

Abstract
The global rise in lung cancer burden, together with its poor survival and resistance to classical chemotherapy underscores the need for identification of critical molecular events involved in lung carcinogenesis. Here, we have applied quantitative profiling of DNA methylation states in a panel of five cancer-associated genes (CDH1, CDKN2A, GSTP1, MTHFR and RASSF1A) to a large case-control study of lung cancer. Our analyses revealed a high frequency of aberrant hypermethylation of MTHFR, RASSF1A and CDKN2A in lung tumours as compared to control blood samples, whereas no significant increase in methylation levels of GSTP1 and CDH1 was observed, consistent with the notion that aberrant DNA methylation occurs in a tumour-specific and gene-specific manner. Importantly, we found that tobacco smoking, sex, and alcohol intake had a strong influence on the methylation levels of distinct genes (RASSF1A and MTHFR), whereas folate intake, age and histological subtype had no significant influence on methylation states. We observed a strong association between MTHFR hypermethylation in lung cancer and tobacco smoking, whereas methylation levels of CDH1, CDKN2A, GSTP1 and RASSF1A were not associated with smoking, indicating that tobacco smoke targets specific genes for hypermethylation. We also found that methylation levels in RASSF1A, but not the other genes under study, were influenced by sex, with males showing higher levels of methylation. Together, this study identifies aberrant DNA methylation patterns in lung cancer and thus exemplifies the mechanism by which environmental factors may interact with key genes involved in tumour suppression and contribute to lung cancer.
Keywords: DNA methylation, lung cancer, risk factors, tobacco, MTHFR
Introduction
Lung cancer is the leading cause of cancer-related malignancy worldwide, accounting for 30% of all cancer-related deaths (1). It is projected that the lung cancer toll by the year 2010 will be 1.5 million deaths, highlighting the lung cancer burden as a major public-health issue in the coming years (1). The vast majority of lung cancer cases (80–90%) are due to smoking, with a striking dose-response relationship (2). Despite the fact that the cause of most lung cancer is well known, the disease has proven difficult to diagnose early and treat successfully, reflecting limited advances in our understanding of the molecular mechanisms underlying lung carcinogenesis and individual susceptibility to lung cancer.
In addition to genetic factors such as mutations and susceptibility differences in the form of rare high-penetrance genes and genetic polymorphism (3-5), the role of epigenetic changes has been implicated in lung cancer etiology (6-12). DNA methylation is an epigenetic event whose pattern is altered frequently in a wide variety of human cancers, including promoter-specific hypermethylation as well as genome-wide hypomethylation (13, 14). Aberrant DNA methylation within CpG islands is among the earliest and most common alterations in human cancers, leading to abnormal expression of a broad spectrum of genes (13, 15-17). While there are now many reports of somatically acquired DNA methylation changes in various genes implicated in lung cancer (13, 14), what triggers these changes is poorly understood.
Various environmental and lifestyle exposures such as tobacco, Helicobacter pylori, plutonium or radon exposure are suspected to be implicated in the development of a wide range of human cancers by eliciting DNA methylation changes (18-20); however, the underlying mechanism and precise epigenetic targets are poorly understood. The major obstacle in establishing a relationship between DNA methylation patterns and exposure to environmental and lifestyle factors in cancer is the fact that case–control studies tend to be too small and lack quantitative measure of methyl-cytosine levels to identify the interactions between DNA methylation changes and specific risk factors.
In the present study, we sought to identify DNA methylation profiles in lung cancer and their association with known or suspected cancer risk factors. For this, we combined the advantages of quantitative measurement of DNA methylation levels in a panel of cancer-associated genes and a large case–control study of lung cancer with adequate statistical power, and identified aberrant DNA methylation of key cellular genes in lung cancer and its association with specific cancer risk factors.
Materials and Methods
Study Population
This study is based on a case–control study on lung cancer conducted at Cancer Research Centre, Moscow (Russia), as part of a larger multicenter case-control study coordinated by the International Agency for Research on Cancer (IARC) (4, 21). Patients newly diagnosed with lung cancer, and a comparable group of hospital-based control subjects without lung cancer, were recruited between February 1998 and October 2002 (Table 1). All lung cancer cases were confirmed histologically and cytologically. A total of 600 lung cancer patients and 600 controls were recruited; among these, tumour tissues were available for 209 lung cancer patients. Of these patients, 172 also provided blood samples. In addition, blood samples from 164 control subjects were included in the analysis for comparison. The blood samples from the control group were sampled to match cases by age, sex and smoking status. Informed consent was obtained from all patients, and the study was approved by the IARC institutional review committee.
Table 1.
Patients and tumours information
| Blood (n=336) | ||||
|---|---|---|---|---|
|
Tumours (n=209) |
Cases
(n=172) |
Controls
(n=164) |
Total
(n=336) |
|
| Sex | ||||
| Male | 171 | 107 | 115 | 222 |
| Female | 38 | 65 | 49 | 114 |
| Age | ||||
| ≤40 | 2 | 2 | 1 | 3 |
| 41-50 | 23 | 15 | 26 | 41 |
| 51-60 | 72 | 52 | 53 | 105 |
| 61-70 | 83 | 64 | 63 | 127 |
| 71+ | 29 | 39 | 21 | 60 |
| Histology | ||||
| Squamous cell carcinoma | 121 | 63 | - | 63 |
| Adenocarcinoma | 58 | 55 | - | 55 |
| Mixed | 12 | 12 | - | 12 |
| Other/unspecified | 18 | 42 | - | 42 |
| Control | - | - | 164 | 164 |
| Tobacco (packs-years) | ||||
| Never | 36 | 63 | 61 | 124 |
| 0-20 | 28 | 8 | 20 | 28 |
| 20-40 | 88 | 22 | 45 | 67 |
| 40-60 | 51 | 50 | 29 | 79 |
| 60+ | 6 | 29 | 9 | 38 |
| Folate intake | ||||
| Low | 119 | 88 | 77 | 165 |
| Medium | 50 | 35 | 40 | 75 |
| High | 40 | 49 | 47 | 96 |
| Alcohol intake (g per day) | ||||
| 0-138 (low) | 18 | 35 | 43 | 78 |
| 139-889 (medium) | 90 | 55 | 61 | 116 |
| 890+ (high) | 50 | 34 | 24 | 58 |
| ND | 51 | 48 | 36 | 84 |
Cell lines and culture conditions
Human lung cancer cell lines used for the analysis of DNA methylation and MTHFR expression were as follows: A549 (lung carcinoma, ATCC #: CCL-185), H1299 (carcinoma, non-small cell lung cancer, ATCC #: CRL-5803), H1975 (adenocarcinoma; non-small cell lung cancer, ATCC #: CRL-5908) and H1650 (adenocarcinoma; bronchoalveolar carcinoma, Stage3B, ATCC #: CRL-5883). Cells were maintained in standard media and conditions recommended by ATCC.
Cancer samples and DNA extraction
Primary tumours of lung and corresponding blood samples were used for the analysis. For each of the lung cancers, one paraffin block was selected, and representative tumour areas were marked on Hematoxylin-Eosin stained slides from these blocks. Where indicated, samples of lung tissues classified as normal tissues adjacent to the cancerous tissue were also used for the analysis. Adjacent nonmalignant lung tissue was available from 51 cases. Four to five thicker sections (8 μm) per tumour were prepared, and paraffin was removed by incubating slides in Xylene (two times 5 min) followed by incubation in 100%-95%-70% ethanol (3 min each) and water. Tumour areas were carefully scraped and brought into the DNA Extraction buffer (TE pH9 with 0.1μg/μl of Proteinase K and 0.25% of Nonidet P40) and incubated at 56°C for at least 24 hours. For obtaining DNA from cell lines, cell pellets were directly resuspended in DNA extraction buffer. Samples were then heated for 10 min at 95°C to inactivate Proteinase K, spun and supernatant frozen at -20°C. Buffy coat separated from blood samples was used for extraction of genomic DNA by automated equipment (Autopure LS by Gentra Systems, Minneapolis, MN). DNA extraction from paraffin-embedded tumour samples was carried out by EX-WAX™ DNA extraction kit (Chemicon). DNA concentration was quantified with Quant'iT PicoGreen dsDNA reagent (Molecular Probes), an ultrasensitive assay for fluorescent detection of nucleic acids. Sample DNA concentrations were calculated based on a standard curve established with Lambda DNA. The concentrations were then adjusted to 25 ng/μl.
Bisulfite conversion
For methylation analyses, genomic DNA is modified by treatment with sodium bisulfite, which converts all unmethylated cytosines to uracil, then to thymidine during the subsequent PCR step (22). Briefly, 1 μg of DNA in 50 μl distilled H2O was incubated with 5.5 μl of 2 M NaOH at 37°C for 10 minutes, followed by 16 hours treatment at 50°C after adding 30 μl of freshly-prepared 10 mM hydroquinone (Sigma) and 520 μl of freshly-prepared 10mM sodium-bisulfite (Sigma) at pH 5.0. The bisulfite-treated DNA was purified using a DNA Wizard cleanup kit (Promega), by following manufacturer's instructions. The purified DNA was denatured at room temperature for 5 minutes with 5.5 μL of 3M NaOH, followed by ethanol precipitation with 33μL of 10M NH4Ac and 170μL of ethanol. After washing with 70% ethanol, the DNA pellet was resuspended in 50μL TE pH 7.5.
Pyrosequencing assays
To quantify the percentage of methylated cytosine in individual CpG sites, bisulfite-converted DNA was pyrosequenced using a pyrosequencing system (PSQ™ 96MA, Biotage, Sweden). This method treats each individual CpG site as a C/T polymorphism and generates quantitative data for the relative proportion of the methylated versus the unmethylated allele. In our selection of relevant genes that are potential targets of DNA hypermethylation associated with lung cancers, we were guided by two criteria: 1) genes that may have an association with lung cancer based on their supposed biological function, and 2) genes that are proposed to be the frequent targets of hypermethylation in cancer or involved in DNA methylation process itself.
We have established pyrosequencing assays for quantitative measurement of DNA methylation levels in the promoter region of 5 genes (CDH1, CDKN2A, GSTP1, MTHFR, and RASSF1A) in tumour and blood samples (Supplementary Table 1). Quantitative measurement of DNA methylation states are of special importance for the establishment of relationship between epigenetic states and environmental/lifestyle factors that are likely to induce subtle and cumulative changes that may culminate in phenotypic traits after repetitive exposure over a long period of time. Pyrosequencing offers a highly reliable, quantitative, and high-throughput method for analysis of DNA methylation at multiple CpG sites with built-in internal control for completeness of bisulfite treatment (23). Specific pyrosequencing primers were designed to focus on a series of 5 to 8 ‘target’ CpG dinucleotides in the promoter region of the cyclin-dependent kinase inhibitor 2A (CDKN2A, p16INK4A), E-cadherin (CDH1), glutathione S-transferase pi (GSTP1), methylentetrahydrofolate reductase (MTHFR), the Ras-association domain family 1 (RASSF1A) gene, and for the LINE-1 repetitive sequence (Supplementary Table 1 and Supplementary Figure 1). We were careful to include a non-CpG cytosine in the region for pyrosequencing, as it provides the internal control of the completeness of bisulfite treatment (Supplementary Table 1), the criteria considered to be of critical importance for reliability of DNA methylation analysis. Hot-start PCR was performed with HotStarTaq Master Mix kit (Qiagen), and pyrosequencing was carried out in accordance with the manufacturer's protocol (Biotage). The target CpGs were evaluated by converting the resulting pyrograms to numerical values for peak heights (Supplementary Figure 2). Percentage of methylation were calculated as the mean of all CpG analyzed at a given gene promoter.
Gene expression analysis
Total RNA was extracted using RNeasy mini kit (Qiagen). Between 500ng and 1.5μg of total RNA was used for reverse transcription using 200 units of M-MLV Reverse Transcriptase (Invitrogen kit, Carlsbad, CA). 2 μl of the reverse transcriptase reaction was subsequently subjected to PCR amplification using PCR primers designed to generate a DNA fragment 150-250 bp in length. The sequences of primers were as follows: for the MTHFR gene, 5′-CGAACTGCTGAGGGAGCTGT-3′ and 5′-ATGGCCCGTGATCTCCTCCA-3′; for GAPDH, 5′-GTCCACTGGCGTCTTCAC-3′ and 5′-CAGGAGGCATTGCTGATG-3′. PCR reaction was performed in a total volume of 20 ul containing 1 ul of cDNA product, 0.15 mM dNTP, 2.5 mM of MgCl2, 1X PCR buffer, 1.5 unit of Taq DNA polymerase (GoTaq; Promega). PCR products were resolved on 2% agarose gel and the intensity of the bands were quantified by Quantity One 4.6.6 (BioRad).
Statistical Analysis
All methylation data were generated without knowledge of the exposure status and case-control status of the subjects and histological features of the samples analysed. To compare methylation levels in lung tumour samples and blood samples, we used the Wilcoxon rank-sum test that allows the comparison of two groups of independent but continuous samples. Multivariate linear regression analysis was performed to test whether any of the risk factors (smoking, alcohol and folate status) and demographic or clinical characteristics (sex, age and histology) were associated with DNA methylation. Statistical significance of the mean differences between the non-smokers and the other smoking categories were determined by multiple comparison using Dunnett's test. Analyses were performed using SAS software, version 9.1 (SAS Institute Inc, Cary, NC). A P-value of less than 0.01 was considered statistically significant. Cumulative tobacco consumption was calculated by multiplying smoking duration (in years) by smoking intensity (in the equivalent of cigarette packs) and expressed as pack-years. Folate intake was estimated from the food frequency questionnaire which included 23 food items as previously described (24). To assess DNA hypermethylation frequency, we calculated the percentage of tumour samples with methylation levels above 95% quantile levels in blood samples. The statistical significance for differential methylation in lung tumours, normal appearing adjacent lung tissue and blood was calculated using Newman-Keuls' test.
Results
Patient characteristics
A total of 209 lung cancer cases and 164 control subjects were included in the study (Table 1). Among these (cases and controls), 23% were women and 77% were men. Among the cases of lung cancer, 18% were women and 82% were men. Most smokers were men (96%), whereas most non-smokers were women (86%). Cases and control subjects were age-matched with average age of 60 years for both cases and controls. A majority of the cases (74%) were between 51 and 70 years of age.
The predominant tumour histology types were squamous cell carcinoma (SCC) (58%) and adenocarcinoma (28%). SCC was predominant in smokers (68%), whereas adenocarcinoma was the main histology type in never-smokers (70%). SCCs were equally distributed among former and current smokers, whereas both SCC and adenocarcinoma were equally distributed over all age groups. We categorised the subjects as never smokers (26%), 0–20 pack-years (py)(13%), 20–40 py (36%), 40–60 py (21%), and over 60 py (4%). Mixed adeno-squamous (ADC-SCC) type was identified in 6% of the cases. Based on folate intake the subjects were categorized into 4 groups: <4 (25%), 4–5.16 (28%), 5.16–6.14 (24%), and >6.14 (23%). Alcohol consumption (grams per day of ethanol) groups were categorised into low (0–138 g/day, 16%), medium (139–889 g/day, 40%), and high (>890 g/day, 20%).
Methylation profiles of CDH1, CDKN2A, GSTP1, MTHFR and RASSF1A genes in lung cancer
Results of quantitative analysis of methylation status in lung tumours and blood samples are shown in Figure 1 and Supplementary Figure 2 and 3. Analysis of all the cases for methylation levels showed that one gene (MTHFR) exhibited higher levels of methylation (≥25%), two genes (CDH1, and GSTP1) show rather low levels of methylation or were virtually unmethylated (<10%), and the 2 other genes (CDKN2A and RASSF1A) exhibited intermediate levels of methylation (Figure 1). The multivariant pair-wise comparison of DNA methylation revealed no strong correlation between any gene pairs (data not shown). Comparison of mean methylation levels of all CpG sites in tumours and blood samples from cases and controls revealed a highly significant increase in methylation levels in tumours for CDKN2A (p<0.01), MTHFR (p<0.01) and RASSF1A (p<0.01), a moderate but significant decrease for CDH1 (p=0.01), whereas methylation states at the GSTP1 gene were indistinguishable between tumours and blood samples (p=0.2) (Figure 1 and Supplementary Figure 3).
Figure 1.
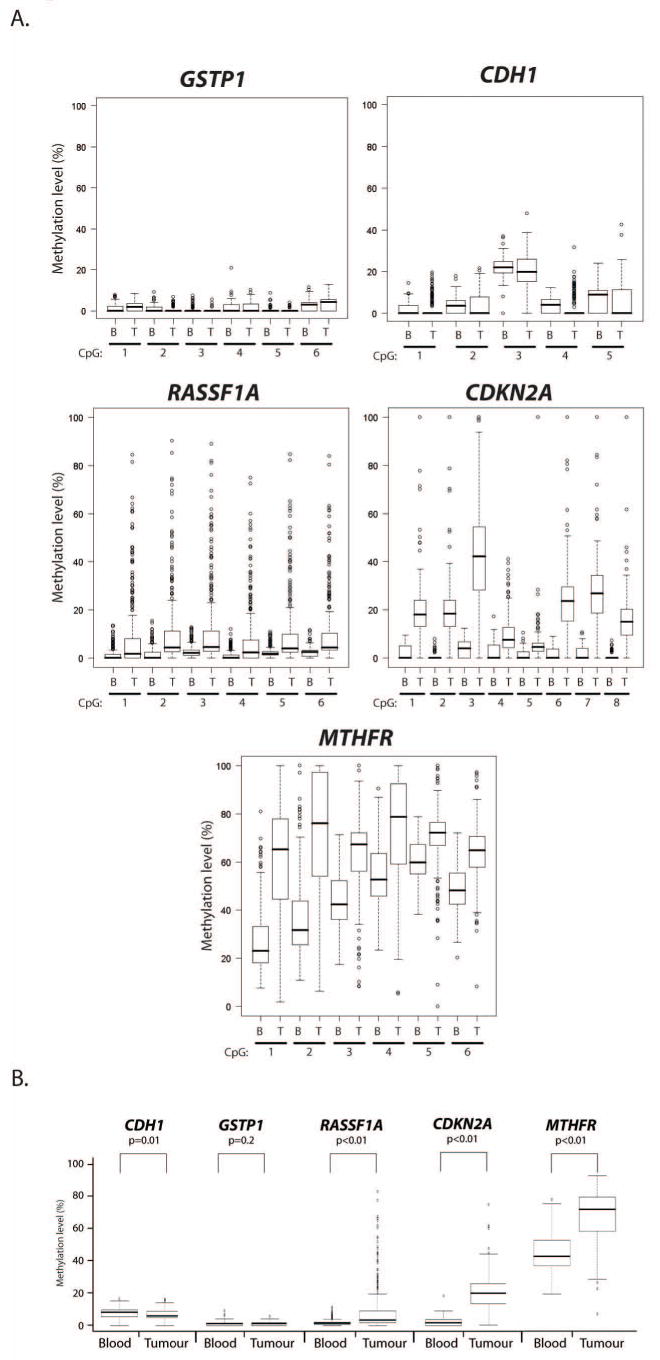
Graphical representation (boxplots) comparing the DNA methylation levels in lung tumours and blood samples. (A) Boxplots of the results obtained by the analysis of individual genes and CpG sites. (B) Boxplots of the summary results obtained by the analysis of mean levels of all CpG sites for a given gene and the level of statistical significance for differential methylation in tumours compared to blood samples.
Analysis of DNA methylation frequency (defined as the percentage of tumour samples with methylation levels above 95% quantile levels in control blood samples) showed that one gene (CDKN2A) was most frequently methylated (91%), two genes (MTHFR and RASSF1A) exhibited intermediate methylation frequency (39% and 36%, respectively), and the two remaining genes (GSTP1 and CDH1) were virtually unmethylated (≤1%) in lung tumours (Supplementary Table 2 and Supplementary Figure 3). When analysed individually, 99% of the lung cancer samples had methylation of at least one of these genes; 34% had one gene methylated, 60% had two genes methylated, 14% had three genes methylated, and none had four or more genes methylated.
Association between methylation levels, clinicopathologic features and risk factors exposure
We initially analysed associations between the methylation of CDH1, CDKN2A, GSTP1, MTHFR, and RASSF1A genes (both mean levels of all CpG sites or individual CpG sites) separately and available epidemiological and clinical information including smoking status, folate intake, alcohol consumption, sex, age and histological subtype of the tumour. The most statistically significant factors were then included in a multivariable analysis. No association was found between methylation levels (measured as mean levels of all CpG sites) of any gene analysed in blood samples of cases and controls and any risk factor included in the analysis (data not shown).
Associations between methylation levels of CDH1, CDKN2A, GSTP1, MTHFR and RASSF1A in lung cancer and clinical features and risk factor exposures are shown in Table 2. Lung cancers from men exhibited higher methylation levels of RASSF1A than those from women (7.5% vs 17.9%, P<0.01), whereas methylation levels of CDH1, CDKN2A, GSTP1 and MTHFR were not found to correlate with sex. No association was found between methylation status of any gene analysed and age or histological subtypes of the tumour. We found no association between methylation of any gene and alcohol intake, with the exception of RASSF1A, which exhibited a significantly lower level of methylation in the intermediate group (5.1%, P<0.01) than in the low-intake (17%) or high-intake (23.3%) group (Table 2).
Table 2.
DNA methylation levels at five genes analysed in lung cancer, stratified by sex, age, histology, tobacco consumption, and alcohol intake
| Gene | |||||
|---|---|---|---|---|---|
|
CDH1 Mean (P) |
CDKN2A Mean (P) |
GSTP1 Mean (P) |
MTHFR Mean (P) |
RASSF1A Mean (P) |
|
|
| |||||
| Sex | |||||
| Women | 8.8 (-) | 19.0 (-) | 1.36 (-) | 70.4 (-) | 7.5 (-) |
| Men | 5.0 (0.05) | 18.0 (0.83) | 1.63 (0.56) | 64.9 (0.10) | 17.9 (<0.01) |
| Age | |||||
| <=40 | 2.6 (-) | 8.35 (-) | 1.5 (-) | 70.1 (-) | 8.3 (-) |
| 40-44 | 7.2 (0.4) | 24.7 (0.10) | 2.0 (0.90) | 54.0 (0.12) | 3.2 (0.79) |
| 45-49 | 6.9 (0.4) | 20.7 (0.17) | 0.9 (0.78) | 70.3 (1.00) | 18.5 (0.38) |
| 50-54 | 8.5 (0.2) | 19.8 (0.18) | 1.7 (1.00) | 67.5 (0.96) | 15.1 (0.61) |
| 55-59 | 7.2 (0.4) | 17.8 (0.32) | 1.7 (1.00) | 66.7 (0.91) | 13.1 (0.79) |
| 60-64 | 7.5 (0.4) | 16.5 (0.43) | 1.3 (0.98) | 72.2 (0.98) | 14.2 (0.70) |
| 65-69 | 7.8 (0.3) | 25.4 (0.03) | 1.2 (0.97) | 73.1 (0.94) | 13.3 (0.78) |
| 70+ | 7.4 (0.4) | 14.4 (0.62) | 1.7 (1.0) | 67.4 (0.95) | 16.1 (0.53) |
| Histology | |||||
| Squamous cell carcinoma | 6.3 (-) | 16.4 (-) | 1.6 (-) | 70.7 (-) | 11.5 (-) |
| Adenocarcinoma | 6.9 (0.93) | 15.1 (0.82) | 1.5 (1.00) | 67. 8 (0.22) | 14.6 (0.25) |
| Tobacco | |||||
| Pack years - Never | 4.7 (-) | 19.5 (-) | 1.9 (-) | 51.6 (-) | 19.8 (-) |
| 0-20 | 8.1 (0.29) | 18.8 (0.99) | 1.0 (0.21) | 68.0 (<0.01) | 8.7 (0.58) |
| 20-40 | 8.3 (0.21) | 19.2 (1.00) | 0.9 (0.13) | 75.6 (<0.01) | 9.3 (0.07) |
| 40-60 | 7.5 (0.39) | 21.3 (0.91) | 1.6 (0.91) | 64.5 (<0.01) | 13.4 (1.00) |
| 60+ | 7.5 (0.39) | 12.5 (0.33) | 2.0 (0.99) | 78.7 (<0.01) | 12.3 (1.00) |
| Current vs former - Never | 4.7 (-) | 18.9 (-) | 1.9 (-) | 50.9 (-) | 19.9 (-) |
| Ex-smoker | 6.9 (0.47) | 19.8 (0.97) | 0.7 (0.05) | 63.8 (<0.01) | 10.4 (0.08) |
| Current smoker | 8.5 (0.13) | 19.0 (1.00) | 1.2 (0.25) | 72.1 (<0.01) | 9.9 (0.05) |
| Smoking intensity - Never | 3.3 (-) | 19.5 (-) | 1.9 (-) | 50.5 (-) | 9.4 (-) |
| 0-12 cig/day | 9.0 (0.04) | 21.4 (0.82) | 0.9 (0.15) | 75.9 (<0.01) | 5.1 (0.58) |
| 13-16 cig/day | 8.4 (0.06) | 19.7 (1.00) | 1.0 (0.19) | 78.6 (<0.01) | 11.1 (0.07) |
| 17-19 cig/day | 8.4 (0.06) | 19.8 (1.00) | 1.3 (0.45) | 66.3 (<0.01) | 8.95 (0.99) |
| 20+ cig/day | 6.7 (0.26) | 15.4 (0.45) | 1.6 (0.81) | 68.0 (<0.01) | 8.7 (0.99) |
| Folates intake | |||||
| Low | 6.8 (-) | 17.4 (-) | 1.4 (-) | 65.4 (-) | 9.1 (-) |
| Medium | 6.1 (0.72) | 18.2 (0.90) | 1.4 (0.53) | 65.7 (0.99) | 11.9 (0.21) |
| High | 7.8 (0.53) | 18.5 (0.86) | 1.6 (0.34) | 69.0 (0.11) | 10.1 (0.91) |
| Alcohol intake (g/day) | |||||
| 0-138 | 7.7 (-) | 21.2 (-) | 1.1 (-) | 71.4 (-) | 17.3 (-) |
| 139-889 | 7.0 (0.89) | 19.0 (0.59) | 1.5 (0.34) | 65.8 (0.03) | 5.3 (<0.01) |
| 890-3119 | 7.1 (0.92) | 19.3 (0.72) | 1.5 (0.53) | 66.8 (0.12) | 4.9 (<0.01) |
| 3120+ | 5.8 (0.77) | 14.2 (0.17) | 1.8 (0.34) | 66.6 (0.40) | 23.3 (0.33) |
While increased levels of DNA methylation of the CDKN2A and RASSF1A genes were observed in tumours of both smokers and non-smokers, the methylation level of the MTHFR gene was significantly higher in current smokers (72.1%, P<0.01) and ex-smokers (63.8%, P<0.01) than in never smokers (51.6%) (Table 2). No consistent dose-response association between MTHFR methylation levels and the number of cigarettes per day smoked was observed. Smoking may affect methylation status of some (but not the other) CpG sites in the same gene; therefore we compared smoking status and methylation levels across CpG sites of the MTHFR gene, and found that within the 6 CpGs of MTHFR analysed, CpGs 1–4 exhibited significantly greater methylation levels in tumours of smokers than in those who never smoked (Supplementary Table 3). By contrast, methylation levels in the two remaining CpG sites (CpG 5 and 6) did not show appreciable difference between smokers and non-smokers. Methylation levels of any CpG site of the MTHFR gene in blood samples of cases and controls were indistinguishable between smokers and never smokers (Supplementary Table 3). Therefore, smoking was associated with a significant increase in DNA methylation of specific CpG sites of MTHFR in lung cancer.
Methylation of the 4 CpGs that are differentially methylated in smokers and non-smokers was not significantly associated with pack-years smoked (Supplementary Table 3). These results indicate that hypermethylation of specific CpG sites of MTHFR in lung tumours increases with exposure to tobacco smoke, and that this increase does not occur in a dose-dependent fashion. However, methylation levels of the MTHFR gene were significantly higher in tumours of current smokers (72.1%, P<0.01) than in ex-smokers (63.8%).
MTHFR methylation and its association with expression of the gene and global hypomethylation in lung cancer
Unscheduled DNA hypermethylation has been associated with gene silencing, therefore we sought to determine whether levels of MTHFR methylation correlate with expression of the gene. To this end, we took advantage of lung cancer cell lines because of the ease of analysis of gene expression in these cells. To determine MTHFR mRNA levels and MTHFR methylation, we performed RT-PCR analysis and pyrosequencing assay, respectively, using identical samples of lung cancer cell lines. The results have revealed that cell lines with high levels of MTHFR methylation (A549, H1299, and H1650) exibit significantly lower level of MTHFR mRNA compared to the cell line with low levels of MTHFR methylation (H1975) (Figure 2). These results demonstrate an inverse correlation between levels of MTHFR methylation and gene expression and thus suggest that hypermethylation of MTHFR gene at the studied CpG sites may have functional significance and may result in partial or complete silencing of the gene.
Figure 2.
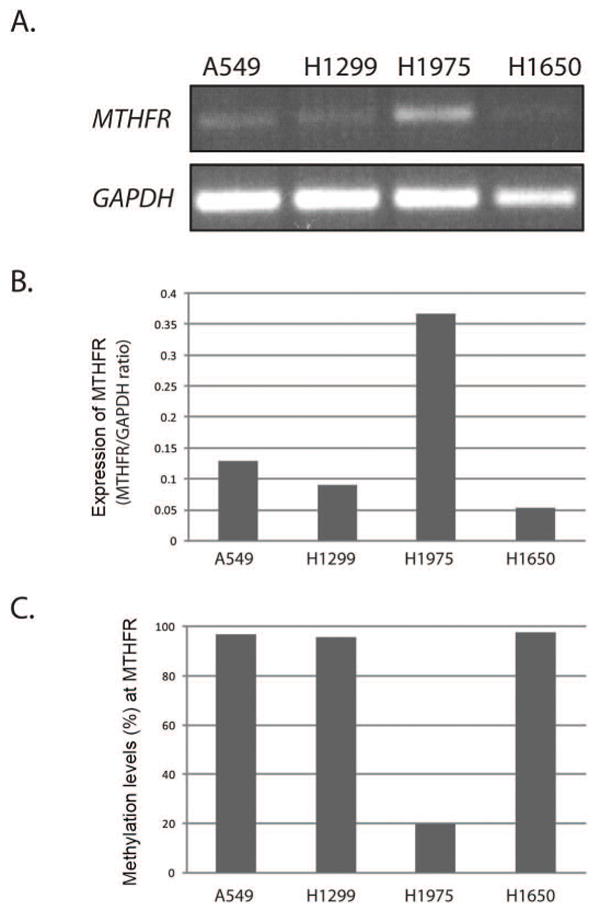
MTHFR expression and DNA methylation analysis in lung cancer cell lines. (A) Four lung cancer cell lines (A549, H1299, H1975, and H1650) were grown in under culture conditions recommended by ATCC and total RNA was analyzed by RT-PCR. Equal RNA loading was controlled by GAPDH (B) Quantification of MTHFR expression levels in (A) was carried out by densitometric analysis and normalization versus GAPDH. (C) Cells were grown as in (A) and DNA extracted was used for analysis of MTHFR methylation. The results shown are representative of 3 independent experiments.
The comparison between DNA methylation levels in cancer tissue and non-tumourigenic adjacent tissues might enforce the association between the exposure to environmental factors and methylation of an individual gene. Therefore, we next examined MTHFR methylation levels in normal appearing adjacent lung tissues. As shown in Supplementary Figure 4A, levels of MTHFR methylation in normal appearing adjacent tissues were comparable to those in blood samples, but significantly lower in comparison to the methylation levels seen in lung tumours. These findings further confirm the notion that MTHFR hypermethylation is a tumour specific event.
The MTHFR gene product plays a role in the maintenance of methionine pool (25, 26), and the inactivation of MTHFR in mice results in significant decrease of global 5-methylcytosine content (27), therefore downregulation of the MTHFR gene induced by gene hypermethylation may impair DNA methylation reactions leading to global hypomethylation. To test whether MTHFR hypermethylation is indeed associated with global hypomethylation, we examined the methylation levels of LINE-1 sequences, a highly repeated and widely interspersed human retrotransposons commonly used as a surrogate for global demethylation (28-30), in lung tumours and corresponding blood samples. We found that the average level of LINE-1 methylation in blood samples and adjacent non-malignant lung tissues was 76% (73-77%) and 73% (67-75%), respectively, whereas it was 63% (41-72%) in lung tumour tissues (Supplementary Figure 4B), an average loss of ∼12% of methylation in the tumour, consistent with global demethylation of the tumour cell genome (28). Interestingly, the calculation of the correlation coefficient between of LINE-1 methylation and methylation levels of the genes under study revealed that in tumour samples a low but consistent inverse correlation is observed between LINE-1 and MTHFR methylation levels (-0.16), whereas no correlation between methylation levels of LINE-1 and RASSF1A or CDKN2A genes was found (Figure 3). These results demonstrate a tumour specific global hypomethylation which inversely correlates with MTHFR methylation levels, suggesting that MTHFR hypermethylation and associated silencing of the gene may promote genome-wide demethylation in tumour cells.
Figure 3.
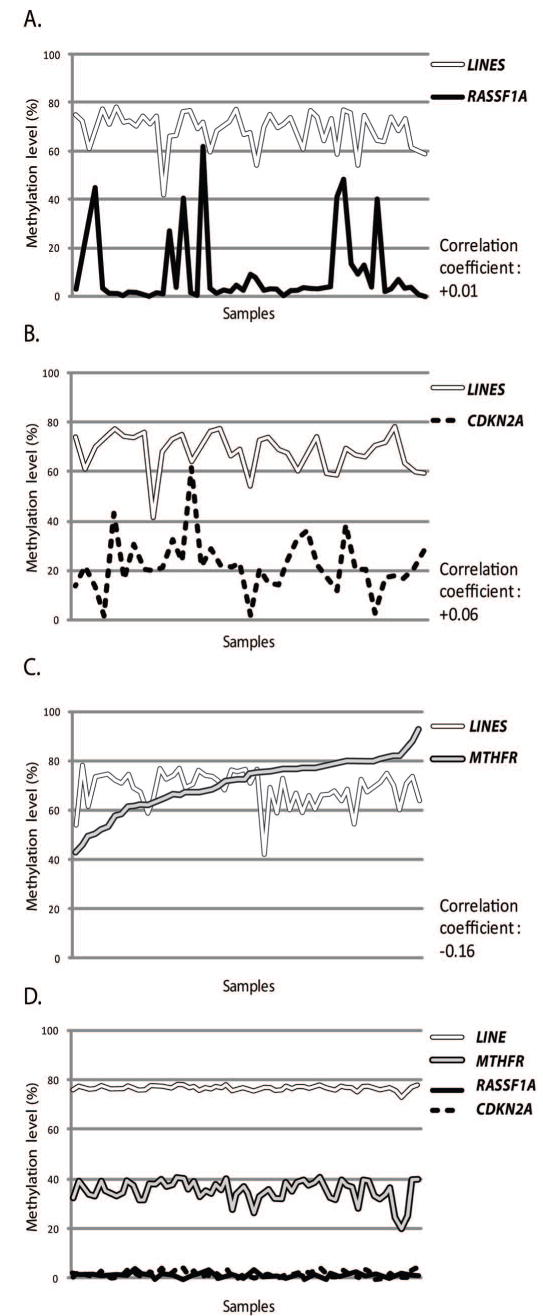
The methylation levels of LINE-1 and its association with hypermethylation of MTHFR in lung tumours. Lung tumour DNA and corresponding blood DNA were evaluated for LINE-1 methylation (surrogate for global methylation levels). The levels of methylation obtained for LINE-1 were compared to methylation levels obtained for RASSF1A (A), CDKN2A (B) and MTHFR (C) in the same lung tumour samples and the correlation coefficient was calculated. As a control, methylation levels obtained for LINE-1, MTHFR, RASSF1A and CDKN2A in blood samples are also shown (D). Note a marked inverse correlation (correlation coefficient, -0.16) between LINE-1 methylation (global methylation) and MTHFR methylation in lung cancer (C).
Discussion
In this study, we have established pyrosequencing assays for analysis of DNA methylation levels in the promoter region of 5 genes (CDH1, CDKN2A, GSTP1, MTHFR and RASSF1A) and combined these quantitative assays with a large series of lung tumour samples and corresponding blood (control) samples. We found a high frequency of aberrant hypermethylation of MTHFR, RASSF1A and CDKN2A, but not CDH1 and GSTP1, in lung tumours compared with corresponding normal control samples. These results are consistent with the notion that epigenetic changes mediated by hypermethylation are tumour-specific events (13, 17).
In lung tumours, the CpG island of CDKN2A is the most frequently hypermethylated among the genes analyzed (91%), in agreement with previous reports showing a high frequency of CDKN2A methylation in variety of human neoplasia including lung cancers (6-10). The second most frequently hypermethylated CpGs in lung tumours were those of the MTHFR gene. MTHFR methylation levels were strongly elevated in a large fraction of lung cancer samples (39%). The MTHFR gene product is critical in maintaining an adequate methionine pool, and is believed to be important in the process of DNA methylation itself (31). Comparing lung tumours and normal (blood) tissues, there was an increase in hypermethylation of all 6 CpG sites of MTHFR (Supplementary Table 3).
We also observed a high frequency of hypermethylation of the RASSF1A gene (36%), a candidate tumour suppressor gene that has been implicated as a pivotal gatekeeper of cell-cycle progression (32). These results are consistent with previous studies showing that RASSF1A is commonly inactivated by hypermethylation in a broad spectrum of human tumours (33-35). De novo methylation of the RASSF1A promoter is one of the most frequent epigenetic inactivation events detected in both non-small-cell lung cancer (NSCLC) and small-cell lung cancer (SCLC) types, and leads to silencing of RASSF1A expression (34, 36). In contrast, we found no significant increase in methylation levels of GSTP1 (0.6%) and CDH1 (1.2%) in lung tumours as compared to blood samples, supporting the notion that unscheduled hypermethylation and associated gene silencing does not occur randomly, but rather in a gene-specific manner (13, 37). It is believed that the hypermethylation of tumor suppressor genes and other cancer-associated genes would be selected and fixed in tumor cells in accordance with the degree of growth advantage caused by their inactivation. Therefore, hypermethylation of MTHFR, RASSF1A or CDKN2A may confer a growth advantage to cancer cells and contribute to the cancer phenotype.
Our analyses that compared tumour histology and methylation levels of any of the genes analysed showed no significant correlation with histological subtypes, consistent with previous studies where prevalence of hypermethylation was indistinguishable between major histological subtypes of lung cancer (10, 38). Our study is not in agreement with the studies by Van der Weyden et al. (36) and Toyooka et al. (39), who reported differences in the methylation patterns of squamous cell carcinoma and adenocarcinoma. This discrepancy might be due, in part, to differences in exposure and techniques used for analysis of DNA methylation. Furthermore, the pair-wise comparison of DNA methylation levels revealed no strong correlation between any gene pairs, indicating that methylation status of any single gene was largely independent of methylation status of other genes, in agreement with previous studies (39). The absence of concordant hypermethylation in multiple genes also suggests that the methylator phenotype (CIMP) observed in a subset of several cancer types such as colorectal cancer is unlikely to be present in lung cancer, a notion consistent with previous studies on different cancer types (40).
Our data showed that methylation levels in RASSF1A, but not the other 4 genes under study, were correlated with sex, with males showing higher levels of methylation than females. There have been few studies on the age and sex differences in DNA methylation levels and patterns. Quantitative profiling of DNA methylation in a small panel of genes suggested that sex is a strong predictor of methylation levels, with males showing higher methylation levels (41). In contrast, high-resolution methylation profiling of three human chromosomes (6, 20 and 22) did not find a significant attributable effect of age and sex on methylation levels (42). Our results suggest that sex is a strong predictor of methylation levels in some genes (such as RASSF1A) in lung tumours, but not in normal tissue (lymphocytes), consistent with the notion that male sex is associated with higher methylation levels in specific genes (41).
To date there have been few reports on associations between DNA methylation changes and alcohol consumption in human cancer, notably liver cancer, head and neck cancer, and colorectal cancer (19, 43, 44). Our data revealed lower levels of RASSF1A methylation in lung tumours from medium alcohol intake groups as compared to those from light or heavy drinkers, whereas no association was found between alcohol consumption and DNA methylation levels of the other four genes under study. These findings suggest that alcohol consumption may be inversely associated with DNA methylation levels in specific genes, although there was no clear dose-response pattern of MTHFR methylation across alcohol intake groups.
Importantly, we found strong association between MTHFR methylation levels and tobacco smoking status, whereas methylation levels in RASSF1A and CDKNA2 were not associated with smoking status, indicating that tobacco exposure targets specific genes for hypermethylation. Interestingly, there was no consistent dose-response pattern of MTHFR methylation across smoking groups, suggesting that even short periods of smoking are sufficient to induce aberrant methylation of MTHFR. It is possible that tobacco smoking and methylation levels of RASSF1A in lung cancer are correlated with certain tobacco consumption threshold above which other changes (such as gene mutations) induced by carcinogens in tobacco smoke may circumvent the need for gene silencing by DNA hypermethylation in tumours. Another noteworthy observation is that methylation levels of MTHFR were significantly higher in tumours of current smokers then in ex-smokers, suggesting that methylation levels may decrease after smoking cessation.
The finding that the methylation of MTHFR correlates with tobacco smoking in lung cancer patients is intriguing. MTHFR has been shown to play a critical role in maintaining an adequate methionine pool as well as ensuring that the homocysteine concentration does not reach toxic levels (31). The enzyme MTHFR catalyses the synthesis of methyonine and consequently is required for its metabolite, S-adenosylmethionine (SAM), which is critical for DNA methylation reactions, and its expression and activity may alter DNA methylation states and contribute to cancer risk (25, 26). Consistent with this notion, MTHFR hypermethylation in lung cancer cell lines correlates with gene expression and is associated with global hypomethylation in lung cancer (this study), whereas the disruption of the MTHFR gene in mice results in global DNA hypomethylation and susceptibility to several disorders (27). Furthermore, the presence of polymorphism in the MTHFR gene (C667T, alanine to valine), a common germ-line variant associated with lower enzymatic activity, has been implicated in cancer risk (45-49). Therefore, our results showing higher levels of MTHFR methylation in lung cancer among smokers than in never-smokers suggest that tobacco smoking may target MTHFR through an epigenetic mechanism.
Previous studies have unequivocally demonstrated that DNA hypermethylation is associated with gene silencing, and that genes with high levels of methylcytosine in their promoter region are usually transcriptionally silent (13). Therefore, tobacco-mediated deregulation of MTHFR gene expression may silence the gene and thus disrupt the DNA methylation process itself. While further studies are required to test the functional impact of MTHFR hypermethylation, it is possible that tobacco-mediated hypermethylation of MTHFR, and consequently partial or complete silencing of the gene, triggers global hypomethylation, a phenomenon almost universally observed in human cancer (13, 17). Consistent with this hypothesis, we found a negative correlation between MTHFR methylation and MTHFR gene expression and also between MTHFR methylation and LINE-1 methylation, used as a surrogate of genome-wide hypomethylation, whereas no correlation between LINE-1 methylation and methylation of any other gene studied was observed. However, this does not rule out the possibility that different components of tobacco smoke may induce promoter-specific hypermethylation of genes other than MTHFR, and also that other pathways may be involved in tumour specific global hypomethylation.
The precise mechanism that underlies targeting of MTHFR for hypermethylation by tobacco smoke in lung cancer remains unclear. Generally, it has been proposed that several factors related to the DNA methylation process, such as DNMT activity and proximity to a methylation center, as well as locus-specific factors including transcription factor motifs, histone marks and local chromatin structure, could be involved in the differential susceptibility of methylation among the genes (33). Furthermore, the preexisting methylation status of CpG islands may also contribute to the differential vulnerability to aberrant methylation (50). In this respect, it is noteworthy that among the genes analysed in our study, only MTHFR exhibited appreciable levels of preexisting methylation in normal tissues. These observations may reflect a higher affinity of methylcytosine for putative tobacco smoke epimutagens compared with unmethylated cytosines. It is thus tempting to speculate that pre-existing methylation of MTHFR may represent a hotspot for tobacco compounds that deregulate local methylation process, possibly through DNMT activity (Figure 4). An analogous mechanism has been reported for preferential binding of benzo[a]pyrene, a highly carcinogenic polycyclic aromatic hydrocarbon (PAH) present in cigarette smoke, to methylated CpG sites, forming major mutational hotspots in human lung cancer (51, 52). Another possibility is that tobacco smoke compounds may remove cis-acting elements that block the spread of methylation from a methylation center, thus exposing neighboring CpG sites to aberrant methylation (Figure 4). In addition, histone modifications including histone acetylation and methylation maintain local chromatin structure and are believed to play important roles in protecting against unscheduled DNA methylation (53). Therefore, different agents from tobacco smoke may disrupt histone modification patterns and thus expose CpG sites to hypermethylation (19). Further studies are required to elucidate the causal involvement of smoking in the epigenetic inactivation of specific genes in smokers.
Figure 4.
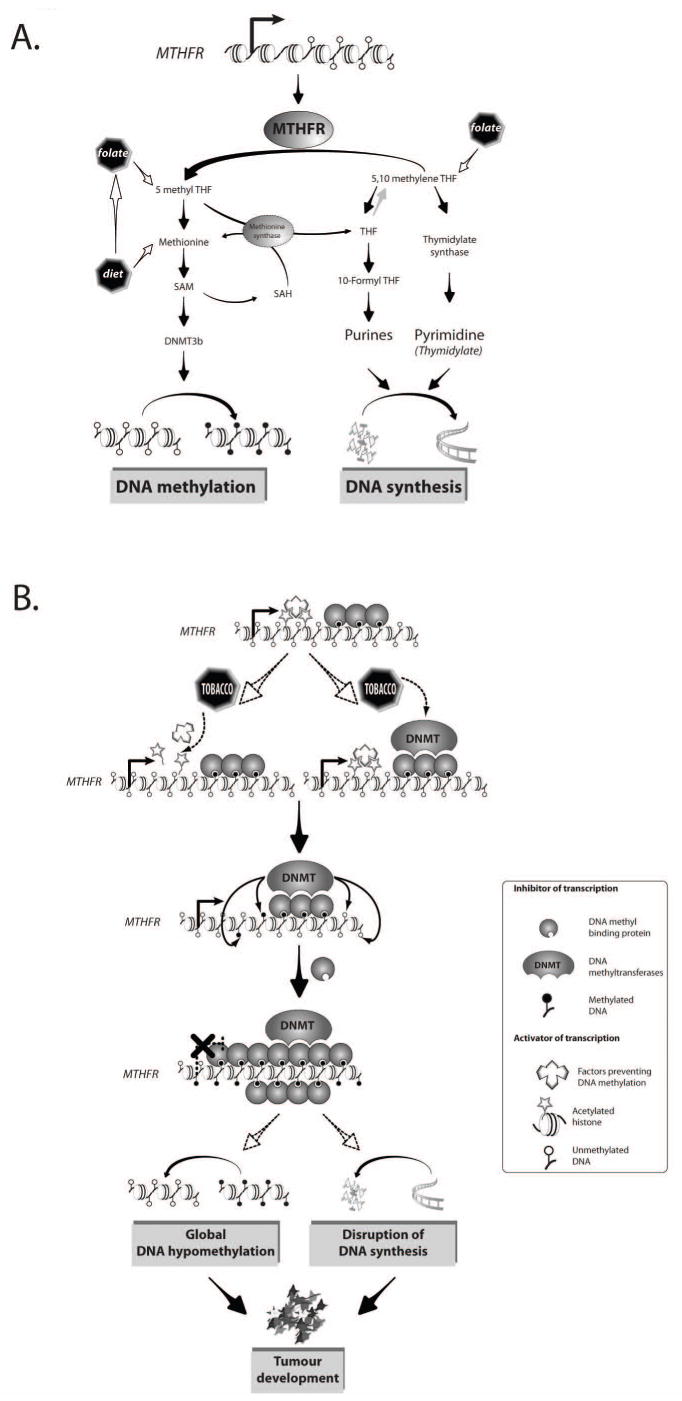
Interaction of tobacco with methyltransfer and DNA methylation. (A) A simplified representation of the cellular pathways involving MTHFR and other key enzymes regulating the metabolism of the methyl group (DNA methylation) and DNA synthesis. (B) A model for the effect of tobacco exposure on the metabolism of the methyl group and the process of DNA methylation. The pre-existing methylation of MTHFR may represent a hotspot for putative tobacco epimutagenes contributing to the differential vulnerability to aberrant methylation. In this model, a higher affinity of methylcytosine for certain tobacco smoke compound(s) deregulates local methylation process through either an enhanced DNMT activity or by removing cis-acting elements (e.g. histone acetylation) that block the spread of methylation from neighboring methyled CpG sites. This leads to abnormal hypermethylation of and silencing of the MTHFR gene, resulting in the deregulation of methyl group metabolism. Thus, tobacco-mediated hypermethylation of MTHFR, and consequent partial or complete silencing of the gene, may trigger global hypomethylation and/or deregulation of DNA synthesis, both of which may contribute to cancer development.
In summary, this study provides evidence of gene-specific and sex-specific differences in methylation patterns in lung cancers arising in tobacco smokers and alcohol drinkers, and thus exemplifies the mechanism by which environmental factors may interact with key genes involved in methyl-donors/acceptors and tumour suppression. In light of these findings, it is reasonable to propose that deregulation of MTHFR and folate metabolism by carcinogens in tobacco smoke may be an underlying mechanism in lung cancer. While further studies are required to test the functional impact of MTHFR and RASSF1A methylation changes in lung cancer, this information could facilitate the development of more accurate risk models. Recent studies have suggested that, in lung cancer patients, aberrant hypermethylation of specific genes can be detected in samples obtained through noninvasive sampling methods such as plasma or sputum. Therefore, analysis of promoter methylation in specific genes may provide a biomarker valuable for identification of individuals with an elevated risk of lung cancer.
Supplementary Material
Supplementary Figure 1. Schematic representation of the chromosomal loci of the genes analyzed. The CG density is indicated by vertical bars. Exons are indicated by black box and arrows indicate the orientation of each gene. Location of the analyzed sequence for each gene is represented by empty box and is obtained from the UCSC genome browser. Sets of primers (F - forward; R – reverse) for each gene analyzed are indicated by arrows. The LINE-1 sequences analyzed are repetitive elements in the genome and therefore are not shown here (accession number M80343).
Supplementary Figure 2. Examples of pyrogrames obtained from the analysis of DNA extracted from lung tumour (upper panel) and blood sample (lower panel) of the same patient. Representative programs of GSTP1, RASSF1A, CDH1, CDKN2A, MTHFR, and LINE-1 are shown.
Supplementary Figure 3. Summary of the analysis of DNA methylation in 5 genes and LINE-1 in lung tumours and blood controls.
Supplementary Figure 4. Differential methylation of MTHFR and LINE-1 in lung tumours and normal appearing adjacent tissues. (A) Level of methylation obtained for MTHFR in lung tumours (n=209), normal appearing adjacent tissues (n=51) and blood (n=336) are summarized. (B) Levels of LINE-1 methylation in lung tumours (n=55), normal appearing adjacent tissues (n=51) and blood (n=64). The statistical significance for differential methylation in tissues analyzed was calculated using Newman-Keuls' test. The groups exhibiting statistically different methylation levels at P<0.01 are indicated (***).
Acknowledgments
We thank Dr Triantafillos Liloglou for sharing with us his unpublished data on LINE-1 methylation analysis and Dr Rabih Murr for critical reading of the manuscript and helpful discussions. T. Vaissière is supported by a PhD fellowship from la Ligue National (Française) Contre le Cancer. The work in the IARC Epigenetics Group is supported by grants from the National Institutes of Health/National Cancer Institute (NIH/NCI), United States; the Association pour la Recherche sur le Cancer (ARC), France; la Ligue Nationale (Française) Contre le Cancer, France; the European Network of Excellence Environmental Cancer Risk, Nutrition and Individual Susceptibility (ECNIS), and the Swiss Bridge Award (to Z.H.). The authors of this article are partners of European Cancer Risk, Nutrition and Individual Susceptibility, a network of excellence operating within the European Union 6th Framework Program, Priority 5: “Food Quality and Safety” (contract no. 513943).
References
- 1.Parkin DM, Bray FI, Devesa SS. Cancer burden in the year 2000. The global picture. Eur J Cancer. 2001;37 8:S4–66. doi: 10.1016/s0959-8049(01)00267-2. [DOI] [PubMed] [Google Scholar]
- 2.Tobacco smoke and involuntary smoking. Vol. 83. IARC Monographs; Lyon (France): 2004. IARC monographs on the evaluation of carcinogenic risks of chemicals to humans. [PMC free article] [PubMed] [Google Scholar]
- 3.Schwartz AG, Prysak GM, Bock CH, Cote ML. The molecular epidemiology of lung cancer. Carcinogenesis. 2007;28:507–18. doi: 10.1093/carcin/bgl253. [DOI] [PubMed] [Google Scholar]
- 4.Hung RJ, McKay JD, Gaborieau V, et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452:633–7. doi: 10.1038/nature06885. [DOI] [PubMed] [Google Scholar]
- 5.Thorgeirsson TE, Geller F, Sulem P, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–42. doi: 10.1038/nature06846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim DH, Nelson HH, Wiencke JK, et al. p16(INK4a) and histology-specific methylation of CpG islands by exposure to tobacco smoke in non-small cell lung cancer. Cancer Res. 2001;61:3419–24. [PubMed] [Google Scholar]
- 7.Belinsky SA, Nikula KJ, Palmisano WA, et al. Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis. Proc Natl Acad Sci U S A. 1998;95:11891–6. doi: 10.1073/pnas.95.20.11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palmisano WA, Divine KK, Saccomanno G, et al. Predicting lung cancer by detecting aberrant promoter methylation in sputum. Cancer Res. 2000;60:5954–8. [PubMed] [Google Scholar]
- 9.Kersting M, Friedl C, Kraus A, Behn M, Pankow W, Schuermann M. Differential frequencies of p16(INK4a) promoter hypermethylation, p53 mutation, and K-ras mutation in exfoliative material mark the development of lung cancer in symptomatic chronic smokers. J Clin Oncol. 2000;18:3221–9. doi: 10.1200/JCO.2000.18.18.3221. [DOI] [PubMed] [Google Scholar]
- 10.Jarmalaite S, Kannio A, Anttila S, Lazutka JR, Husgafvel-Pursiainen K. Aberrant p16 promoter methylation in smokers and former smokers with nonsmall cell lung cancer. Int J Cancer. 2003;106:913–8. doi: 10.1002/ijc.11322. [DOI] [PubMed] [Google Scholar]
- 11.Feng Q, Hawes SE, Stern JE, et al. DNA methylation in tumor and matched normal tissues from non-small cell lung cancer patients. Cancer Epidemiol Biomarkers Prev. 2008;17:645–54. doi: 10.1158/1055-9965.EPI-07-2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Risch A, Plass C. Lung cancer epigenetics and genetics. Int J Cancer. 2008;123:1–7. doi: 10.1002/ijc.23605. [DOI] [PubMed] [Google Scholar]
- 13.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–28. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 14.Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8:286–98. doi: 10.1038/nrg2005. [DOI] [PubMed] [Google Scholar]
- 15.Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer. 2003;3:253–66. doi: 10.1038/nrc1045. [DOI] [PubMed] [Google Scholar]
- 16.Belinsky SA. Gene-promoter hypermethylation as a biomarker in lung cancer. Nat Rev Cancer. 2004;4:707–17. doi: 10.1038/nrc1432. [DOI] [PubMed] [Google Scholar]
- 17.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–53. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 18.Belinsky SA, Klinge DM, Liechty KC, et al. Plutonium targets the p16 gene for inactivation by promoter hypermethylation in human lung adenocarcinoma. Carcinogenesis. 2004;25:1063–7. doi: 10.1093/carcin/bgh096. [DOI] [PubMed] [Google Scholar]
- 19.Herceg Z. Epigenetics and cancer: towards an evaluation of the impact of environmental and dietary factors. Mutagenesis. 2007;22:91–103. doi: 10.1093/mutage/gel068. [DOI] [PubMed] [Google Scholar]
- 20.Maekita T, Nakazawa K, Mihara M, et al. High levels of aberrant DNA methylation in Helicobacter pylori-infected gastric mucosae and its possible association with gastric cancer risk. Clin Cancer Res. 2006;12:989–95. doi: 10.1158/1078-0432.CCR-05-2096. [DOI] [PubMed] [Google Scholar]
- 21.Hung RJ, Brennan P, Canzian F, et al. Large-scale investigation of base excision repair genetic polymorphisms and lung cancer risk in a multicenter study. J Natl Cancer Inst. 2005;97:567–76. doi: 10.1093/jnci/dji101. [DOI] [PubMed] [Google Scholar]
- 22.Frommer M, McDonald LE, Millar DS, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci U S A. 1992;89:1827–31. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tost J, Dunker J, Gut IG. Analysis and quantification of multiple methylation variable positions in CpG islands by Pyrosequencing. Biotechniques. 2003;35:152–6. doi: 10.2144/03351md02. [DOI] [PubMed] [Google Scholar]
- 24.Hung RJ, Hashibe M, McKay J, et al. Folate-related genes and the risk of tobacco-related cancers in Central Europe. Carcinogenesis. 2007;28:1334–40. doi: 10.1093/carcin/bgm067. [DOI] [PubMed] [Google Scholar]
- 25.Paz MF, Avila S, Fraga MF, et al. Germ-line variants in methyl-group metabolism genes and susceptibility to DNA methylation in normal tissues and human primary tumors. Cancer Res. 2002;62:4519–24. [PubMed] [Google Scholar]
- 26.Kawakami K, Ruszkiewicz A, Bennett G, Moore J, Watanabe G, Iacopetta B. The folate pool in colorectal cancers is associated with DNA hypermethylation and with a polymorphism in methylenetetrahydrofolate reductase. Clin Cancer Res. 2003;9:5860–5. [PubMed] [Google Scholar]
- 27.Chen Z, Karaplis AC, Ackerman SL, et al. Mice deficient in methylenetetrahydrofolate reductase exhibit hyperhomocysteinemia and decreased methylation capacity, with neuropathology and aortic lipid deposition. Hum Mol Genet. 2001;10:433–43. doi: 10.1093/hmg/10.5.433. [DOI] [PubMed] [Google Scholar]
- 28.Chalitchagorn K, Shuangshoti S, Hourpai N, et al. Distinctive pattern of LINE-1 methylation level in normal tissues and the association with carcinogenesis. Oncogene. 2004;23:8841–6. doi: 10.1038/sj.onc.1208137. [DOI] [PubMed] [Google Scholar]
- 29.Yang AS, Estecio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004;32:e38. doi: 10.1093/nar/gnh032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Estecio MR, Gharibyan V, Shen L, et al. LINE-1 hypomethylation in cancer is highly variable and inversely correlated with microsatellite instability. PLoS ONE. 2007;2:e399. doi: 10.1371/journal.pone.0000399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jung AY, Poole EM, Bigler J, Whitton J, Potter JD, Ulrich CM. DNA methyltransferase and alcohol dehydrogenase: gene-nutrient interactions in relation to risk of colorectal polyps. Cancer Epidemiol Biomarkers Prev. 2008;17:330–8. doi: 10.1158/1055-9965.EPI-07-2608. [DOI] [PubMed] [Google Scholar]
- 32.Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet. 2000;25:315–9. doi: 10.1038/77083. [DOI] [PubMed] [Google Scholar]
- 33.Kim DH, Kim JS, Ji YI, et al. Hypermethylation of RASSF1A promoter is associated with the age at starting smoking and a poor prognosis in primary non-small cell lung cancer. Cancer Res. 2003;63:3743–6. [PubMed] [Google Scholar]
- 34.Pfeifer GP, Dammann R. Methylation of the tumor suppressor gene RASSF1A in human tumors. Biochemistry (Mosc) 2005;70:576–83. doi: 10.1007/s10541-005-0151-y. [DOI] [PubMed] [Google Scholar]
- 35.Dammann R, Yang G, Pfeifer GP. Hypermethylation of the cpG island of Ras association domain family 1A (RASSF1A), a putative tumor suppressor gene from the 3p21.3 locus, occurs in a large percentage of human breast cancers. Cancer Res. 2001;61:3105–9. [PubMed] [Google Scholar]
- 36.van der Weyden L, Adams DJ. The Ras-association domain family (RASSF) members and their role in human tumourigenesis. Biochim Biophys Acta. 2007;1776:58–85. doi: 10.1016/j.bbcan.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet. 2006;7:21–33. doi: 10.1038/nrg1748. [DOI] [PubMed] [Google Scholar]
- 38.Kikuchi S, Yamada D, Fukami T, et al. Hypermethylation of the TSLC1/IGSF4 promoter is associated with tobacco smoking and a poor prognosis in primary nonsmall cell lung carcinoma. Cancer. 2006;106:1751–8. doi: 10.1002/cncr.21800. [DOI] [PubMed] [Google Scholar]
- 39.Toyooka S, Maruyama R, Toyooka KO, et al. Smoke exposure, histologic type and geography-related differences in the methylation profiles of non-small cell lung cancer. Int J Cancer. 2003;103:153–60. doi: 10.1002/ijc.10787. [DOI] [PubMed] [Google Scholar]
- 40.Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer. 2004;4:988–93. doi: 10.1038/nrc1507. [DOI] [PubMed] [Google Scholar]
- 41.Sarter B, Long TI, Tsong WH, Koh WP, Yu MC, Laird PW. Sex differential in methylation patterns of selected genes in Singapore Chinese. Hum Genet. 2005;117:402–3. doi: 10.1007/s00439-005-1317-9. [DOI] [PubMed] [Google Scholar]
- 42.Eckhardt F, Lewin J, Cortese R, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006;38:1378–85. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Engeland M, Weijenberg MP, Roemen GM, et al. Effects of dietary folate and alcohol intake on promoter methylation in sporadic colorectal cancer: the Netherlands cohort study on diet and cancer. Cancer Res. 2003;63:3133–7. [PubMed] [Google Scholar]
- 44.Giovannucci E, Rimm EB, Ascherio A, Stampfer MJ, Colditz GA, Willett WC. Alcohol, low-methionine--low-folate diets, and risk of colon cancer in men. J Natl Cancer Inst. 1995;87:265–73. doi: 10.1093/jnci/87.4.265. [DOI] [PubMed] [Google Scholar]
- 45.Hubner RA, Lubbe S, Chandler I, Houlston RS. MTHFR C677T has differential influence on risk of MSI and MSS colorectal cancer. Hum Mol Genet. 2007;16:1072–7. doi: 10.1093/hmg/ddm055. [DOI] [PubMed] [Google Scholar]
- 46.Chen J, Giovannucci E, Kelsey K, et al. A methylenetetrahydrofolate reductase polymorphism and the risk of colorectal cancer. Cancer Res. 1996;56:4862–4. [PubMed] [Google Scholar]
- 47.Slattery ML, Potter JD, Samowitz W, Schaffer D, Leppert M. Methylenetetrahydrofolate reductase, diet, and risk of colon cancer. Cancer Epidemiol Biomarkers Prev. 1999;8:513–8. [PubMed] [Google Scholar]
- 48.Ma J, Stampfer MJ, Christensen B, et al. A polymorphism of the methionine synthase gene: association with plasma folate, vitamin B12, homocyst(e)ine, and colorectal cancer risk. Cancer Epidemiol Biomarkers Prev. 1999;8:825–9. [PubMed] [Google Scholar]
- 49.Toffoli G, Gafa R, Russo A, et al. Methylenetetrahydrofolate reductase 677 C-->T polymorphism and risk of proximal colon cancer in north Italy. Clin Cancer Res. 2003;9:743–8. [PubMed] [Google Scholar]
- 50.Vertino PM, Yen RW, Gao J, Baylin SB. De novo methylation of CpG island sequences in human fibroblasts overexpressing DNA (cytosine-5-)-methyltransferase. Mol Cell Biol. 1996;16:4555–65. doi: 10.1128/mcb.16.8.4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith LE, Denissenko MF, Bennett WP, et al. Targeting of lung cancer mutational hotspots by polycyclic aromatic hydrocarbons. J Natl Cancer Inst. 2000;92:803–11. doi: 10.1093/jnci/92.10.803. [DOI] [PubMed] [Google Scholar]
- 52.Yoon JH, Smith LE, Feng Z, Tang M, Lee CS, Pfeifer GP. Methylated CpG dinucleotides are the preferential targets for G-to-T transversion mutations induced by benzo[a]pyrene diol epoxide in mammalian cells: similarities with the p53 mutation spectrum in smoking-associated lung cancers. Cancer Res. 2001;61:7110–7. [PubMed] [Google Scholar]
- 53.Vaissiere T, Sawan C, Herceg Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat Res. 2008;659:40–8. doi: 10.1016/j.mrrev.2008.02.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Schematic representation of the chromosomal loci of the genes analyzed. The CG density is indicated by vertical bars. Exons are indicated by black box and arrows indicate the orientation of each gene. Location of the analyzed sequence for each gene is represented by empty box and is obtained from the UCSC genome browser. Sets of primers (F - forward; R – reverse) for each gene analyzed are indicated by arrows. The LINE-1 sequences analyzed are repetitive elements in the genome and therefore are not shown here (accession number M80343).
Supplementary Figure 2. Examples of pyrogrames obtained from the analysis of DNA extracted from lung tumour (upper panel) and blood sample (lower panel) of the same patient. Representative programs of GSTP1, RASSF1A, CDH1, CDKN2A, MTHFR, and LINE-1 are shown.
Supplementary Figure 3. Summary of the analysis of DNA methylation in 5 genes and LINE-1 in lung tumours and blood controls.
Supplementary Figure 4. Differential methylation of MTHFR and LINE-1 in lung tumours and normal appearing adjacent tissues. (A) Level of methylation obtained for MTHFR in lung tumours (n=209), normal appearing adjacent tissues (n=51) and blood (n=336) are summarized. (B) Levels of LINE-1 methylation in lung tumours (n=55), normal appearing adjacent tissues (n=51) and blood (n=64). The statistical significance for differential methylation in tissues analyzed was calculated using Newman-Keuls' test. The groups exhibiting statistically different methylation levels at P<0.01 are indicated (***).