

Abstract
Multiple investigations in vivo have shown that melatonin (MEL) has a neuroprotective effect in the treatment of spinal cord injury (SCI). This study investigates the role of MEL as an intervening agent for ameliorating Ca2+-mediated events, including activation of calpain, following its administration to rats sustaining experimental SCI. Calpain, a Ca2+-dependent neutral protease, is known to be involved in the pathogenesis of SCI. Rats were injured using a standard weight-drop method that induced a moderately severe injury (40 g.cm force) at T10. Sham controls received laminectomy only. Injured animals were given either 45 mg/kg MEL or vehicle at 15 min post-injury by intraperitoneal injection. At 48 hr post-injury, spinal cord (SC) samples were collected. Immunofluorescent labelings were used to identify calpain expression in specific cell types, such as neurons, glia, or macrophages. Combination of terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end labeling (TUNEL) and double immunofluorescent labelings was used to identify apoptosis in specific cells in the SC. The effect of MEL on axonal damage was also investigated using antibody specific for dephosphorylated neurofilament protein (dNFP). Treatment of SCI animals with MEL attenuated calpain expression, inflammation, axonal damage (dNFP), and neuronal death, indicating that MEL provided neuroprotective effect in SCI. Further, expression and activity of calpain and caspse-3 were examined by Western blotting. The results indicated a significant decrease in expression and activity of calpain and caspse-3 in SCI animals after treatment with MEL. Taken together, this study strongly suggested that MEL could be an effective neuroprotective agent for treatment of SCI.
Keywords: apoptosis, calpain, macrophages, melatonin, neurons, spinal cord injury
Introduction
Spinal cord injury (SCI) can result in severe disability, sensory disorder, paralysis, other neurologic deficits, and even death. Unfortunately, about 250,000 people suffer from SCI in the United States, with almost 11,000 new cases occurring every year [1]. The pathophysiology of SCI is initiated by a primary injury because of mechanical compression of the spinal cord (SC) and propagated by a secondary injury, which largely induces apoptotic cell death [2]. This secondary injury is thought to include several mechanisms including ischemia, reperfusion, and increase in intracellular Ca2+ (ic[Ca2+]) leading to activation of pro-apoptotic proteases, such as calpains and caspases for induction of apoptotic cascades [3]. Because secondary injury cascades begin several hours after primary injury and evolve over several days, this apoptotic death is believed to be a potential target for drug therapy [4].
Following injury, increase in ic[Ca2+] leads to rises in the expression and activity of the Ca2+-activated neutral protease calpain; and calpain activity has been shown to play an important role in cell injury and tissue destruction following SCI [2]. While calpain plays a role in normal cell functions [5], overactivation of this protease leads to degradation of cytoskeletal and myelin proteins and also the activation of pro-apoptotic factors such as Bax, calcineurin, and caspase-3 [6–10]. Overactivation of calpain has been noted in neurons and glial cells exposed to toxins in vitro [11, 12], and similar results are noted in vivo in the central nervous system (CNS) tissue following traumatic brain injury [13] and SCI [2, 13]. Apoptosis in neurons, both in culture and in the penumbra of the SCI animals, can be reversed by treatment with calpain inhibitors [14, 15].
Because SCI induces numerous cell death cascades, pharmacotherapy may require either a combination of agents or a single agent with multiple effects. Currently, the only approved treatment for SCI is methylprednisolone [16], and the efficacy of this therapy has been controversial [17]. Our current investigation outlines the use of melatonin (MEL) as a therapy for experimentally induced SCI in rats.
MEL is an indoleamine, naturally produced in the pineal gland and other tissues, and it has been shown to possess several properties that may be responsible for its neuroprotective effects [18–24]. In animal models of SCI, MEL has been found highly effective in reducing the level of lipid peroxidation following injury [25, 26]. This effect may be due to facts that MEL is powerful antioxidant [18, 19, 27] and it stimulates several antioxidants, including catalase, glutathione reductase, peroxidase, and superoxide dismutase [19–21, 23]. MEL has also been shown to attenuate glutamate-mediated Ca2+-influx and inflammation by inhibiting the levels of pro-inflammatory cytokines [19, 21, 28]. Because of its multi-faceted properties, MEL has been tested in several studies involving CNS insult [21, 23, 24, 29]. The abilities of MEL to inhibit apoptosis in brain cells [30] as well is in other tissue [31, 32] have been well documented.
Protein degradation following SCI leads to neuronal death and destruction of axons and myelin. As calpain has been implicated in this process, it may be possible to preserve the axon–myelin structural unit and protect cells by inhibiting increases in calpain expression. The intention of our experiments was to determine whether MEL treatment could reduce calpain expression following SCI and thereby attenuate cytoskeletal protein degradation, preserve the structure of axons and myelin, and limit neuronal death in SCI.
Our results indicated that MEL treatment indeed inhibited inflammation by reducing the activation of astrocytes, microglia, and macrophages. Calpain expression and caspase-3 activation were also significantly reduced, and neuronal death was attenuated following administration of MEL in acute SCI rats. Furthermore, MEL treatment was also associated with reduced axonal degeneration in the SCI. These experimental data suggest that MEL as a single agent or perhaps in combination with other agents may have therapeutic potential for the treatment of SCI. Preliminary data of this study has been presented earlier [33].
Material and methods
Animal care and injury induction
Male Sprague–Dawley rats were purchased from Charles River Laboratories (Wilmington, MA, USA) and were provided with water and food pellets ad libitum. All care, surgery, and injury induction were performed in accordance with the “Guide for the Care and Use of Laboratory Animals” of the US Department of Health and Humans Services (National Institutes of Health, Bethesda, MD, USA) and were approved by the Institutional Animal Care and Use Committee (IACUC) at the Medical University of South Carolina (Charleston, SC, USA). Induction of SCI was performed following the standardized weight-drop method as previously described [34]. Briefly, rats were anesthetized and a laminectomy was performed with the aid of a dissecting microscope at the T10. Rats were then placed in a stereotactic device, and the spinal column was immobilized. An impounder (0.3 cm in diameter) was placed gently on the dura and SCI was induced by directly dropping a 5 g weight from a height of 8 cm onto the impounder. Sham animals underwent laminectomy only. An additional sham group received MEL but was not injured in order to compare the effects of MEL treatment in intact animals and in injured animals. Animals with SCI were randomly divided into two groups: the MEL group received 45 mg/kg MEL (Helsinn Chemical Co., Biasca, Switzerland) by intraperitoneal injection at 15 min after SCI, and the vehicle group received equal volumes of dimethyl sulfoxide (DMSO, Sigma Chemical, St. Louis, MO, USA). We used the DMSO to dissolve MEL. At 48 hr post-injury, all rats were anesthetized and sacrificed by decapitation. The original laminectomy was extended and two 1-cm SCI segments were excised, including the lesion and the caudal penumbra.
Double immunofluorescent labelings for calpain expression in specific cell types
For double immunofluorescent labelings, SC tissue was immediately frozen (−70°C) in Optima Cutting Temperature (OCT) media (Fisher Scientific, Suwanee, GA, USA) upon harvest. A cryostat (Reichert-Jung, Leica, Deerfield, IL, USA) was used to produce 5 μm sections, which were fixed to slides in 95% ethanol for 10 min and then rinsed thrice in phosphate-buffered saline (PBS, pH 7.4) for 10 min. Fixation and all subsequent steps were conducted at room temperature (RT). Sections were blocked with 2% (v/v) normal horse and goat sera (Sigma-Aldrich) for 30 min. The samples were incubated for 1 hr with an appropriate dilution of a primary IgG antibody such as polyclonal calpain (1:100) [35], monoclonal NeuN (1:100, clone A60; Chemicon, Temecula, CA, USA), monoclonal CD11b (1:100, clone OX42; Invitrogen, Carlsbad, CA, USA), monoclonal glial fibrillary acidic protein (GFAP; 1:400, clone GA5; Chemicon), and pan monoclonal dNFP (1:100, SMI-311; Covance Princeton, NJ, USA). Slides were then rinsed thrice in PBS for 5 min each followed by incubation with fluorescein isothiocyanate (FITC)-conjugated anti-mouse secondary IgG antibody (Vector Laboratories, Burlingame, CA, USA) and Texas-Red conjugated anti-rabbit secondary IgG antibody (Vector). Then, the sections were rinsed twice in PBS for 5 min each, once in distilled water for 5 min, and mounted with coverslips using Vectashield medium (Vector). Imaging was performed using fluorescence microscopy with ImagePro Plus software (Media Cybergenetics, Silver Spring, MD). Observations were made in the ventral and dorsal horns of the gray matter.
Combination of terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end labeling (TUNEL) and double immunofluorescent labelings
We reported this method previously [36]. Briefly, SC tissue was frozen (−70°C) in OCT (Fisher Scientific) immediately after harvest. Five-micrometer sections of SC were fixed to slides in 95% ethanol for 10 min and post-fixed in 4% methanol-free formaldehyde for 15 min at RT. Sections were rinsed twice in PBS for 5 min each and then incubated with equilibration buffer (Promega, Madison, WI, USA) for 10 min at 37°C. The TUNEL reaction mixture [21.5 μL equilibration buffer, 2.5 μL digoxigenin (DIG)-labeling mix, and 1 μL TdT (25 units/μL)] was then added, and the slides were incubated at 37°C in a humid atmosphere for 1 hr. The reaction was stopped by placing slides in a 2× NaCl/Na-citrate buffer (300 mm NaCl, 30 mm Na-citrate, pH 7.4) bath for 15 min at RT. After washing with PBS to remove any unincorporated DIG, tissues were blocked in 2% sheep and horse sera for 1 hr (Sigma-Aldrich) and then incubated with NeuN antibody (Chemicon) at a 1:100 dilution for 1 hr. Following washing, slides were incubated with secondary antibodies at a 1:75 dilution for 30 min. The secondary antibodies used were: FITC-labeled horse anti-rabbit IgG (Vector) and rhodamine-labeled sheep anti-DIG IgG (Roche Molecular Biochemicals, Indianapolis, IN, USA). Slides were once again washed and then mounted with Vectashield medium (Vector). Images were captured using ImagePro Plus software (Media Cybernetics) at 200× magnification. TUNEL-positive neurons in dorsal and ventral horn were quantified according to our previously reported method [37].
Western blotting
This method was previously described [38–40]. Following harvest, the SC segments were homogenized in a buffer (50 mm Tris–HCl, pH 7.4, 1 mm phenylmethylsulfonyl fluoride, and 5 mm EGTA). Protein concentration was determined using Coomassie Plus Protein Assay Reagent (Pierce, Rockford, IL, USA) by a spectrophotometer at 595 nm (Spectronic, Rochester, NY, USA). Equal volume of sample buffer (62.5 mm Tris–HCl, pH 6.8, 2% SDS, 5 mm β-mercaptoethanol, 10% glycerol) was added to each sample, and samples were then boiled for 5 min. Samples were diluted to 1 mg/mL protein and equal volumes of samples were loaded onto 4–20% gradient gels, which were electrophoresed in an electrophoresis apparatus (Bio-Rad, Hercules, CA, USA) at 200 V. Resolved proteins were then transferred to a nylon membrane (Millipore, Bedford, MA, USA) using an electroblotting apparatus (Genie, Idea Scientific, Minneapolis, MN, USA). The membranes were then blocked for 1 hr in 5% powdered nonfat milk dissolved in a Tris/Tween solution (20 mm Tris–HCl, pH 7.6, 0.1% Tween 20 in saline), and primary IgG antibodies were diluted in blocking buffer and overnight incubated with the membrane. Primary antibodies were polyclonal calpain [35] and polyclonal caspase-3 (1:200, Biotech, Santa Cruz, CA, USA). Membranes were then incubated with a secondary IgG antibody (MP Biomedicals, Solon, OH, USA): goat anti-rabbit for calpain; goat anti-mouse for caspase-3 at a 1:2000 dilution for 1 hr. Between steps, membranes were washed three times in Tris/Tween solution. Membranes were incubated with enhanced chemiluminescence (ECL) reagents (Amersham Pharmacia, Buckinghamshire, UK) and ECL-treated blots were immediately exposed to X-OMAT AR films (Eastman Kodak, Rochester, NY, USA). Autoradiograms were scanned on a UMAX PowerLook 1000 Scanner (UMAX Technologies, Fremont, CA, USA) using Photoshop software (Adobe Systems, Seattle, WA, USA), and optical density (OD) of each band was determined using Quantity One software (Bio-Rad).
Quantitative data of immunofluorescent images and semi-quantitative analysis of mean fluorescence intensity (MFI) of TUNEL staining were performed using the National Institute of Health ImageJ software (NIH, Bethesda, MD, USA).
Statistical analysis
Data were expressed as mean ± S.E.M. of separate experiments (n ≥ 3) and compared by one-way analysis of variance (ANOVA) followed by Fisher's post hoc test. A significant difference between two treatments (P < 0.05) was determined by ANOVA using the StatView software (Abacus Concepts, Berkeley, CA, USA).
Results
We used Western blotting to determine whether MEL treatment of SCI rats influenced the expression and activity of both calpain and caspase-3 (Fig. 1).
Fig. 1.
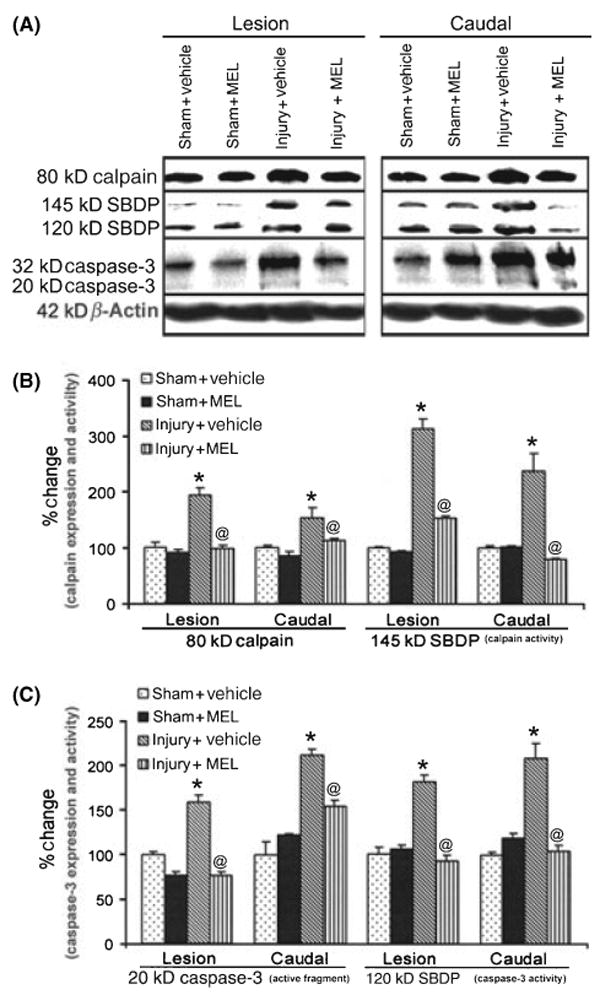
The effects of melatonin (MEL) on expression and activity of calpain and caspase-3 following acute spinal cord (SC) injury. (A) Representative Western blots using lesion and caudal penumbra SC segments from sham and SCI rats treated with vehicle (dimethyl sulfoxide) or MEL (45 mg/kg) for 48 hr. (B) Determination of OD of bands representing calpain expression and activity. (C) Determination of OD of bands representing caspase-3 active fragment and activity. Data presented as percentage change in comparison with sham + vehicle set at 100% (n = 3). Sham + vehicle vs. injury + vehicle (*P ≤ 0.05); and injury + vehicle vs. injury + MEL (@P ≤ 0.05).
Calpain expression (Fig. 1A,B), as detected by an antibody against calpain, was significantly increased to 201 ± 24% in the lesion of the rats following SCI, compared with calpain expression set at 100% in sham-vehicle rats. An increasing trend of calpain expression was also observed in the caudal penumbra in SCI rats. Caspase-3 expression (Fig. 1A,C), as detected by an antibody that also recognized the 20-kDa active caspase-3, was significantly increased in the lesion (160 ± 14%) and caudal penumbra (170 ± 19%) of SCI animals. Treatment with MEL significantly reduced both calpain and 20-kD active-caspase-3 expression in the lesion of SCI rats to the levels similar to those seen in sham animals. Calpain and caspase-3 activities were also analyzed using α-spectrin antibody that recognized the calpain-cleaved 145-kD spectrin breakdown product (SBDP) and the caspase-3-cleaved 120-kD SBDP, respectively. The data suggested that both calpain and caspase-3 activities (as measured in the generation of 145-kD SBDP and 120-kD SBDP, respectively) were reduced in the lesion and caudal penumbra in rats treated with MEL, compared with sham rats. Treatment with MEL had no significant effect on expression and activity of calpain or caspase-3 in sham animals.
We previously demonstrated calpain upregulation in neurons and glial cells following SCI [36, 41–43]. In the present study, the effects of MEL treatment on calpain expression in neurons were assessed by double immunofluorescent labelings of SC tissue harvested at 48 hr post-injury (Fig. 2). Tissue was examined using both a calpain antibody and neuron-specific (NeuN) antibody. At the lesion and caudal penumbral sites, sham animals showed little calpain expression and prominent NeuN expression, and similar results were seen in tissue from sham animals treated with MEL (i.e., sham + MEL). Vehicle-treated SCI animals showed decreased levels of NeuN expression and increased levels of calpain expression in dorsal and ventral sections of the SC lesion segment. In sections taken from the caudal penumbra of vehicle-treated SCI animals, healthy neuronal morphology was noted, however, these sections continued to demonstrate appreciably increased calpain expression, compared with sham animals. Compared to injury + vehicle animals, MEL treatment appeared to reduce calpain expression in the ventral caudal penumbra but not in other areas.
Fig. 2.
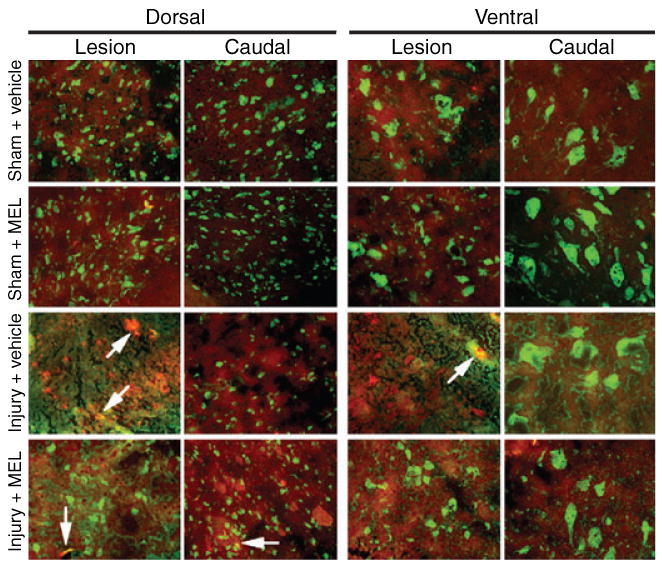
The effects of melatonin (MEL) on calpain expression in neurons following acute spinal cord (SC) injury. Double immunofluorescent labelings were performed using lesion and caudal penumbra SC sections from sham and SCI rats treated with vehicle or MEL (45 mg/kg) for 48 hr. Calpain expression in neurons was assessed using calpain antibody (red) and NeuN antibody (green). Arrows show co-staining (n = 3, 200× magnification).
As apoptosis begins as early as 6 hr post-injury [44, 45], we also examined DNA fragmentation in neurons in SC segments (Fig. 3). No TUNEL-positive cells were found in the lesion or caudal penumbra of sham animals, while the SCI animals treated with vehicle (i.e., injury + vehicle) showed extensive TUNEL-labeling of neurons and other cells in both dorsal and ventral regions of the SC (Fig. 3A). Compared with vehicle-treated SCI animals, MEL-treated SCI animals showed similar TUNEL-labeling in the ventral area of the lesion segment; however, less TUNEL-labeling of neuronal and non-neuronal cells in all other areas of the lesion and caudal penumbra. Semi-quantitative analysis of the mean fluorescent intensity (MFI) per unit area of neurons showed significantly higher levels of TUNEL-positive neurons in both dorsal and ventral horn of lesion and caudal sections from SCI rats, compared with corresponding sections from sham animals (Fig. 3B). SC sections from injured rats treated with MEL (i.e., injury + MEL) showed significant decrease in TUNEL-positive neurons.
Fig. 3.
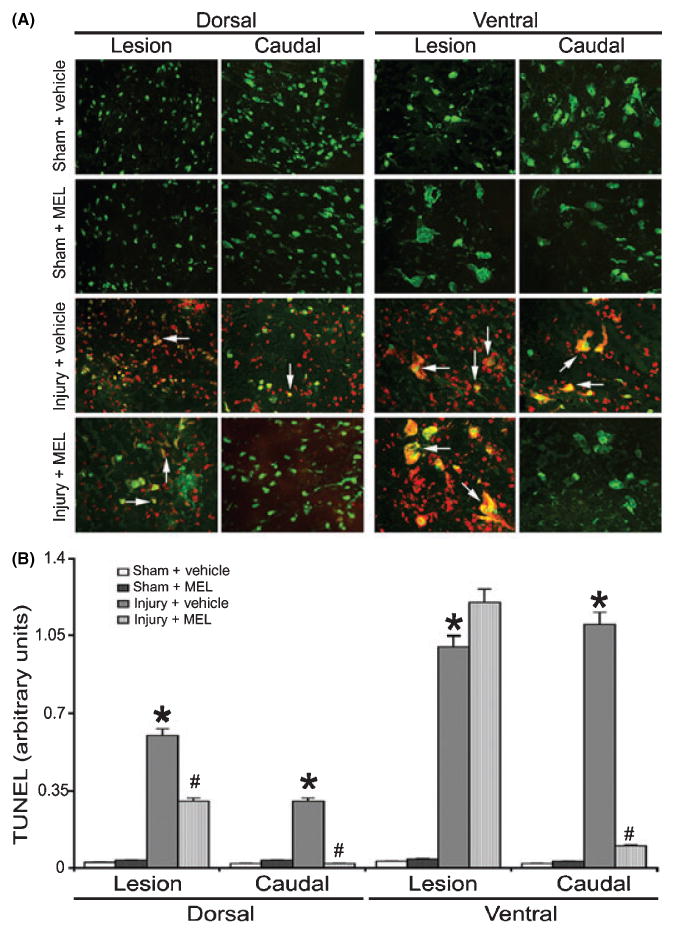
The effects of melatonin (MEL) on neuronal death following acute spinal cord (SC) injury. (A) Double immunofluorescent labelings for detecting apoptotic death in neurons were performed using lesion and caudal penumbra SC sections from sham and SCI rats treated with vehicle or MEL (45 mg/kg) for 48 hr. Apoptotic death in neurons was assessed by TUNEL staining (red) and NeuN staining (green). Arrows show co-staining (n = 3, 200× magnification). (B) Quantification of TUNEL-positive cells. Sham + vehicle vs. injury + vehicle (*P ≤ 0.05); and injury + vehicle vs. injury + MEL (#P < 0.05).
In order to examine the inflammatory response following SCI, macrophage infiltration was examined along with calpain expression using double immunofluorescent labelings with calpain and CD11b (OX42) antibodies (Fig. 4). The OX42 clone identifies both activated and inactivated microglia and macrophages. SC sections from sham animals and sham animals treated with MEL demonstrated little calpain expression and basal levels of OX42-positive cells. The lesion and caudal penumbra of vehicle-treated SCI animals showed increased numbers of OX42-positive cells and increased calpain expression in the white matter. MEL treatment of SCI animals did not appear to reduce the number of OX42-positive cells in the lesion segment; however, qualitatively fewer OX42-positive cells were seen in the caudal penumbra, compared with vehicle-treated SCI animals. Calpain expression was also marginally reduced in the SCI rats treated with MEL.
Fig. 4.
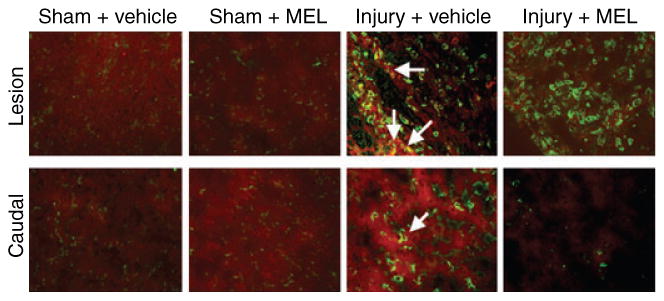
The effects of melatonin (MEL) on infiltration of inflammatory cells following acute spinal cord (SC) injury. Double immunofluorescent labelings of macrophages/microglia (inflammatory cells) and calpain expression were performed using lesion and caudal penumbra SC sections from sham and SCI rats treated with vehicle or MEL (45 mg/kg) for 48 hr. Macrophages/microglia and calpain expression were assessed using OX42 antibody (green) and calpain antibody (red), respectively. Arrows show co-staining (n = 3, 200× magnification).
In order to assess increased astrogliosis following acute SCI, SC sections from the lesion and caudal penumbra were examined using double immunofluorescent labelings with calpain and astrocytic marker GFAP antibodies (Fig. 5). In sham animals, basal levels of calpain expression and GFAP-positive cells were noted. Compared with sham animals, injured animals treated with only vehicle showed notable increases in calpain and GFAP stainings in both the lesion and caudal penumbra, and co-staining was also noted. In the lesion, MEL-treated SCI animals demonstrated low calpain and GFAP levels similar to those seen in vehicle-treated SCI animals. However, sections from the caudal penumbra in MEL-treated SCI animals demonstrated calpain and GFAP levels similar to sham animals and demonstrably less than vehicle-treated SCI animals, indicating that MEL treatment attenuated the degree of post-SCI astrocystosis in the penumbra.
Fig. 5.
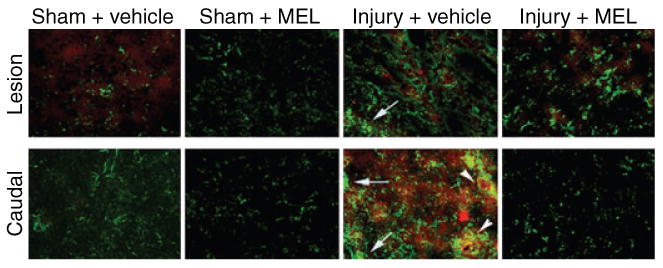
The effects of melatonin (MEL) on astrogliosis following acute spinal cord (SC) injury. Double immunofluoresecent labelings for astrogliosis and calpain expression were performed using lesion and caudal penumbra SC sections from sham and SCI rats treated with vehicle or MEL (45 mg/kg) for 48 hr. Astrogliosis and calpain expression were assessed using GFAP antibody (green) and calpain antibody (red), respectively. Arrows depict astrogliosis. Arrowheads show co-staining (n = 3, 200× magnification).
We also examined the effects of MEL treatment on axonal damage using an antibody against dephosphorylated neurofilament protein (dNFP), which was increased after axonal injury (Fig. 6). Sham + vehicle and Sham + MEL animals showed little dNFP staining in the lesion and caudal penumbra (Fig. 6A). The lesion and caudal penumbra from SCI animals treated with vehicle (DMSO) alone demonstrated markedly increased expression of dNFP (Fig. 6B), indicating increased axonal damage. In contrast, MEL treatment reduced dNFP expression in both lesion and caudal penumbra (Fig. 6B), suggesting that MEL treatment attenuated axonal damage in SCI.
Fig. 6.
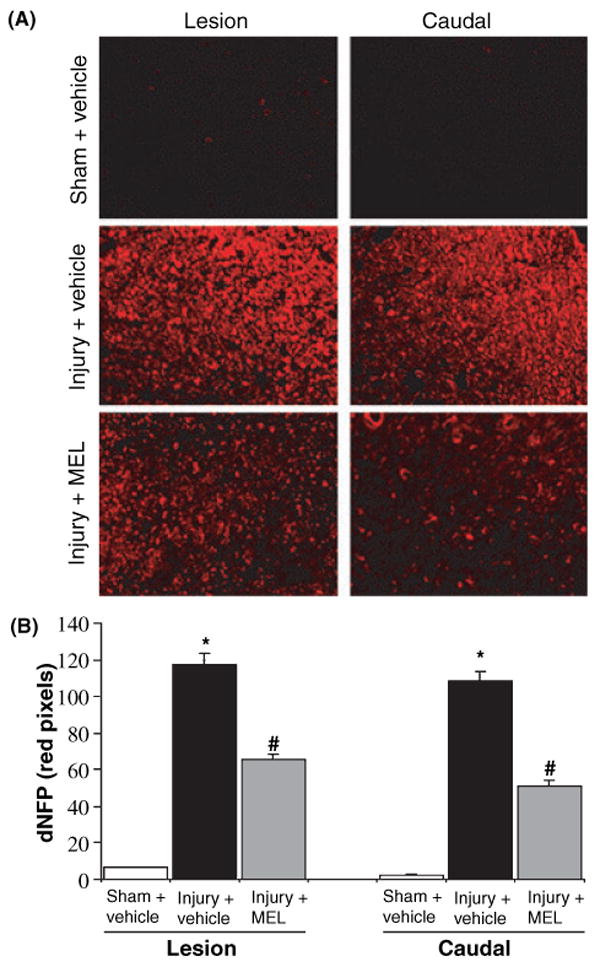
The effects of melatonin (MEL) on axonal damage following acute spinal cord (SC) injury. (A) Single immunofluorescent labeling for axonal damage was performed using lesion and caudal penumbra SC sections from sham and SCI rats treated with vehicle or MEL (45 mg/kg) for 48 hr. Use of dephosphorylated neurofilament protein (dNFP) antibody (red) detected axonal damage (n = 3, 200× magnification). (B) Determination of dNFP. Data were analyzed with NIH ImageJ software. Significant differences in the number of pixels were indicated: sham + vehicle vs. injury + vehicle (*P ≤ 0.05); and injury + vehicle vs. injury + MEL (#P < 0.05).
Discussion
In addition to many normal physiologic roles of MEL, the neuroprotective properties of this pineal gland hormone have been investigated in neurodegenerative diseases and injuries [29]. Investigations of the chemical properties of MEL, both in vivo and in vitro, indicate that MEL and its metabolites are potent antioxidants that directly scavenge both reactive oxygen species (ROS) and reactive nitrogen species (RNS) in the cells [18, 46, 47]. Besides, MEL stimulates many antioxidant enzymes. As MEL easily crosses the blood–brain barrier, reduces inflammation, ROS, RNS, and lipid peroxidation; it has also been implicated to have high neuroprotective capabilities [48]. Thus, the use of MEL as a potential therapeutic agent in the pathologic processes involving inflammation and ROS has generated a great deal of interest. Our current studies have shown that MEL has beneficial effects as a therapeutic agent in acute SCI in rats. Treatment with MEL decreased inflammation and calpain expression in neurons and demonstrated fewer reactive astroctytes, macrophages, and microglia. Moreover, MEL attenuated axonal damage and cell death by inhibition of calpain and caspase-3 activities.
Elevated levels of ic[Ca2+] have been found in the SC lesion and penumbra in SCI rats [42] (Sribnick et al., unpublished). As increases in ic[Ca2+] activate calpain and MEL reduces glutamate-induced Ca2+ influx, we investigated the effect of MEL on calpain expression and activity in SCI rats. Increased expression of calpain in neurons following SCI has been proposed to contribute to continued axonal damage and neuronal cell death [43]. In addition to degradation of NFP and other cytoskeletal proteins, activated calpain plays a direct role in the apoptotic cascade through caspase-3 activation [2]. Calpain has previously been implicated in cell death in the pathophysiology of CNS trauma such as traumatic brain injury and SCI and diseases such as Alzheimer's disease and Parkinson's disease [2, 37, 49–52]. The current findings of increased calpain activity at 48 hr after injury are in agreement with the previously demonstrated calpain activity in the lesion and penumbra [42, 53]. Similar changes were seen after SCI animals were treated with estrogen, though a lesser effect was found with methylprednisolone treatment [53–55]. Treatment with MEL, in our studies, significantly decreased calpain expression in neurons, both in the lesion and caudal penumbra. This indicates protection of cytoskeletal proteins for maintaining structural integrity of cell membrane. MEL is known to stabilize membranes and thus prevents cell damage and death [56]. As both calpain and caspase-3 activities are known to participate in cell death, the implication is that reduction of their activities by MEL may have protected neurons in the SC following injury.
Present findings also confirm the earlier work on neuroprotection by estrogen and calpain inhibitor in SCI [2, 53, 55]. In addition to decreases in calpain expression and activation, we found MEL treatment also reduced neuronal death in SCI rats. The extensive loss of neurons seen in the lesion of dorsal and ventral sections of the SC is most likely due to necrosis after primary injury. MEL treatment attenuated some of this loss of neurons in lesion of the dorsal region, which was likely to be apoptotic in nature, or rescued the neurons that were only partially damaged. The loss of neuron in the ventral region of the lesion was not protected by MEL. This may be due to the facts that motoneurons are more sensitive to damage and they are necrotic, which is irreversible, in the lesion. Also, MEL may not be reaching the lesion due to impairment of blood supply. Nonetheless, MEL treatment significantly attenuated neuron death in the penumbra (periphery of the lesion). This protection is most likely due to inhibition of calpain and caspase-3 activities by MEL in the lesion and penumbra as both proteases are involved in cell death mechanisms.
As inflammatory response in SCI is one of the damaging factors and MEL has been found to have anti-inflammatory properties [21, 28], we have examined whether anti-inflammatory effect of MEL provides neuroprotection to SCI rats by reducing the activation of glial and inflammatory cells. MEL treatment reduced infiltration and activation of inflammatory cells in the lesion and penumbra and limited the increased expression of calpain in microglia, macrophages, and astrocytes. These OX42-positive cells represent infiltrating macrophages, not resident microglia, as very little staining is seen in sham spinal cord. While microglia should be present in uninjured SC, the level of calpain expression was barely detectable as shown by double immuofluorescent labeling. Infiltration of macrophages (neutrophils) and OX42- and ED2-positive cells with increased calpain activity occurs in the lesion during 12–24 hr after SCI [41]. Activation of astrocytes, macrophages, and microglia has long been postulated to enhance degeneration of SC because of release of toxic factors (including pro-inflammatory cytokines), inhibition of axonal growth, and initiation of secondary degeneration processes [41, 45, 57]. MEL reportedly reduces the level of inflammatory cytokines, inflammation, and lesion volume in cortical injury [21, 28, 29]. In the present study, MEL treatment demonstrated a significantly decreased calpain expression and activity in astrocytes, macrophages, and microglia in the lesion and penumbra. The number of these cells was also decreased, particularly in the penumbra of MEL treated SCI rats. The decreased reactivity of astrocytes, macrophages, and microglia by MEL might ameliorate the axonal injury, cell death, and further damage to the penumbra of the SCI rats.
Axonal damage, neuronal death, and myelin breakdown have been demonstrated in lesion and penumbra of SCI; hence, attenuation of cell death and preservation of the axon–myelin structural unit is essential for recovery of function [2, 3, 42, 53, 55]. Such preservation of axonal integrity and protection of neurons has been found in estrogen-treated SCI animals [55]. In this study, a significant decrease in axonal damage, particularly in the penumbra region, was noted in the MEL-treated SCI animals. Treatment with MEL alone and in combination with methylprednisolone has been previously suggested to protect neurons and restore function in SCI [58–60]. However, the current study is focused on the MEL efficacy on inflammatory and proteolytic mechanisms that are involved in axonal damage and neuron death. This implies that MEL, by protecting cells and reducing axonal damage, may help restore conduction and improve motor function in SCI rats.
The effects of MEL and its metabolites as potent free radical scavengers and antioxidants have been well documented [18, 20–22, 47]. The neuroprotective effects of MEL, as demonstrated in the present study, may also be because of its antioxidant properties. MEL stimulates intracellular antioxidant enzymes, including glutathione reductase and superoxide dismutase [20, 29, 61]. As it reduces ROS and lipid peroxidation, MEL has been used as a therapeutic agent in Alzheimer's disease, Huntington's disease, Parkinson's disease, and stroke [23, 29]. MEL has been found to reduce amyloid-β toxicity to neurons [62, 63]. It is neuroprotective in stroke and traumatic brain injury [24]. MEL provides neuroprotection by preventing mitochondrial damage and apoptosis [64, 65]. The ability of MEL to reduce ROS and lipid peroxidation may alleviate damage caused by secondary injury processes in SCI [25, 26, 66]. Beneficial effect of combination of methylprednisolone and MEL in SCI may have resulted from ultrastructural preservation of axons and mitochondria [58].
As destruction of SC following injury is multifactorial, treatment of one damaging pathway is unlikely to provide significant beneficial effect. Thus, treatment with a combination of different protective agents or with an agent with multi-active properties may have more potential for function recovery following SCI. MEL is one such agent with multiple effects, and its efficacy has been tested in other diseases and neurotrauma models. Based on the results obtained from this study, we suggest that MEL-mediated neuroprotection is partly because of inhibition of calpain activity involved in cell death and axonal damage as well as a decrease in inflammatory responses, which are also increased in SCI. In this study, we have tested efficacy of MEL in an acute SCI model. Further work is necessary to examine efficacy of MEL administering at different times following acute SCI as well as chronic SCI for clinical relevance. Nonetheless, the present study suggests that MEL is potential therapeutic agent for treatment of SCI.
Acknowledgments
This work was supported in part by the R01 (NS-31622, NS-38146, NS-45967, NS-41088, NS-57811, and CA-91460) grants and the Medical Scientist Training Program (MSTP) grant (GM08716) from the NIH (Bethesda, MD, USA), and also the Spinal Cord Injury Research Fund (SCIRF-0803, SCIRF-1205, and SCIRF-0607) grants from the state of South Carolina.
References
- 1.National Science Statistical Center. National Spinal Cord Injury Center; Birmingham, AL: 2006. Spinal cord injury: Facts and FIgures at a glance. [Google Scholar]
- 2.Ray SK, Hogan EL, Banik NL. Calpain in the pathophysiology of spinal cord injury: neuroprotection with calpain inhibitors. Brain Res Brain Res Rev. 2003;42:169–185. doi: 10.1016/s0165-0173(03)00152-8. [DOI] [PubMed] [Google Scholar]
- 3.Ray SK, Banik NL. Chaplain and its involvement in the path physiology of CNS injuries and diseases: therapeutic potential of chaplain inhibitors for prevention of neurodegeneration. Curr Drug Targets CNS Neurol Disord. 2003;2:173–189. doi: 10.2174/1568007033482887. [DOI] [PubMed] [Google Scholar]
- 4.Dumont RJ, Verma S, Okonkwo DO, et al. Acute spinal cord injury, part II: contemporary pharmacotherapy. Clin Neuropharmacol. 2001;24:265–279. doi: 10.1097/00002826-200109000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Zimmerman UJ, Boring L, Pak JH, et al. The calpain small subunit gene is essential: its inactivation results in embryonic lethality. IUBMB Life. 2000;50:63–68. doi: 10.1080/15216540050176610. [DOI] [PubMed] [Google Scholar]
- 6.Wu HY, Tomizawa K, Oda Y, et al. Critical role of calpain-mediated cleavage of calcineurin in excitotoxic neurodegeneration. J Biol Chem. 2004;279:4929–4940. doi: 10.1074/jbc.M309767200. [DOI] [PubMed] [Google Scholar]
- 7.Blomgren K, Zhu C, Wang X, et al. Synergistic activation of caspase-3 by m-calpain after neonatal hypoxia–ischemia: a mechanism of “pathological apoptosis”? J Biol Chem. 2001;276:10191–10198. doi: 10.1074/jbc.M007807200. [DOI] [PubMed] [Google Scholar]
- 8.Wood DE, Thomas A, Devi LA, et al. Bax cleavage is mediated by calpain during drug-induced apoptosis. Oncogene. 1998;17:1069–1078. doi: 10.1038/sj.onc.1202034. [DOI] [PubMed] [Google Scholar]
- 9.Ray SK, Neuberger TJ, Deadwyler G, et al. Calpain and calpastatin expression in primary oligodendrocyte culture: preferential localization of membrane calpain in cell processes. J Neurosci Res. 2002;70:561–569. doi: 10.1002/jnr.10414. [DOI] [PubMed] [Google Scholar]
- 10.Nath R, Raser KJ, Staord D, et al. Non-erythroid α-spectrin breakdown by calpain and interleukin-1β-converting-enzyme-like protease(s) in apoptotic cells: contributory roles of both protease families in neuronal apoptosis. Biochem J. 1996;3:683–690. doi: 10.1042/bj3190683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sribnick EA, Ray SK, Banik NL. Estrogen prevents glutamate-induced apoptosis in c6 glioma cells by a receptor-mediated mechanism. Neuroscience. 2006;137:197–209. doi: 10.1016/j.neuroscience.2005.08.074. [DOI] [PubMed] [Google Scholar]
- 12.Das A, Sribnick EA, Wingrave JM, et al. Calpain activation in apoptosis of ventral spinal cord 4.1 (VSC4.1) motoneurons exposed to glutamate: calpain inhibition provides functional neuroprotection. J Neurosci Res. 2005;81:551–562. doi: 10.1002/jnr.20581. [DOI] [PubMed] [Google Scholar]
- 13.Mccracken E, Hunter AJ, Patel S, et al. Calpain activation and cytoskeletal protein breakdown in the corpus callosum of head-injured patients. J Neurotrauma. 1999;16:749–761. doi: 10.1089/neu.1999.16.749. [DOI] [PubMed] [Google Scholar]
- 14.Ray SK, Wilford GG, Crosby CV, et al. Diverse stimuli induce calpain overexpression and apoptosis in C6 glioma cells. Brain Res. 1999;829:18–27. doi: 10.1016/s0006-8993(99)01290-1. [DOI] [PubMed] [Google Scholar]
- 15.Ray SK, Matzelle DD, Wilford GG, et al. Cell death in spinal cord injury (SCI) requires de novo protein synthesis: calpain inhibitor E-64-d provides neuroprotection in SCI lesion and penumbra. Ann N Y Acad Sci. 2001;939:436–449. doi: 10.1111/j.1749-6632.2001.tb03655.x. [DOI] [PubMed] [Google Scholar]
- 16.Bracken MB, Shepard MJ, Collins WF, JR, et al. Methylprednisolone or naloxone treatment after acute spinal cord injury: 1-year follow-up data. Results of the Second National Acute Spinal Cord Injury Study. J Neurosurg. 1992;76:23–31. doi: 10.3171/jns.1992.76.1.0023. [DOI] [PubMed] [Google Scholar]
- 17.Hurlbert RJ. Methylprednisolone for acute spinal cord injury: an inappropriate standard of care. J Neurosurg. 2000;93:1–7. doi: 10.3171/spi.2000.93.1.0001. [DOI] [PubMed] [Google Scholar]
- 18.Tan DX, Chen LD, Poeggeler B, et al. Melatonin: a potent, endogenous hydroxyl radical scavenger. Endocrine J. 1993;1:57–60. [Google Scholar]
- 19.Allegra M, Reiter RJ, Tan DX, et al. The chemistry of melatonin's interaction with reactive species. J Pineal Res. 2003;34:1–10. doi: 10.1034/j.1600-079x.2003.02112.x. [DOI] [PubMed] [Google Scholar]
- 20.Reiter RJ, Tan DX, Manchester LC, Qi W. Biochemical reactivity of melatonin with reactive oxygen and nitrogen species: a review of the evidence. Cell Biochem Biophys. 2001;34:237–256. doi: 10.1385/CBB:34:2:237. [DOI] [PubMed] [Google Scholar]
- 21.Cuzzocrea S, Reiter RJ. Pharmacological actions of melatonin in acute and chronic inflammation. Curr Top Med Chem. 2002;2:153–165. doi: 10.2174/1568026023394425. [DOI] [PubMed] [Google Scholar]
- 22.Reiter RJ, Burkhardt S, Cabrera J, Garcia JJ. Beneficial neurobiological effects of melatonin under conditions of increased oxidative stress. Curr Med Chem – Central Nervous System Agents. 2002;2:45–58. [Google Scholar]
- 23.Reiter RJ, Tan DX, Leon J, et al. When melatonin gets on your nerves: its beneficial actions in experimental models of stroke. Exp Biol Med (Maywood, NJ) 2005;230:104–117. doi: 10.1177/153537020523000205. [DOI] [PubMed] [Google Scholar]
- 24.Beni SM, Kohen R, Reiter RJ, et al. Melatonin-induced neuroprotection after closed head injury is associated with increased brain antioxidants and attenuated late-phase activation of NF-κB and AP-1. FASEB J. 2004;18:149–151. doi: 10.1096/fj.03-0323fje. [DOI] [PubMed] [Google Scholar]
- 25.Erten SF, Kocak A, Ozdemir I, et al. Protective effect of melatonin on experimental spinal cord ischemia. Spinal Cord. 2003;41:533–538. doi: 10.1038/sj.sc.3101508. [DOI] [PubMed] [Google Scholar]
- 26.Genovese T, Mazzon E, Muia C, et al. Attenuation in the evolution of experimental spinal cord trauma by treatment with melatonin. J Pineal Res. 2005;38:198–208. doi: 10.1111/j.1600-079X.2004.00194.x. [DOI] [PubMed] [Google Scholar]
- 27.Fujimoto T, Nakamura T, Ikeda T, Takagi K. Potent protective effects of melatonin on experimental spinal cord injury. Spine. 2000;25:769–775. doi: 10.1097/00007632-200004010-00003. [DOI] [PubMed] [Google Scholar]
- 28.Guerrero JM, Reiter RJ. Melatonin–immune system relationships. Curr Top Med Chem. 2002;2:167–179. doi: 10.2174/1568026023394335. [DOI] [PubMed] [Google Scholar]
- 29.Maldonado MD, Murillo-Cabezas F, Terron MP, et al. The potential of melatonin in reducing morbidity–mortality after craniocerebral trauma. J Pineal Res. 2006;42:1–11. doi: 10.1111/j.1600-079X.2006.00376.x. [DOI] [PubMed] [Google Scholar]
- 30.Jou MJ, Peng TI, Reiter RJ, et al. Visualization of the antioxidative effects of melatonin at the mitochondrial level during oxidative stress-induced apoptosis of rat brain astrocytes. J Pineal Res. 2004;37:55–70. doi: 10.1111/j.1600-079X.2004.00140.x. [DOI] [PubMed] [Google Scholar]
- 31.Lin AM, Fang SF, Chao PL, Yang CH. Melatonin attenuates arsenite-induced apoptosis in rat brain: involvement of mitochondrial and endoplasmic reticulum pathways and aggregation of α-synuclein. J Pineal Res. 2007;43:163–171. doi: 10.1111/j.1600-079X.2007.00456.x. [DOI] [PubMed] [Google Scholar]
- 32.Radogna F, Paternoster L, Albertini MC, et al. Melatonin antagonizes apoptosis via receptor interaction in U937 monocytic cells. J Pineal Res. 2007;43:154–162. doi: 10.1111/j.1600-079X.2007.00455.x. [DOI] [PubMed] [Google Scholar]
- 33.Yallapragada AV, Stucki BL, Sribnick EA, et al. Society for Neuroscience. Washington, DC: 2004. Melatonin attenuated calpain expression and neuronal death in the lesion and caudal penumbra following spinal cord injury in rats. Program No. 108.6. 2004 Abstract Viewer/Itinerary Planner. Online. [Google Scholar]
- 34.Perot PL, Jr, Lee WA, Hsu CY, et al. Therapeutic model for experimental spinal cord injury in the rat: I. Mortality and motor deficit. Cent Nerv Syst Trauma. 1987;4:149–159. doi: 10.1089/cns.1987.4.149. [DOI] [PubMed] [Google Scholar]
- 35.Chakrabarti AK, Yoshida Y, Powers JM, et al. Calcium-activated neutral proteinase in rat brain myelin and subcellular fractions. J Neurosci Res. 1988;20:351–358. doi: 10.1002/jnr.490200309. [DOI] [PubMed] [Google Scholar]
- 36.Ray SK, Schaecher KE, Shields DC, et al. Combined TUNEL and double immunofluorescent labeling for detection of apoptotic mononuclear phagocytes in autoimmune demyelinating disease. Brain Res Brain Res Protoc. 2000;5:305–311. doi: 10.1016/s1385-299x(00)00027-1. [DOI] [PubMed] [Google Scholar]
- 37.Samantaray S, Knaryan V, Banik NL. Parkinsonian neurotoxin rotenone activated calpain and caspase-3 leading to motoneuron degeneration in spinal cord of Lewis rats. Neuroscience. 2007;146:741–755. doi: 10.1016/j.neuroscience.2007.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sur P, Sribnick EA, Wingrave JM, et al. Estrogen attenuates oxidative stress-induced apoptosis in C6 glial cells. Brain Res. 2003;971:178–188. doi: 10.1016/s0006-8993(03)02349-7. [DOI] [PubMed] [Google Scholar]
- 39.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 40.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shields DC, Schaecher KE, Hogan EL, Banik NL. Calpain activity and expression increased in activated glial and inflammatory cells in penumbra of spinal cord injury lesion. J Neurosci Res. 2000;61:146–150. doi: 10.1002/1097-4547(20000715)61:2<146::AID-JNR5>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 42.Wingrave JM, Schaecher KE, Sribnick EA, et al. Early induction of secondary injury factors causing activation of calpain and mitochondria-mediated neuronal apoptosis following spinal cord injury in rats. J Neurosci Res. 2003;73:95–104. doi: 10.1002/jnr.10607. [DOI] [PubMed] [Google Scholar]
- 43.Li Z, Hogan EL, Banik NL. Role of calpain in spinal cord injury: Increased mcalpain immunoreactivity in spinal cord after compression injury in the rat. Neurochem Int. 1995;27:425–432. doi: 10.1016/0197-0186(95)00024-3. [DOI] [PubMed] [Google Scholar]
- 44.Banik NL, Shields DC, Ray SK, Hogan EL. The pathophysiological role of calpain in spinal cord injury. In: Wang KKW, Yuen PW, editors. The pharmacology and toxicology of calcium dependent proteases. Taylor and Francis Publishers; Washington D.C.: 1999. pp. 211–227. [Google Scholar]
- 45.Gomes-Leal W, Corkill DJ, Freire MA, et al. Astrocytosis, microglia activation, oligodendrocyte degeneration, and pyknosis following acute spinal cord injury. Exp Neurol. 2004;190:456–467. doi: 10.1016/j.expneurol.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 46.Reiter RJ. Oxidative damage in the central nervous system: protection by melatonin. Prog Neurobiol. 1998;3:359–384. doi: 10.1016/s0301-0082(98)00052-5. [DOI] [PubMed] [Google Scholar]
- 47.Tan DX, Manchester LC, Terron MP, et al. One molecule, many derivatives: a never-ending interaction of melatonin with reactive oxygen and nitrogen species? J Pineal Res. 2007;42:28–42. doi: 10.1111/j.1600-079X.2006.00407.x. [DOI] [PubMed] [Google Scholar]
- 48.Tan DX, Reiter RJ, Manchester LC, et al. Chemical and physical properties and potential mechanisms: melatonin as a broad spectrum antioxidant and free radical scavenger. Curr Top Med Chem. 2002;2:181–197. doi: 10.2174/1568026023394443. [DOI] [PubMed] [Google Scholar]
- 49.Chera B, Schaecher KE, Rocchini A, et al. Immunofluorescent labeling of increased calpain expression and neuronal death in the spinal cord of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice. Brain Res. 2004;1006:150–156. doi: 10.1016/j.brainres.2004.01.065. [DOI] [PubMed] [Google Scholar]
- 50.Crocker SJ, Smith PD, Jackson-Lewis V, et al. Inhibition of calpains prevents neuronal and behavioral deficits in an MPTP mouse model of Parkinson's disease. J Neurosci. 2003;23:4081–4091. doi: 10.1523/JNEUROSCI.23-10-04081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Posmantur R, Hayes RL, Dixon CE, Taft WC. Neurofilament 68 and neurofilament 200 protein levels decrease after traumatic brain injury. J Neurotrauma. 1994;11:533–545. doi: 10.1089/neu.1994.11.533. [DOI] [PubMed] [Google Scholar]
- 52.Saatman KE, Murai H, Bartus RT, et al. Calpain inhibitor AK295 attenuates motor and cognitive deficits following experimental brain injury in the rat. Proc Natl Acad Sci USA. 1996;93:3428–3433. doi: 10.1073/pnas.93.8.3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sribnick EA, Wingrave JM, Matzelle DD, et al. Estrogen attenuated markers of inflammation and decreased lesion volume in acute spinal cord injury in rats. J Neurosci Res. 2005;82:283–293. doi: 10.1002/jnr.20622. [DOI] [PubMed] [Google Scholar]
- 54.Raynaud F, Marcilhac A. Implication of calpain in neuronal apoptosis. A possible regulation of Alzheimer's disease. FASEB J. 2006;273:3437–3443. doi: 10.1111/j.1742-4658.2006.05352.x. [DOI] [PubMed] [Google Scholar]
- 55.Sribnick EA, Matzelle DD, Ray SK, Banik NL. Estrogen treatment of spinal cord injury attenuates calpain activation and apoptosis. J Neurosci Res. 2006;84:1064–1075. doi: 10.1002/jnr.21016. [DOI] [PubMed] [Google Scholar]
- 56.Saija A, Tomaino A, Trombetta D, et al. Interaction of melatonin with model membranes and possible implications in its photoprotective activity. Eur J Pharm Biopharm. 2002;53:209–215. doi: 10.1016/s0939-6411(01)00239-9. [DOI] [PubMed] [Google Scholar]
- 57.Fawcett JW, Asher RA. The glial scar and central nervous system repair. Brain Res Bull. 1999;49:377–391. doi: 10.1016/s0361-9230(99)00072-6. [DOI] [PubMed] [Google Scholar]
- 58.Cayli SR, Kocak A, Yilmaz U, et al. Effect of combined treatment with melatonin and methylprednisolone on neurological recovery after experimental spinal cord injury. Eur Spine J. 2004;13:724–732. doi: 10.1007/s00586-003-0550-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kaptanoglu E, Tuncel M, Palaoglu S, et al. Comparison of the effects of melatonin and methylprednisolone in experimental spinal cord injury. J Neurosurg. 2000;93:77–84. doi: 10.3171/spi.2000.93.1.0077. [DOI] [PubMed] [Google Scholar]
- 60.Genovese T, Mazzon E, Crisafulli C, et al. Effects of combination of melatonin and dexamethasone on secondary injury in an experimental mice model of spinal cord trauma. J Pineal Res. 2007;43:140–153. doi: 10.1111/j.1600-079X.2007.00454.x. [DOI] [PubMed] [Google Scholar]
- 61.Mccutcheon EP, Selassie AW, Gu JK, Pickelsimer EE. Acute traumatic spinal cord injury, 1993–2000a population-based assessment of methylprednisolone administration and hospitalization. J Trauma. 2004;56:1076–1083. doi: 10.1097/01.ta.0000082312.71894.d4. [DOI] [PubMed] [Google Scholar]
- 62.Olivieri G, Otten U, Meier F, et al. β-Amyloid modulates tyrosine kinase B receptor expression in SH-SY5Y neuroblastoma cells: influence of the antioxidant melatonin. Neuroscience. 2003;120:659–665. doi: 10.1016/s0306-4522(03)00342-7. [DOI] [PubMed] [Google Scholar]
- 63.Lahiri DK, Chen D, Lahiri P, et al. Melatonin, metals, and gene expression: Implications in aging and neurodegenerative disorders. Ann N Y Acad Sci. 2004;1035:216–230. doi: 10.1196/annals.1332.014. [DOI] [PubMed] [Google Scholar]
- 64.Leon J, Acuna-Castroviejo D, Sainz R, et al. Melatonin and mitochondrial function. Life Sci. 2004;75:765–790. doi: 10.1016/j.lfs.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 65.Reiter RJ, Tan DX, Manchester LC, El-Sawi MR. Melatonin reduces oxidant damage and promotes mitochondrial respiration: implications for aging. Ann N Y Acad Sci. 2002;959:238–250. doi: 10.1111/j.1749-6632.2002.tb02096.x. [DOI] [PubMed] [Google Scholar]
- 66.Liu JB, Tang TS, Yang HL, Xiao DS. Anti-oxidation of melatonin against spinal cord injury in rats. Chinese Med J. 2004;117:571–575. [PubMed] [Google Scholar]