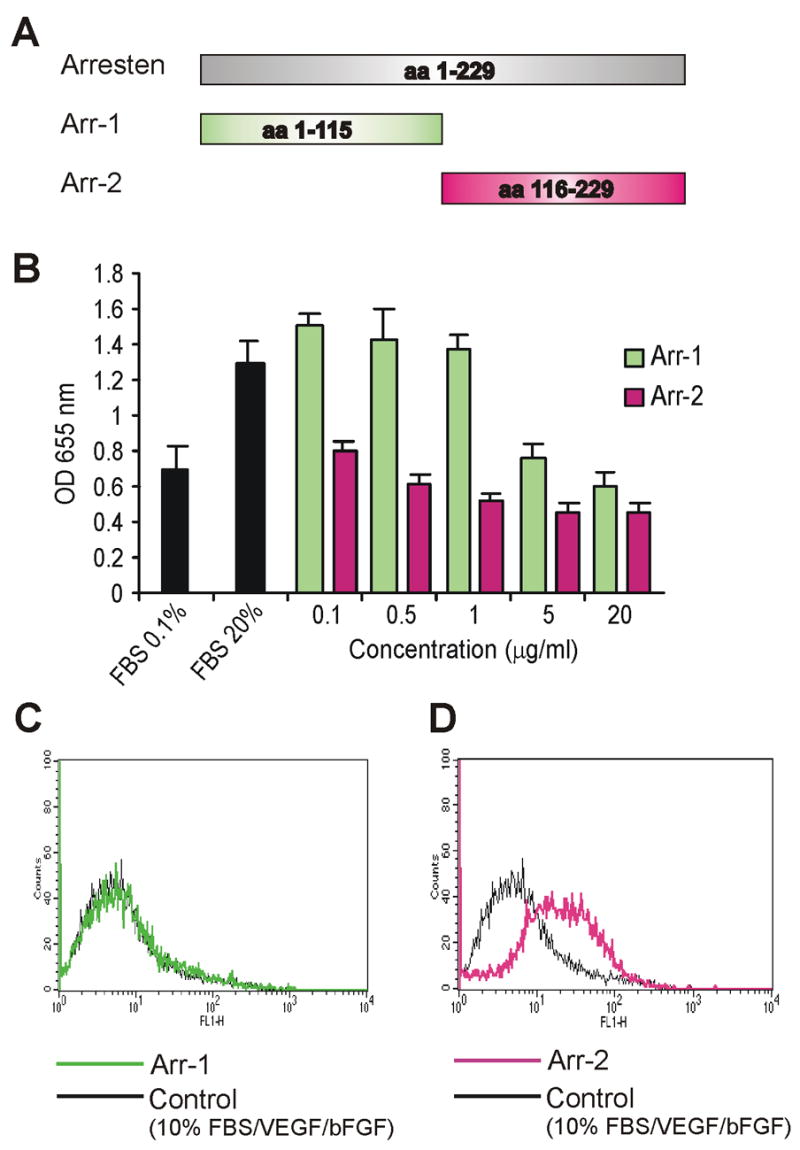
A. Two peptides derived from the full-length arresten were generated by deletion mutagenesis and named, Arr-1 (amino acids 1–115) and Arr-2 (amino acids 116–229). B. Equimolar concentrations (0, 0.1, 0.5, 1, 5 and 20 μg/ml or 0, 7, 35, 70, 350 and 1400 nM, respectively) of arresten peptides Arr-1 and Arr-2 were used for a C-PAE proliferation assay. The Arr-2 peptide decreased proliferation to half of the control (supplemented with 20 % FBS) at a concentration of 0.5 μg/ml, whereas a concentration of 20 μg/ml of Arr-1 was needed for the same inhibitory effect. C–D. The Annexin V-FITC assay was performed on C-PAE cells treated with either Arr-1 or Arr-2 (0.5 μg/ml). The FL-1-height represents the annexin fluorescence intensity as a log scale. The Arr-2 peptide induced a shift of fluorescence intensity indicating increased apoptosis, whereas Arr-1 caused no shift.
Integrin α1 is required for the decreased microvascular endothelial cell attachment, survival and Matrigel neovascularization by arresten
We have previously shown that human arresten inhibits tumor angiogenesis in mice [9] via α1β1 integrin [11]. However, the in vitro experiments of these previous studies demonstrated arresten binding to α1β1 integrin on larger blood vessels, and thus here we wished to study arresten-integrin α1β1 interactions on microvascular endothelial cells that are more important in the context of tumor angiogenesis. In addition, microvascular endothelial cells express integrin α1β1 significantly more than the larger vessels [14]. In order to analyze the capacity of primary microvascular lung endothelial cells (MLEC) to attach in the presence or absence of α1β1 integrin blocking antibodies, we isolated cells from both wild type and α1 integrin deficient mice. On arresten coating the α1β1 integrin blocking antibody had no effect on the attachment of α1 integrin deficient MLEC (mean 107% of control, SE ±12%), but the attachment of the wild type MLEC was reduced to half in the presence of the α1β1 integrin blocking antibody (56% of control ±5%) (Figure 5A). The control IgG did not inhibit the attachment of the cells (Figure 5A). The MLEC attachment on fibronectin ensured that the observed decrease in cell attachment onto arresten by the α1β1 integrin blocking antibody, was in fact due to the disruption of the arresten-α1β1 integrin interaction (Figure 5A).
Figure 5. Integrin α 1 is essential for the EC interactions with arresten in attachment, survival and Matrigel neovascularization assays.

A The attachment of wild type and α 1 integrin deficient MLECs on different substrata in the presence of blocking antibodies. 96-well plates were coated with 10 μg/ml arresten or fibronectin. MLECs (1 ×105 cells/ml) were incubated with the α 1 β 1 integrin antibody or control IgG or equivalent amount of PBS (control) for 15 min. The cells were allowed to attach to the wells for 45 min at 37ºC, un-attached cells were washed and the remaining cells were incubated in full media over night. The cells were then stained with methylene blue, and the OD655 was read on a microplate reader. The change in cell adhesion is presented as % of control. The bars represent mean ± SE (triplicate wells in three separate experiments). Cell attachment onto BSA coated wells was used as a negative control. The α 1 β 1 integrin antibody significantly prevented wild type cells from adhering to arresten coated plates, whereas it had no effect on the adhesion of α 1 integrin deficient MLECs on these plates. B. For the MTT survival assay the MLECs (0.4 ×104 cells per 96-well) were grown in 0.1 % gelatin coated dishes for 24 h, after which the cells were treated with 0, 5, 50 or 500 nM of arresten or T7 peptide for 24 h. MTT staining was performed and the purple color was measured on a microplate reader at 570 nm. The curves represent mean ± SE (triplicate wells in two separate experiments). The loss of cell viability is presented as % of controls. Arresten treatment of α 1 integrin deficient MLECs had no effect on cell viability (ko arr), whereas it decreased the viability of wild type cells (wt arr). The T7 peptide decreased viability of both cell types (wt and ko T7). C. Matrigel solution mixed with 3% DMSO, bFGF (100 ng/ml), VEGF (50 ng/ml), heparin (20 U/ml) and either PBS (control), T7 peptide (200 μM) or arresten (200 μM), was injected subcutaneously into flanks of littermate wild type and α 1 integrin deficient mice. Plugs were subsequently removed, embedded in paraffin, sectioned and stained with hematoxylin and eosin. Blood vessels from 10 high power fields per each plug were counted and averaged. The bars represent the average of 9 or 10 mice per each group ± SE. Arresten treatment of α 1 integrin deficient mice showed no inhibition of neovascularization of the Matrigel plugs. * p-value < 0.05, ** p-value < 0.01, *** p-value < 0.001.
If the pro-apoptotic action of arresten is mediated through the α1β1 integrin receptor, then cells deficient of α1β1 integrin should not respond to arresten treatment. In these studies the T7 peptide, corresponding to the active site of tumstatin, was used as control, since it is well established that it inhibits angiogenesis via binding to integrin αvβ3 [27], and thus the lack of α1 integrin should not interfere in its efficacy. We studied the effect of arresten and T7 on the survival of wild type and α1 integrin deficient MLEC using the MTT cell viability assay that measures the mitochondrial functionality. Arresten inhibited cell survival of only the wild type cells, whereas T7 had an inhibitory effect on both wild type and α1 integrin deficient cells (Figure 5B). Endothelial cell survival was 34% (SE ± 2%) of the control (100%), when MLECs from wild type mice were treated with 500 nM arresten. Treatment with 500 nM arresten had no significant effect on the α1 integrin deficient MLEC survival, 88% (SE ± 7%) of control. T7 peptide decreased the survival of both wild type and integrin α1 null MLECs. The differences in the anti-survival response by arresten between wild type and null cells were statistically significant at concentration of 50 nM (p < 0.01) and 500 nM (p < 0.001).
We used the Matrigel plug assay [28] to study the in vivo effect of arresten on the formation of new capillaries in wild type and α1 integrin deficient mice. Matrigel was injected subcutaneously into the flanks of mice in the presence of bFGF, VEGF, heparin and 3% DMSO, with or without 200 μM arresten or T7 peptide. In wild type mice the number of blood vessels decreased to 60% of control with arresten treatment, and to 56 % of control with T7 treatment (Figure 5C). Interestingly, in the α1 integrin deficient mice, the T7 peptide reduced the number of blood vessels to 72 % of control, but arresten even slightly increased the formation of blood vessels to 115 % of control, although the increase was not statistically significant (Figure 5C). All these results clearly demonstrate that α1 integrin is indeed important for the anti-angiogenic function of arresten.
The RGD (Arg-Gly-Asp) sequence present in many ECM proteins is important for the binding to certain integrins. However, integrin α1 β 1 does not bind to the RGD sequence [12]. To address whether arresten might function via some RGD-dependent integrin in addition to integrin α 1 β 1, a synthetic cyclic RGD peptide and a cyclic control peptide RAD (Arg-Ala-Asp) were used in cell attachment assays. Vitronectin coating was used as positive control, since the binding between vitronectin and integrin αvβ3 is known to be RGD dependent [12]. As expected, in the presence of the RGD peptide the cell attachment to vitronectin was reduced almost to half (not shown). However, neither the RGD peptide nor the negative control peptide had any effect on the endothelial cell attachment on an arresten-coated surface (not shown). This suggests that RGD-dependent integrins are not important for the function of arresten.
Arresten does not inhibit tumor growth in integrin α1 knock-out mice due to the lack of integrin α1 expression in the tumor vasculature
It has previously been shown that colon and renal carcinoma tumors, implanted subcutaneously into α 1 integrin deficient mice, are smaller and less vascularized than the same tumors in wild type mice [20, 29]. We have recently demonstrated that integrin α 1 β 1 is in fact essential for the anti-angiogenic function of arresten in subcutaneous teratocarcinoma tumor SV129-mouse model [11]. To determine whether this phenomenon is universal or depends on which tumor cell line or mouse strain was used, we studied how arresten treatment affected colon carcinoma cell growth in α 1 integrin deficient and wild type Balb/c mice. We also wished to characterize the localization of integrins α 1 and β 3 in the tumor and tumor vessels. Half a million colon carcinoma CT26 cells were injected subcutaneously onto the flanks of these mice. Arresten and T7 peptide treatments were started when the tumors had reached the size of 50 mm3. Equimolar amounts of arresten (10 mg/kg) or T7 (1 mg/kg) were injected intravenously daily for 16 days. As expected, the tumors grew slower in the α 1 integrin deficient mice, when compared to the wild type mice (Figure 6A). T7 peptide had a potent inhibitory effect on tumor growth in both the α 1 integrin deficient and the wild type mice. Consistent with our previous findings, arresten treatment did not inhibit tumor growth in the α 1 integrin deficient mice, whereas it had potent effect on tumor growth in wild type mice (Figure 6A). To determine if the vascular density in the tumors of the α 1 integrin deficient mice was altered, the number of CD31 positive blood vessels was counted. The wild type mice demonstrated a statistically significant decrease in the number of tumor blood vessels when treated with arresten or T7 peptide. The vascular density of the integrin α 1 null mice was slightly reduced compared to the control and again arresten treatment of the α 1 integrin deficient mice had no effect on the vascular density of tumors (Figure 6B). In wild type tumors there was a weak to moderate integrin α 1 immunostaining that partially co-localized with the CD31-positive blood vessels (Figure 6C). On the other hand, integrin α 1 staining was very weak or almost completely absent in the tumors of integrin α 1 deficient mice, and this staining never colocalized with the CD31-positive vessels suggesting that tumor stroma (derived from wild type mouse colon carcinoma) produces small amounts of integrin α 1 (Figure 6C). We used β 3 integrin staining as control, and found moderate staining in the tumor blood vessels both in wild type mice and integrin α 1 deficient mice (data not shown).
Figure 6. Expression of integrin α 1 in tumors with treatment of arresten or T7 peptide in wild type and α 1 integrin deficient tumor-bearing mice.

A CT26 colon carcinoma cells were injected subcutaneously into flanks of wild type and integrin α 1 deficient Balb/c mice. When the tumors reached the size of 50 mm3 equimolar amounts of recombinant arresten (278 μg), T7 peptide (30 μg) or PBS (control) was intravenously injected into the tumor bearing mice daily for 16 days. The data presented is representative of two independent experiments and shown as the mean of final tumor size at day 16 ± SE (* p-value < 0.05). In the α 1 integrin deficient mice only T7 peptide significantly reduced tumor growth, whereas no significant effect was observed upon arresten treatment. B. The blood vessels were stained with CD31, counted and averaged for each experimental group (mean ± SE, * p-value < 0.05, *** p-value < 0.001). C. Double staining of integrin α 1 (green) and CD31 (red) of CT26 colon carcinoma tumors implanted on wild type and integrin α 1 deficient Balb/c mice. Mild integrin α 1 expression can be observed in wild type tumors both in the tumor stroma and in the vasculature (co-localizing with the CD31 staining). CT26 tumors on integrin α 1 deficient mice show no integrin α 1 expression in the vasculature (host-derived) whereas some weak expression is detected in the tumor cells. The CD31 staining shows reduced number of blood vessels upon arresten and T7 peptide treatment in the wild type mice. In the integrin α 1 deficient mice, only T7 reduces the growth of vasculature, arresten has no effect on the number of blood vessels. Magnification 400×.
DISCUSSION
Basement membrane- and especially collagen-derived endogenous inhibitors of angiogenesis have raised a lot of interest as endogenous inhibitors of angiogenesis in the recent years. We have studied the mechanism of arresten, an inhibitor of angiogenesis and tumor growth, derived from the α 1 integrin chain of type IV collagen. We show here, with various methods, that arresten potently induces endothelial cell (EC) apoptosis. This pro-apoptotic effect is mediated by decreasing the expression of anti-apoptotic signaling molecules Bcl-2 and Bcl-xL. The active anti-angiogenic site of arresten is found within the last 113 amino acids of the molecule. We also confirm our earlier finding, that arresten binds to α 1 β 1 integrin, and furthermore that α 1 β 1 integrin is a functionally relevant receptor for the anti-angiogenic function of arresten regardless of what mouse strain or tumor cell line is used. In wild type mice α 1 β 1 integrin localizes both in tumor blood vessels and tumor cells themselves, but in integrin α 1 deficient mice only weak staining is observed on the tumor stroma, and no staining on the host-derived blood vessels.
It is important to note that even though the collagen derived endogenous angiogenesis inhibitors are molecules of about the same size, come from similar parent molecules and have amino acid sequence homologies [6, 7, 8, 9], they all function via distinct mechanisms, bind different cell surface receptors, and affect different parts of the angiogenic process. Endostatin, a fragment of type XVIII collagen, binds to α 5 β 1 integrin, and inhibits EC migration by interfering with cell signaling pathways through ERK1 and p38. Tumstatin, a fragment of the α 3 chain of type IV collagen, on the other hand, binds to αvβ 3 integrin and inhibits the PI3-kinase/Akt/mTOR/4EBP1 signaling pathway, resulting in decreased EC proliferation [10]. The receptors of canstatin, a fragment of the α 2-chain of type IV collagen, are integrins α 3 β 1, αvβ 3 and αvβ 5 [30]. It inhibits Akt activation and induces Fas-dependent apoptosis in EC [31]. We have discovered that arresten binds to α 1 β 1 integrin and inhibits phosphorylation of FAK. Inhibition of FAK leads to inhibition of Raf/MEK/ERK1/2/p38 MAPK pathways that lead to inhibition of hypoxia inducible factor (HIF-1 α) and VEGF expression, resulting in inhibition of EC migration, proliferation, and tube formation [11]. Here we found that arresten also induced endothelial cell apoptosis by regulating mitochondrial Bcl-family signaling molecules. As arresten decreases angiogenesis in several ways it is possible that this molecule affects several distinct cell-signaling pathways.
Studies with canstatin show that within the same molecule, different parts of the molecule can participate in the inhibition of angiogenesis in distinct ways. The C-terminal domain of canstatin mainly associated with the specific inhibition of EC proliferation, whereas N-terminal canstatin is associated with the potential apoptosis-inducing activity on EC [32, 33]. A similar phenomenon is also evident for tumstatin, it has two separate binding sites to αvβ 3 integrin, the N-terminal one being associated with the anti-angiogenic properties and the C-terminal one being associated with the anti-tumor activity [27, 34]. Therefore, it is possible that also arresten might have several receptor binding sites, and thus other receptors, in addition to α 1 β 1 integrin, that are able to affect the anti-angiogenic and anti-tumor properties of this molecule. Our findings show that arresten induces apoptosis both in endothelial cells and in tumor cells in a mouse tumor model, although in vitro the pro-apoptotic effect was EC specific. This effect on the tumor cells might be an indirect effect induced by the lack of blood vessels promoting tumor cell apoptosis or alternatively arresten might actually have a direct effect on the tumor cells in vivo. It is possible that arresten is differentially processed in vivo and in vitro resulting in exposure of novel cryptic receptor binding sites. In addition, we have previously shown that arresten possess two binding sites on endothelial cell (C-PAE) surface, a high-affinity and a low-affinity site. In addition to the high affinity binding to α 1 β 1 integrin, arresten also binds to HSPG on the EC, but it is not yet known whether this binding is of functional significance [9]. Recent studies have shown that HSPGs are important in the regulation of angiogenesis. Particularly perlecan has both angiogenic and anti-angiogenic effects [35], and it has been shown that endorepellin, the C-terminal part of perlecan inhibits angiogenesis by binding to α 2 β 1 integrin [36]. The ability of arresten to bind to both integrins and HSPGs is not unique among the matrix-derived angiogenesis inhibitors. Endostatin has also been shown to bind both to integrins (α 5 β 1 and αvβ 3 integrin), and to a HSPG (glypican-1) [10, 37, 38, 39]. The minimal recognition sequence of integrin α 1 β 1 is GFOGER (Gly-Phe-HydroxyPro-Gly-Glu-Arg), which has to be in a triple helical conformation [40, 41]. Recently, it was found that in the triple helical type IV collagen molecule, the critical amino acids for the α 1 β 1 integrin binding were Asp461 in the α 1(IV) chain and Arg 461 in the α 2(IV) chain [42]. When arresten is cleaved from the type IV collagen, it is no longer in the triple helical form, and thus the GFOGER recognition sequence cannot be relevant for the binding of arresten. However, although remaining unknown, most likely the EC recognition sequence of arresten resides within the last 113 amino acids, shown here to possess the anti-proliferative and pro-apoptotic property of this molecule.
Taken together, this shows that the extracellular matrix, the soluble small molecules derived from the extracellular matrix and the cell surface receptors form an extremely complicated network. Xu et al. have shown that the cleavage of triple helical collagen IV results in the exposure of functionally important cryptic site associated with angiogenesis [43]. Intact type IV collagen and especially the NC1 degradation products of type IV collagen can have opposite effects on EC behavior. Whereas intact type IV collagen promotes cell migration and proliferation via binding to α 1 β 1 and α 2 β 1 integrin, the binding of cleavage products to the same receptors leads to inhibition of angiogenesis [44, 45]. The absence of α 1 β 1 integrin, or any other part of this network, can disturb an intricate balance, and especially in pathological conditions cause surprising effects. In this regard, our results interestingly show that there were slightly more blood vessels in the arresten containing Matrigel plugs when implanted on α 1 integrin deficient mice, and that the proliferation of VEGF stimulated wild type MLECs was increased in the presence of 50 nM arresten (not shown). Furthermore, when the tumors in α 1 integrin deficient mice were treated with arresten, they surprisingly grew somewhat faster. Even though these results were not statistically significant, the trend was consistent in three distinct angiogenesis assays. What could explain this finding? The most obvious reason for this accelerated growth of blood vessels and tumors upon arresten treatment in the α 1 integrin deficient mice would be an up-regulation of another receptor on the EC surface that compensates for the lack of α 1 β 1 integrin. Integrin α 2 β 1 has the same substrate specificity as α 1 β 1 integrin, and therefore would be a likely candidate for compensation. However, it has been shown that no compensatory up-regulation of α 2 integrin in the α 1 integrin deficient tissue takes place [19]. Since arresten also binds to HSPG, the absence of α 1 integrin could render more arresten available to bind HSPG, despite of the lower affinity. Additionally, the increased amount of arresten could block the direct binding of VEGF or other growth factors to HSPGs, thus resulting in increased amounts of bio-available unbound growth factors, which then could bind receptors leading to pro-angiogenic effects on the EC.
Tumors implanted into the α 1 integrin deficient mice grow slower than those in wild type mice. It was suggested that this might be due to the up-regulation of matrix metalloproteases (MMPs), leading to an increased amount of angiostatin, an inhibitor of angiogenesis that is proteolytically derived from plasminogen [29]. It thus appears that although the lack of α 1 integrin does not impair developmental angiogenesis, it does affect pathological angiogenesis as shown in our work. Hamano et al. [25] showed a similar phenomenon with tumstatin. The tumor suppressive effects of tumstatin require αvβ 3 integrin to be expressed on pathological, but not on physiological angiogenic blood vessels. We show here that the tumor blood vessels in the wild type mice are positive for integrin α 1 immunostaining, but in the integrin α 1 deficient mice, there are no integrin α 1 positive vessels, only some weak staining in the tumor stroma (tumor cells were derived from a wild type mouse carcinoma). VEGF is known to significantly induce the expression of α 1 β 1 integrin on the EC surface. Blocking the function of α 1 β 1 integrin by antibodies selectively inhibits VEGF-driven angiogenesis in vivo, without any effects on the pre-existing vasculature. Therefore, it has been suggested that α 1 β 1 integrin is of particular importance in the pathological angiogenesis [22], and our results support this notion. Interestingly, arresten binding to integrin α 1 β 1 inhibits HIF-1 α synthesis and thus leads to inhibition on VEGF expression [11].
In conclusion, we have further analyzed the anti-angiogenic mechanism of arresten, a type IV collagen derived endogenous inhibitor of angiogenesis and tumor growth. We show that arresten significantly induces EC apoptosis by down-regulating the expression of anti-apoptotic Bcl-family signaling molecules. The anti-angiogenic property of arresten is located within the last 113 amino acids of this molecule. In addition, we confirmed our earlier finding that arresten binds to α 1 β 1 integrin, and that α 1 β 1 integrin is a functionally necessary receptor for the anti-angiogenic and anti-tumorigenic nature of arresten in the microvascular ECs in the tumor.
Acknowledgments
Julie Lively is warmly acknowledged for her advice in the MLEC preparation, Eeva-Maija Kiljander for technical assistance and Mary Soubasakos for her help in maintaining the mice. Humphrey Gardner is gratefully thanked for the integrin α 1 deficient mice and integrin α 1 blocking antibody. This study was supported by NIH grants DK 55001 (RK), DK 51711 (RK), grants from the Academy of Finland (PN and TS), Finnish Cancer Organizations (TS), Emil Aaltonen Foundation (PN and MS), Finnish Dental Society Apollonia (TS), Maud Kuistila Foundation (PN and MS), Oulu University Pharmacy Foundation (PN), Cancer Society of Northern Finland (PN), Sigrid Juselius Foundation (MS and TS), Finnish Medical Society Duodecim (MS), Faculty of Medicine, University of Oulu (PN), and research funds of the Program in Matrix Biology at the BIDMC (RK).
ABBREVIATIONS
- BSA
bovine serum albumin
- CPAE
calf pulmonary aortic endothelial cells
- cRGD
cyclic Arg-Gly-Asp
- DMSO
dimethylsulpoxide
- EC
endothelial cell
- FAK
focal adhesion kinase
- FITC
fluorescein isothiosyanate
- HIF
hypoxia inducible factor
- HMVEC
human microvascular endothelial cells
- HSPG
heparin sulphate proteoglycan
- HUVEC
human umbilical vein endothelial cell
- MAPK
mitogen activated protein kinase
- MLEC
microvascular lung endothelial cell
- NC1
non-collagenous domain
- PBS
phosphate buffered saline
- RAD
Arg-Ala-Asp
- RT
room temperature
- TNF-α
tumor necrosis factorα
- VEGF
vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J. Angiogenesis and apoptosis. Semin Cancer Biol. 2003;13:159–167. doi: 10.1016/s1044-579x(02)00133-5. [DOI] [PubMed] [Google Scholar]
- 3.Madri JA. Extracellular matrix modulation of vascular cell behaviour. Transpl Immunol. 1997;5:179–183. doi: 10.1016/s0966-3274(97)80035-4. [DOI] [PubMed] [Google Scholar]
- 4.Paulsson M. Basement membrane proteins: structure, assembly, and cellular interactions. Crit Rev Biochem Mol Biol. 1992;27:93–127. doi: 10.3109/10409239209082560. [DOI] [PubMed] [Google Scholar]
- 5.Prockop DJ, Kivirikko KI. Collagens: molecular biology, diseases, and potentials for therapy. Annu Rev Biochem. 1995:64403–434. doi: 10.1146/annurev.bi.64.070195.002155. [DOI] [PubMed] [Google Scholar]
- 6.O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 7.Maeshima Y, Colorado PC, Torre A, Holthaus KA, Grunkemeyer JA, Ericksen MB, Hopfer H, Xiao Y, Stillman IE, Kalluri R. Distinct antitumor properties of a type IV collagen domain derived from basement membrane. J Biol Chem. 2000;275:21340–21348. doi: 10.1074/jbc.M001956200. [DOI] [PubMed] [Google Scholar]
- 8.Kamphaus GD, Colorado PC, Panka DJ, Hopfer H, Ramchandran R, Torre A, Maeshima Y, Mier JW, Sukhatme VP, Kalluri R. Canstatin, a novel matrix-derived inhibitor of angiogenesis and tumor growth. J Biol Chem. 2000;275:1209–1215. doi: 10.1074/jbc.275.2.1209. [DOI] [PubMed] [Google Scholar]
- 9.Colorado PC, Torre A, Kamphaus G, Maeshima Y, Hopfer H, Takahashi K, Volk R, Zamborsky ED, Herman S, Sarkar PK, Ericksen MB, Dhanabal M, Simons M, Post M, Kufe DW, Weichselbaum RR, Sukhatme VP, Kalluri R. Anti-angiogenic cues from vascular basement membrane collagen. Cancer Res. 2000;60:2520–2526. [PubMed] [Google Scholar]
- 10.Sudhakar A, Sugimoto H, Yang C, Lively J, Zeisberg M, Kalluri R. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc Natl Acad Sci U S A. 2003;100:4766–4771. doi: 10.1073/pnas.0730882100. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 11.Sudhakar A, Nyberg P, Keshamouni VG, Mannam AP, Li J, Sugimoto H, Cosgrove D, Kalluri R. Human α 1 type IV collagen NC1 domain exhibits distinct antiangiogenic activity mediated by α 1 β 1 integrin. J Clin Invest. 2005;115:2801–2810. doi: 10.1172/JCI24813. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Plow EF, Haas TA, Zhang L, Loftus J, Smith JW. Ligand binding to integrins. J Biol Chem. 2000;275:21785–21788. doi: 10.1074/jbc.R000003200. [DOI] [PubMed] [Google Scholar]
- 13.Pozzi A, Wary KK, Giancotti FG, Gardner HA. Integrin alpha1beta1 mediates a unique collagen-dependent proliferation pathway in vivo. J Cell Biol. 1998;142:587–594. doi: 10.1083/jcb.142.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Defilippi P, van Hinsbergh V, Bertolotto A, Rossino P, Silengo L, Tarone G. Differential distribution and modulation of expression of alpha 1/beta 1 integrin on human endothelial cells. J Cell Biol. 1991;114:855–863. doi: 10.1083/jcb.114.4.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu S, Tobias R, McClure S, Styba G, Shi Q, Jackowski G. Removal of endotoxin from recombinant protein preparations. Clin Biochem. 1997;30:455–463. doi: 10.1016/s0009-9120(97)00049-0. [DOI] [PubMed] [Google Scholar]
- 16.Maeshima Y, Yerramalla UL, Dhanabal M, Holthaus KA, Barbashov S, Kharbanda S, Reimer C, Manfredi M, Dickerson WM, Kalluri R. Extracellular matrix-derived peptide binds to alpha(v)beta(3) integrin and inhibits angiogenesis. J Biol Chem. 2001;276:31959–31968. doi: 10.1074/jbc.M103024200. [DOI] [PubMed] [Google Scholar]
- 17.Salo T, Lyons JG, Rahemtulla F, Birkedal-Hansen H, Larjava H. Transforming growth factor-beta1 up-regulates type IV collagenase expression in cultured human keratinocytes. J Biol Chem. 1991;266:11436–11441. [PubMed] [Google Scholar]
- 18.Nyberg P, Heikkila P, Sorsa T, Luostarinen J, Heljasvaara R, Stenman UH, Pihlajaniemi T, Salo T. Endostatin inhibits human tongue carcinoma cell invasion and intravasation and blocks the activation on matrix metalloprotease-2, -9, and -13. J Biol Chem. 2003;278:22404–22411. doi: 10.1074/jbc.M210325200. [DOI] [PubMed] [Google Scholar]
- 19.Gardner H, Kreidberg J, Koteliansky V, Jaenisch R. Deletion of integrin alpha 1 by homologous recombination permits normal murine development but gives rise to a specific deficit in cell adhesion. Dev Biol. 1996;175:301–313. doi: 10.1006/dbio.1996.0116. [DOI] [PubMed] [Google Scholar]
- 20.Pozzi A, LeVine WF, Gardner HA. Low plasma levels of matrix metalloproteinase 9 permit increased tumor angiogenesis. Oncogene. 2002;21:272–281. doi: 10.1038/sj.onc.1205045. [DOI] [PubMed] [Google Scholar]
- 21.Tedjarati S, Baker CH, Apte S, Huang S, Wolf JK, Killion JJ, Fidler IJ. Synergistic therapy of human ovarian carcinoma implanted orthotopically in nude mice by optimal biological dose of pegylated interferon alpha combined with paclitaxel. Clin Cancer Res. 2002;8:2413–2422. [PubMed] [Google Scholar]
- 22.Senger DR, Claffey KP, Benes JE, Perruzzi CA, Sergiou AP, Detmar M. Angiogenesis promoted by vascular endothelial growth factor: regulation through alpha1beta1 and alpha2beta1 integrins. Proc Natl Acad Sci U S A. 1997;94:13612–13617. doi: 10.1073/pnas.94.25.13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mendrick DL, Kelly DM, duMont SS, Sandstrom DJ. Glomerular epithelial and mesangial cells differentially modulate the binding specificities of VLA-1 and VLA-2. Lab Invest. 1995;72:367–375. [PubMed] [Google Scholar]
- 24.Xiang R, Lode HN, Dreier T, Gillies SD, Reisfeld RA. Induction of persistent tumor-protective immunity in mice cured of established colon carcinoma metastases. Cancer Res. 1998;58:3918–3925. [PubMed] [Google Scholar]
- 25.Hamano Y, Zeisberg M, Sugimoto H, Lively JC, Maeshima Y, Yang C, Hynes RO, Werb Z, Sudhakar A, Kalluri R. Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell. 2003;3:589–601. doi: 10.1016/s1535-6108(03)00133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cory S, Huang DC, Adams JM. The Bcl-family: roles in cell survival and oncogenesis. Oncogene. 2003;22:8590–8607. doi: 10.1038/sj.onc.1207102. [DOI] [PubMed] [Google Scholar]
- 27.Maeshima Y, Colorado PC, Kalluri R. Two RGD-independent alpha vbeta 3 integrin binding sites on tumstatin regulate distinct anti-tumor properties. J Biol Chem. 2000;275:23745–23750. doi: 10.1074/jbc.C000186200. [DOI] [PubMed] [Google Scholar]
- 28.Passaniti A, Taylor RM, Pili R, Guo Y, Long PV, Haney JA, Pauly RR, Grant DS, Martin GR. A simple, quantitative method for assessing angiogenesis and antiangiogenic agents using reconstituted basement membrane, heparin, and fibroblast growth factor. Lab Invest. 1992;67:519–528. [PubMed] [Google Scholar]
- 29.Pozzi A, Moberg PE, Miles LA, Wagner S, Soloway P, Gardner HA. Elevated matrix metalloprotease and angiostatin levels in integrin alpha 1 knockout mice cause reduced tumor vascularization. Proc Natl Acad Sci U S A. 2000;97:2202–2207. doi: 10.1073/pnas.040378497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mundel TM, Kalluri R. Type IV collagen-derived angiogenesis inhibitors. Microvasc Res. 2007;74:85–89. doi: 10.1016/j.mvr.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Panka DJ, Mier JW. Canstatin inhibits Akt activation and induces Fas-dependent apoptosis in endothelial cells. J Biol Chem. 2003;278:37632–37636. doi: 10.1074/jbc.M307339200. [DOI] [PubMed] [Google Scholar]
- 32.He GA, Luo JX, Zhang TY, Wang FY, Li RF. Canstatin-N fragment inhibits in vitro endothelial cell proliferation and suppresses in vivo tumor growth. Biochem Biophys Res Commun. 2003;312:801–805. doi: 10.1016/j.bbrc.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 33.He GA, Luo JX, Zhang TY, Hu ZS, Wang FY. The C-terminal domain of canstatin suppresses in vivo tumor growth associated with proliferation of endothelial cells. Biochem Biophys Res Commun. 2004;318:354–360. doi: 10.1016/j.bbrc.2004.04.038. [DOI] [PubMed] [Google Scholar]
- 34.Shahan TA, Ziaie Z, Pasco S, Fawzi A, Bellon G, Monboisse JC, Kefalides NA. Identification of CD47/integrin-associated protein and alpha(v)beta3 as two receptors for the alpha3(IV) chain of type IV collagen on tumor cells. Cancer Res. 1999;59:4584–4590. [PubMed] [Google Scholar]
- 35.Segev A, Nili N, Strauss BH. The role of perlecan in arterial injury and angiogenesis. Cardiovasc Res. 2004;63:603–610. doi: 10.1016/j.cardiores.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 36.Bix G, Fu J, Gonzalez EM, Macro L, Barker A, Campbell S, Zutter MM, Santoro SA, Kim JK, Hook M, Reed CC, Iozzo RV. Endorepellin causes endothelial cell disassembly of actin cytoskeleton and focal adhesions through alpha2beta1 integrin. J Cell Biol. 2004;166:97–109. doi: 10.1083/jcb.200401150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rehn M, Veikkola T, Kukk-Valdre E, Nakamura H, Ilmonen M, Lombardo C, Pihlajaniemi T, Alitalo K, Vuori K. Interaction of endostatin with integrins implicated in angiogenesis. Proc Natl Acad Sci U S A. 2001;98:1024–1029. doi: 10.1073/pnas.031564998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karumanchi SA, Jha V, Ramchandran R, Karihaloo A, Tsiokas L, Chan B, Dhanabal M, Hanai JI, Venkataraman G, Shriver Z, Keiser N, Kalluri R, Zeng H, Mukhopadhyay D, Chen RL, Lander AD, Hagihara K, Yamaguchi Y, Sasisekharan R, Cantley L, Sukhatme VP. Cell surface glypicans are low-affinity endostatin receptors. Mol Cell. 2001;7:811–822. doi: 10.1016/s1097-2765(01)00225-8. [DOI] [PubMed] [Google Scholar]
- 39.Kalluri R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer. 2003;3:422–433. doi: 10.1038/nrc1094. [DOI] [PubMed] [Google Scholar]
- 40.Knight CG, Morton LF, Onley DJ, Peachey AR, Messent AJ, Smethurst PA, Tuckwell DS, Farndale RW, Barnes MJ. Identification in collagen type I of an integrin alpha2 beta1-binding site containing an essential GER sequence. J Biol Chem. 1998;273:33287–33294. doi: 10.1074/jbc.273.50.33287. [DOI] [PubMed] [Google Scholar]
- 41.Knight CG, Morton LF, Peachey AR, Tuckwell DS, Farndale RW, Barnes MJ. The collagen-binding A-domains of integrins alpha(1)beta(1) and alpha(2)beta(1) recognize the same specific amino acid sequence, GFOGER, in native (triple-helical) collagens. J Biol Chem. 2000;275:35–40. doi: 10.1074/jbc.275.1.35. [DOI] [PubMed] [Google Scholar]
- 42.Renner C, Sacca B, Moroder L. Synthetic heterotrimeric collagen peptides as mimics of cell adhesion sites of the basement membrane. Biopolymers. 2004;76:34–47. doi: 10.1002/bip.10569. [DOI] [PubMed] [Google Scholar]
- 43.Xu J, Rodriguez D, Peticlerc E, Kim JJ, Hangai M, Moon YS, Davis GE, Brooks PC. Proteolytic exposure of cryptic site within collagen type IV is required for angiogenesis and tumor growth in vivo. J Cell Biol. 2001;154:1069–1079. doi: 10.1083/jcb.200103111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Petitclerc E, Boutaud A, Prestayko A, Xu J, Sado Y, Ninomiya Y, Sarras MP, Jr, Hudson BG, Brooks PC. New functions for non-collagenous domains of human collagen type IV. Novel integrin ligands inhibiting angiogenesis and tumor growth in vivo. J Biol Chem. 2000;275:8051–8061. doi: 10.1074/jbc.275.11.8051. [DOI] [PubMed] [Google Scholar]
- 45.Kerbel R, Folkman J. Clinical translation of angiogenesis inhibitors. Nat Rev Cancer. 2002;2:727–739. doi: 10.1038/nrc905. [DOI] [PubMed] [Google Scholar]