

Abstract
Mammalian electron transfer flavoproteins (ETF) are heterodimers containing a single equivalent of flavin adenine dinucleotide (FAD). They function as electron shuttles between primary flavoprotein dehydrogenases involved in mitochondrial fatty acid and amino acid catabolism and the membrane-bound electron transfer flavoprotein ubiquinone oxidoreductase. The structure of human ETF solved to 2.1-Å resolution reveals that the ETF molecule is comprised of three distinct domains: two domains are contributed by the α subunit and the third domain is made up entirely by the β subunit. The N-terminal portion of the α subunit and the majority of the β subunit have identical polypeptide folds, in the absence of any sequence homology. FAD lies in a cleft between the two subunits, with most of the FAD molecule residing in the C-terminal portion of the α subunit. Alignment of all the known sequences for the ETF α subunits together with the putative FixB gene product shows that the residues directly involved in FAD binding are conserved. A hydrogen bond is formed between the N5 of the FAD isoalloxazine ring and the hydroxyl side chain of αT266, suggesting why the pathogenic mutation, αT266M, affects ETF activity in patients with glutaric acidemia type II. Hydrogen bonds between the 4′-hydroxyl of the ribityl chain of FAD and N1 of the isoalloxazine ring, and between αH286 and the C2-carbonyl oxygen of the isoalloxazine ring, may play a role in the stabilization of the anionic semiquinone. With the known structure of medium chain acyl-CoA dehydrogenase, we hypothesize a possible structure for docking the two proteins.
Keywords: x-ray crystallography, glutaric acidemia type II
In 1956, Crane and Beinert identified electron transfer flavoprotein (ETF), based on its capacity to transfer reducing equivalents from mammalian fatty acyl-CoA dehydrogenases to various electron acceptors (1, 2). Mammalian ETF is the electron acceptor for nine primary flavoprotein dehydrogenases, including the four straight- chain-specific fatty acyl-CoA dehydrogenases (medium-, short-, long-, and very-long chain dehydrogenase), as well as several dehydrogenases involved in amino acid [isovaleryl- (3), glutaryl- (4), and short/branched chain- (5)] and choline [sarcosine- and dimethylglycine-dehydrogenase (6)] catabolism. In mammalian mitochondria, reducing equivalents from these dehydrogenases are transferred sequentially to ETF, ETF-ubiquinone oxidoreductase (ETF-QO), and ubiquinol-cytochrome c oxidoreductase, complex III (7). Defects in ETF or ETF-QO result in glutaric acidemia type II (GAII), an often fatal disease resulting from the inability to catabolize various fatty acyl-CoAs (8).
Sequences of several ETFs have been reported, and three have been expressed as active proteins (9). They form a closely related family with no apparent homology to other known protein sequences. Sequence similarities among the ETFs are moderate, with the human and Paracoccus ETFs showing the highest similarities (≈50% identity), whereas the ETF from W3A1 is only moderately conserved (≈25% identity) (9). In addition to the flavin nucleotide, human (F.E.F., unpublished work), porcine (10), Paracoccus denitrificans (F.E.F., unpublished work), and W3A1 ETF (11) also contain one equivalent of AMP. The role of the mononucleotide is not entirely clear but has been shown to accelerate the in vitro binding of flavin adenine dinucleotide (FAD) to porcine apo-ETF after denaturation of the holoprotein with guanidine–HCl (10).
Biochemical and biophysical studies have been used to investigate the FAD binding site and the binding site(s) for redox partners on ETF (12). Experiments with FAD analogues indicate that the 8 position of the xylene portion of the isoalloxazine ring is exposed to solvent and that, when substituted with bulky constituents, kcat of medium chain acyl-CoA dehydrogenase in the reductive half-reaction of ETF is relatively unaffected, although the apparent KmETF increases. Irradiation of ETF reconstituted with 8-N3-FAD, a photoaffinity analogue, results in preferential crosslinking of the flavin nucleotide to the β subunit (13). Flavin analogue studies and spectroscopic experiments indicate that there must be amino acid side chains that stabilize anionic charge localized in the region of N1-C2O of the anionic flavin semiquinone and hydroquinone of mammalian ETFs (14). The interaction of ETF with its redox partners is at least partially electrostatic in nature (15), and both porcine medium chain acyl-CoA dehydrogenase and porcine ETF-QO are chemically crosslinked preferentially to the β subunit of porcine ETF (16, 17). Kinetic experiments with a chimeric human/P. denitrificans ETF also suggest that the amino-terminal region of the β subunit is involved with the binding of some, but not all, redox partners of ETF (18). Finally, recent analysis of the primary sequences of known ETF subunits suggest a dinucleotide binding motif, similar to those found in many nucleotide binding proteins, in the carboxyl-terminal region of all α subunits (9, 19).
Definitive interpretations of structure/mechanism investigations are lacking because of the paucity of structural data regarding ETF. To further our understanding on the mechanism of electron transfer among proteins of the ETF-linked branch of the mitochondrial respiratory chain, and to begin to understand the basis of pathophysiology of GAII, we have determined the three-dimensional structure of the human ETF protein to 2.1-Å resolution by x-ray crystallography.
Purification and Crystallization of Human ETF
Human ETF was purified and crystallized as described (20). Briefly, cDNAs encoding ETF subunits were inserted in pBluescript SK+, expressed in Escherichia coli DH5α, and purified following the procedure of Herrick et al. (18). The purified ETF protein was then repetitively concentrated/diluted with 20 mM Hepes (pH 7.0) to a final protein concentration of 15 mg/ml. Crystals were formed by vapor diffusion using the sitting drop technique (21), by mixing equal volumes of the ETF solution with a precipitating solution (15% PEG 1500 in 50 mM Hepes, pH 7.0), and equilibrating against the precipitating solution at 19°C. As previously reported, the crystals of ETF belong to the monoclinic space group P21, with unit cell parameters a = 47, b = 104, c = 64 Å, and β = 110° (20). Assuming one molecule per asymmetric unit, the Matthews coefficient is 2.79 (22). Data set statistics are given in Table 1.
Table 1.
Data collection and phasing statistics
| Parameter | Native | HGI (HGIano)* | HGII (HGIIano)* | K2OsCl6 [K2OsCl6(ano)]* |
|---|---|---|---|---|
| Diffraction data | ||||
| Resolution, Å | 2.1 | 3.0 | 3.0 | 3.0 |
| Rmerge,† % | 6.40 | 5.50 | 8.90 | 4.90 |
| Total observations | 118,435 | 36,438 | 18,252 | 43,114 |
| Unique reflections | 32,383 | 11,199 | 6,581 | 17,078 |
| Completeness, % | 95.6 | 96.4 | 88.7 | 94.7 |
| MIRAS phasing | ||||
| Riso‡(F) | 9.0 | 12.2 | 7.6 | |
| (Fano)§ | 2.8 | 5.3 | 3.3 | |
| Figure of merit¶ | 0.413 (0.253) | 0.335 (0.128) | 0.270 (0.148) | |
| Phasing power‖ | 2.09 (1.02) | 1.82 (0.54) | 1.25 (0.53) | |
| Rcullis** | 0.596 | 0.543 | 0.679 | |
| (Rkraut)‡‡ | (0.117) | (0.175) | (0.127) | |
| Concentration, mM | 1 | 5 | 2 | |
| Soaking time, hr | 1 | 4 | 8 | |
| Number of sites | 1 | 5 | 1 | |
| Final model statistics | ||||
| Final R factor | 17.1% | Rfree | 22.1% | |
| rmsd bond lengths | 0.006 Å | rmsd bond angles | 1.4° | |
| No. water molecules | 300 | |||
HGI, thimerosal treated data set 1; HGII, thimerosal treated data set 2; rmsd, root-mean-square deviation; MIRAS, multiple isomorphous replacement anomalous scattering.
Statistics for anomalous data sets are shown in parentheses.
Rmerge = Σh Σi |Ihi − 〈Ih〉|/Σh Σi |Ihi|, where h are unique reflection indices and i indicates symmetry equivalent indices.
Riso = Σ ||Fph| − |Fp||/Σ |Fp| for isomorphous replacement (iso) data.
Fano = Σ ||F+| − |F−||/Σ |F+| for acentric anomalous scattering (ano) data.
The overall figure of merit was 0.57.
Rcullis = Σh(|FPH − FP| − |fH|)/ΣH |FPH − FP| for all centric data.
Rkraut = Σ (|| FPH+| − |Fphc+|| + ||FPH−| − |Fphc−||)/Σ(|FPH+| + |FPH−|).
Phasing power = frms/Erms, where frms = [(Σ fH2)/n]1/2 and Erms = |Σ (FPH − |FP + fH|)2/n]1/2.
Structure Determination
The crystal structure of human ETF was solved using multiple isomorphous replacement methods together with anomalous scattering data (multiple isomorphous replacement anomalous scattering). Heavy atom screening was carried out by soaking pre-formed crystals for various times in mother liquor containing 1 mM of the mercurial, thimerosal. Two mercury derivatives, one with a single site and the other with five sites, were obtained. A third derivative was obtained using K2OsCl6. The positions of the heavy atoms were determined by inspection of difference Patterson maps, and confirmed by difference Fourier techniques, using x-ray data with I/σ >3.0, between 30- and 3.0-Å resolution. The positions and occupancies of the heavy atoms were first refined using the program heavy (23), and multiple isomorphous replacement phases were calculated and refined using the phases package (24). The correct hand of the heavy atoms was determined from an anomalous difference Fourier map of the multiple-site mercury derivative, using x-ray data between 30 and 3.0 Å. Phasing statistics are given in Table 1. At this stage, the overall figure of merit was 0.57 for 13,386 reflections.
Initial electron density maps were calculated using data between 30 and 3.0 Å. Mini-maps were constructed to locate the molecular envelope and to define the contents of the asymmetric unit. Density modification using the solvent flattening procedure (25) was used to improve the multiple isomorphous replacement phases. At this time, well-defined α-helices and β-strands were easily identified. A polyalanine model was then constructed using the molecular graphics program turbo-frodo (26) on a Silicon Graphics workstation. The direction of the β-strands were confirmed by connectivity to well-defined α-helices. Approximately 500 residues out of 568 total were built into the initial polyalanine model of ETF. Electron density maps were then generated by combining the multiple isomorphous replacement phases and those of the model according to the SIGMAA algorithm (27). Phase combination allowed for completion of the chain tracing, and for identification of the FAD and AMP cofactors. At this stage, the standard crystallographic R factor for all data between 30.0 and 3.0 Å with I/σ >3, was 40.1%. The side chains were identified by location of the mercury sites and identification of several cysteine residues involved in mercury binding. After several rounds of manual fitting of the map, along with additional sequence assignment followed by energy minimization during each cycle using the POWELL minimization procedure in the x-plor package (28), the entire sequence was assigned. At this stage, the crystallographic R factor was 31.5%. The connectivity was confirmed using simulated annealing omit maps. The final crystallographic R factor for all observed data in the resolution range from 8.0 Å to 2.1 Å after individual B factor refinement was 17.1%, with an R-free of 22.1%, including 300 water molecules.
Fig. 1 shows a ribbon diagram of the human ETF structure. Beginning in the α subunit with αQ21, which is the second residue in the mature protein, the density is continuous throughout the entire α subunit. Lysine-226 of the α subunit falls outside the acceptable Ramachandran plot regions. This residue is located at the beginning of helix G in domain II (Fig. 2), and forms crystal packing contacts between neighboring molecules. The β subunit, beginning with βL4, shows continuous density through residue βI255. The average main-chain B factor is 24.0 Å2, with an average side-chain B factor of 29.2 Å2. The average solvent B factor is 33.4 Å2, and those of FAD and AMP are 15.5 Å2 and 10.2 Å2, respectively. Water molecules were assigned such that they were within at least 3.3 Å of a hydrogen bonding partner.
Figure 1.
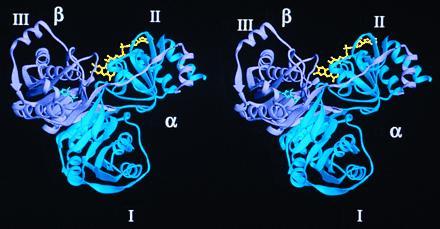
A stereo ribbon diagram of the human ETF structure. FAD (yellow) binds in a cleft formed by the α (blue) and β (lavender) subunits. The AMP (green) is located entirely within the β subunit. All ribbon models were made with the program ribbons (29).
Figure 2.
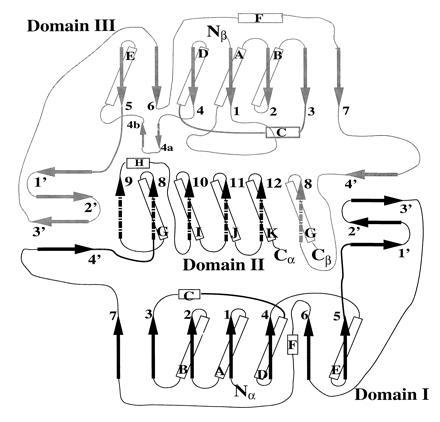
Topology of the ETF protein, indicating a pseudo 2-fold between domains I and III. The α subunit (black), makes up domain I and domain IIα, and the β subunit (gray) consists of domains III and IIβ. α-helices (cylinders), β-strands (arrows), and random coils (lines) are indicated. Broken arrows indicate regions belonging to domain II, whereas solid arrows denote domains I and III.
Description of ETF Polypeptide Fold
The two subunits of ETF form three separate domains: domain I, or the N-terminal portion of the α subunit; domain II, or the C-terminal portion of the α subunit and a small C-terminal portion of the β subunit; and domain III, composed of the majority of the β subunit. As depicted in Fig. 1, the ETF molecule is approximately 65 Å wide, 64 Å high, and 43 Å deep. Domains I and III share identical polypeptide folds, with the exception of a few loops, and are related by a pseudo 2-fold axis, though there is no sequence similarity (Figs. 2 and 3). The rms deviation between the main chain atoms of residues located either in α-helices or β-strands is 2.1 Å, with a 13.6% residue identity between the two domains (Fig. 3). Therefore, in a manner similar to that of sulfite reductase (30), it is likely that a previously unrecognized gene duplication event followed by a truncation of a second FAD-containing domain occurred, leading to the heterodimeric ETF protein of today. This is intriguing because the ETF from Megasphaera elsdenii has a larger β subunit, and binds two molecules of FAD per αβ heterodimer (12). Both domains I and III are comprised of a seven-stranded parallel β-sheet at their core, flanked by solvent exposed α-helices. These domains also contain a three-stranded anti-parallel β-sheet (strands 1′–3′), with a fourth strand (4′) coming from the pseudo-symmetry related domain (Fig. 2). These tight interactions between domains I and III stabilize the quaternary structure of the ETF protein, while forming a “shallow bowl” where domain II rests in the molecule.
Figure 3.
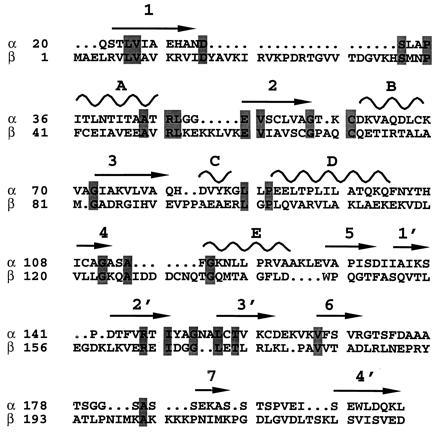
Sequence alignment of domains I and III. The sequence alignment was made based on structural superposition of the two domains. Arrows above the sequence denote β-strands, whereas curves represent α-helices. The secondary structure elements are identified as denoted in Fig. 2. The top lines (α) represent domain I, whereas the bottom lines (β) are from domain III. Residues that are identical between the two domains are shaded. There is a 13.6% residue identity between the two domains, with an rms deviation of 2.1 Å between the Cα atoms of residues located either on helices or strands.
Domains I and II are separated by a hinge, located after strand 7, and including strand 4′ (Fig. 2, lower left-hand corner). Domain II can be further subdivided into domain IIα and IIβ, which are comprised of the C-terminal portions of the α and β subunits, respectively. Domain IIα folds in a manner reminiscent of bacterial flavodoxins (31), though there is no apparent sequence similarity. The fold consists of a five-stranded parallel β-sheet (Fig. 2, strands 8–12) as the core of the domain, flanked by alternating α-helices (helices G–K). The sixth strand of domain II is donated by the β subunit (strand 8) or domain IIβ. Sequence comparisons among members of the ETF family indicate a region of high sequence similarity located in domain IIα (9), the C-terminal portion of the α subunit. This region binds FAD, with most of the FAD binding to domain IIα, whereas the isoalloxazine ring is situated in a crevice between domains II and III, with the xylene portion pointed toward the β subunit (Fig. 4). This is consistent with previous results of crosslinking studies using 8-azido FAD, in which the cofactor analogue was predominantly crosslinked to the β subunit (13). Domain IIβ does not interact with the FAD, but instead wraps around domain IIα, retaining domain IIα in the shallow bowl formed by domains I and III.
Figure 4.
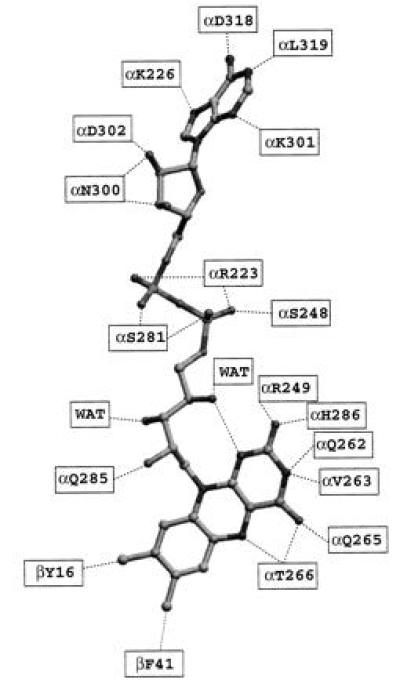
Residues involved in binding the FAD cofactor of ETF. Dotted lines indicate residues (or solvent) that are within hydrogen bonding distance (<3.3 Å) of one another. WAT denotes ordered water molecules. FAD is shown as it is modeled in ETF.
Description of Cofactor Binding Sites
While domain IIα resembles flavodoxin, the location of the flavin ring is not the same between the two proteins. Fig. 4 illustrates interactions between the isoalloxazine ring of the FAD cofactor with ETF. The ribityl chain of FAD is “bent,” with the 4′-OH of the ribityl group forming a hydrogen bond with N1 (3.0 Å) of the isoalloxazine ring. N1 of the αH286 side chain is within 2.9 Å of the C2 carbonyl oxygen atom of the FAD isoalloxazine ring, and may function in stabilization of the anionic semiquinone. Further stabilization may come from the backbone nitrogen atom of αR249, located 3.1 Å away. N3 of the FAD isoalloxazine ring is 3.0 Å from the side chain of αQ262 and 2.8 Å away from the main-chain carbonyl oxygen of αV263. The FAD C4 carbonyl oxygen atom is 3.1 Å away from both the amide nitrogens of αQ265 and αT266. Located 2.8 Å from the N5 of the FAD ring is the hydroxyl group of αT266. This latter interaction may help to modulate the redox potential of the FAD cofactor. Of all the pathogenic mutations known to exist in the ETF protein in patients with GAII, the most frequent mutation is αT266M, which has recently been biochemically characterized and appears to specifically affect the oxidative half reaction between ETF and ETF-QO (D. Salazar and F.E.F., unpublished work). This residue is conserved in all the ETFs sequenced thus far, with the exception of a conservative substitution of Ser for Thr in the W3A1 ETF protein. Located almost perpendicular to, and 3.6 Å away from, the C7- and C8-methyl groups of the isoalloxazine ring are βF41 and βY16, respectively. βY16 is conserved in P. denitrificans ETF, but is replaced by a leucine in the W3A1 protein. It is most likely that this residue was responsible for crosslinking with 8-azido FAD, resulting in the hypothesis that the β subunit contained the FAD binding domain (13). Consistent with the human ETF structure is biochemical data indicating that the C8 portion of the FAD cofactor is exposed to solvent and can accommodate bulky covalent substitutions, including a CoA moiety (13). Lastly, the guanidinium portion of αR249 is 3.8 Å away from the center of the dimethyl benzene portion of the isoalloxazine ring. This interaction may also play a role in stabilization of the anionic semiquinone, and offset the apparently unfavorable interaction of the anionic dehydrogenase interacting with the anionic semiquinone of ETF.
The ADP and ribityl moieties of the FAD are exposed and lie in a channel on the surface of domain II. The strand–loop–helix containing αS281 (strand 10/helix I) helps in binding the pyrophosphate portion of the FAD, along with αS248 at the end of strand 9 and helix H (Fig. 2). Further stabilization of the negative charge of the phosphate groups is achieved by interactions with the guanidinium side chain of αR223, located approximately 3.0 Å away. The adenosine moiety of FAD interacts with the main-chain atoms on loops between strand 11 and helix J, strand 8 and helix G, and strand 12 and helix K.
The crystal structure revealed that the human ETF protein also contains an AMP molecule. The AMP is buried deeply within domain III and may play a structural role. The loop located within domain III between strand 4 and helix E, including strands 4a and 4b, binds the phosphate portion of the AMP. This corresponds to a region that is moderately conserved in all the ETF proteins studied thus far (9). The adenine base interacts with strands 1 and 2, and to a lesser extent with helix D. Structural analysis does not indicate a particular role for the AMP, other than to aid in maintaining the tertiary structure of the β subunit. The fact that the human ETF protein binds AMP may be a structural remnant of a NADH binding site, which is the physiological electron donor for the M. elsdenii ETF protein (12).
Sequence Homology Within the ETF Family of Flavoproteins
Chen and Swenson (9) compared the sequences of three ETFs and the putative gene products of two fixb open reading frames, and identified a consensus sequence in the C-terminal portion of the α subunit that was proposed to bind either the nucleotide moiety of FAD or possibly AMP. We have extended this lineup, including the recently acquired yeast ETF along with two other bacterial ETFs and three additional bacterial FixB proteins (Fig. 5). Based on this alignment, and the structure of the FAD binding domain (Fig. 4), it is striking that all the residues located directly around the isoalloxazine ring are highly preserved throughout all the sequences, with the only substitution being a serine for threonine at position α266 in both the W3A1 ETF protein and the Azotobacter FixB protein. These residues are highly conserved in spite of the wide range of redox potentials, ranging from −0.3 V to +0.2 V. This suggests that the redox potential of the flavin may be influenced more by polarization of the isoalloxazine ring by the apoprotein as a whole rather than by interactions with immediate neighboring residues. These conserved neighboring residues may function in formation and stabilization of the red anionic semiquinone, the common stable reduced species of this family of proteins.
Figure 5.

Sequence alignment of residues involved in binding FAD among members of the ETF α subunit family of proteins. Proteins used in the sequence alignment are: hetf, human ETF (32); petf, P. denitrificans ETF (33); yetf, Saccharomyces cerevisiae hypothetical ETF α subunit; wetf, Methylophilus methylotrophus W3A1 ETF (9); betf, Bradyrhizobium japonicum putative ETF (GenBank accession no. U32230U32230); cetf, Clostridium acetobutylicum putative ETF (34); afix, Azorhizobium caulinodans putative FixB (35); zfix, Azotobacter vinelandii putative FixB, (Protein Identification Resource accession no. S49188S49188); bfix, B. japonicum putative FixB (36); cfix, C. acetobutylicum putative FixB (GenBank accession no. M91817M91817); and rfix, Rhizobium meliloti putative FixB (37). Residues that are identical in at least six of the sequences are shaded. Arrows indicate residues involved in binding the isoalloxazine portion of the FAD, as shown in Fig. 4.
Mutations in ETF Responsible for GAII
Recessively inherited defects of either ETF or ETF-QO cause GAII, which in its most severe form causes death in the first days or weeks of life. Several mutations in the mammalian ETF known to cause GAII have been identified. Mutations are known to occur in both the α and β subunits, but all known mutations in the β subunit result in an unstable polypeptide, of which only one has been identified and characterized (38, 39). The one known β mutation, βR164Q, resides at a subunit interface, and possibly forms an important hydrogen bond necessary for stabilizing the quaternary structure of the intact ETF protein. Numerous mutations have been explicitly identified in the α subunit (40, 41); however, only three lead to the formation of a stable ETF heterodimer with reduced activity. The most prevalent mutation is αT266M, which occurs in approximately 50% of the patients identified thus far (8, 41). As described earlier, this residue is hydrogen-bonded to N5 of the FAD isoalloxazine ring, and possibly functions to help stabilize the FAD cofactor. Two other known mutations are αV157G (40) and αG116R (41). Valine-157 is located on strand 3′ of domain I, in the middle of a β-strand. Because this residue is located on an interface between the α and β subunits, substitution of the valine with glycine may decrease the hydrophobic interactions, thus effecting the association between the two subunits. Glycine-116 is located at the beginning of helix G in domain IIα, in a hydrophobic pocket. Sequence comparisons indicate that in all the sequences known thus far, most contain a glycine at this position. Three exceptions are the ETF from B. japonicum (phe), M. methylotrophus W3A1 (ser), and P. denitrificans (ala). Substitution with the charged arginine residue would be more energetically costly and thus destabilize the tertiary structure of the ETF protein (42). αG116 is also located near the subunit interface, within 10 Å of the previously mentioned βR164, suggesting that maintaining the structural integrity in this region is important for subunit association, and thus stabilization of the quaternary structure of the ETF protein.
Location of Docking Surfaces Between ETF and Medium Chain Acyl-CoA Dehydrogenase
In an attempt to identify possible sites of interaction between ETF and one of its physiological electron donors, medium chain acyl-CoA dehydrogenase (MCAD), we have modeled the docking of ETF to MCAD. The model was made by manual inspection of the two structures, followed by rigid body energy minimization using the x-plor package. For an efficient electron transfer, the most likely complex would be an assembly that minimizes the distance between the two isoalloxazine rings of the flavin cofactors. One possible mode of binding is depicted by the ribbon diagram in Fig. 6. In this model, the surface area of interaction between the two proteins is maximized (≈3400 Å2), while the flavin-flavin distance is minimized (≈19.5 Å). There are at least 13 salt bridges and several hydrogen bonds formed between one ETF molecule and the MCAD dimer. For modeling, the ETF and MCAD proteins were restricted to rigid bodies, and were not allowed to “breath.” It is conceivable that conformational changes occur upon binding, which could decrease the distance between the two FADs by as much as 3–4 Å, and would possibly increase the number of interactions between the two molecules. From this model, electrons would pass from MCAD at the si side of the FAD ring, as postulated from the structure of MCAD with bound product, since the acyl-CoA ligand is “blocking” the re side (44). Kinetic studies have also demonstrated that enoyl-CoA product is present when MCAD binds ETF (43). Another mode of binding between the two proteins that cannot be ruled out would have the ETF molecule slightly tilted followed by a rotation of 180° along an axis that is perpendicular to the ETF/MCAD interaction surface. This model, however, has a flavin–flavin distance of 25 Å, and has only seven salt bridges between the two molecules. Any other attempt to dock the two proteins have flavin–flavin distances greater than 30 Å.
Figure 6.
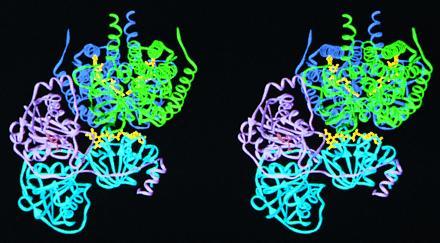
A stereo ribbon diagram depicting the hypothetical docking of ETF to an MCAD dimer. The refined structures of human ETF and porcine MCAD (43) were used. The MCAD dimer (green and dark blue, mainly the green monomer) fits into the groove formed by the α (cyan) and β (lavender) subunits of ETF, in a manner close to the FAD of MCAD. By this mechanism, the closest flavin–flavin distance is approximately 19.5 Å.
The structure of ETF provides a model for this unique family of flavoproteins. Conservation of the residues around the FAD moiety indicate that, whereas these residues may not be directly involved in modulation of the redox potential, they may determine the chemistry of the species formed upon reduction/oxidation of the enzyme-bound FAD. The positively charged arginine moiety, which has been described in stacking with aromatic residues, would be more preferable than an aromatic ring in stabilization of an anionic semiquinone. Furthermore, the histidine residue located near the FAD C2 carbonyl oxygen atom may be important in polarization of the flavin ring and further stabilization of the anionic semiquinone. Determination of the structure of the semiquinone form of ETF should reveal the nature of which residues located near the FAD are responsible for this stabilization. Finally, the model of ETF binding to MCAD suggests possible electron paths for intermolecular electron transfer between the two flavoproteins. Further experiments are in progress to test the validity of this model.
Acknowledgments
We thank Ming Wang for his assistance during data collection. This work was supported by National Institutes of Health Grant GM29076 (J.-J.P.K.) and National Research Service Award Fellowships DK09157 (D.L.R.) and DK49726 (F.E.F.).
Footnotes
Abbreviations: ETF, electron transfer flavoprotein; ETF-QO, ETF-ubiquinone oxidoreductase; GAII, glutaric acidemia type II; FAD, flavin adenine dinucleotide; MCAD, medium chain acyl-CoA dehydrogenase.
References
- 1.Crane F L, Beinert H. J Biol Chem. 1956;218:717–731. [PubMed] [Google Scholar]
- 2.Beinert H. Enzymes. 1963;7:467–476. [Google Scholar]
- 3.Ikeda Y, Tanaka K. J Biol Chem. 1983;258:1077–1085. [PubMed] [Google Scholar]
- 4.Leinich A C, Goodman S I. J Biol Chem. 1986;261:4090–4096. [PubMed] [Google Scholar]
- 5.Ikeda Y, Tanaka K. J Biol Chem. 1983;258:9477–9487. [PubMed] [Google Scholar]
- 6.Frisell W R, MacKenzie C G. J Biol Chem. 1962;237:94–98. [PubMed] [Google Scholar]
- 7.Ruzicka F J, Beinert H. J Biol Chem. 1977;252:8440–8445. [PubMed] [Google Scholar]
- 8.Loehr J P, Goodman S I, Frerman F E. Pediatr Res. 1990;27:311–315. doi: 10.1203/00006450-199003000-00024. [DOI] [PubMed] [Google Scholar]
- 9.Chen D, Swenson R P. J Biol Chem. 1994;269:32120–32130. [PubMed] [Google Scholar]
- 10.Sato K, Nishina Y, Shiga K. J Biochem (Tokyo) 1993;114:215–222. doi: 10.1093/oxfordjournals.jbchem.a124157. [DOI] [PubMed] [Google Scholar]
- 11.DuPlessis E R, Rohlfs R J, Hille R, Thorpe C. Biochem Mol Biol Int. 1994;32:195–199. [PubMed] [Google Scholar]
- 12.Thorpe C. In: Chemistry and Biochemistry of Flavoenzymes. Muller F, editor. Vol. 2. Boca Raton, FL: CRC; 1991. pp. 471–486. [Google Scholar]
- 13.Gorelick R J, Thorpe C. Biochemistry. 1986;25:7092–7098. doi: 10.1021/bi00370a050. [DOI] [PubMed] [Google Scholar]
- 14.Husain M, Stankovich M T, Fox B G. Biochem J. 1984;219:1043–1047. doi: 10.1042/bj2191043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beckmann J D, Frerman F E. J Biol Chem. 1983;258:7563–7569. [PubMed] [Google Scholar]
- 16.Steenkamp D J. Biochem J. 1987;243:519–524. doi: 10.1042/bj2430519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steenkamp D J. Biochem J. 1988;255:869–876. doi: 10.1042/bj2550869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herrick K R, Salazar D, Goodman S I, Finocchiaro G, Bedzyk L A, Frerman F E. J Biol Chem. 1994;269:32239–32245. [PubMed] [Google Scholar]
- 19.Tsai M H, Saier M H., Jr Res Microbiol. 1995;146:397–404. doi: 10.1016/0923-2508(96)80285-3. [DOI] [PubMed] [Google Scholar]
- 20.Roberts D L, Herrick K R, Frerman F E, Kim J-J P. Protein Sci. 1995;4:1654–1657. doi: 10.1002/pro.5560040825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McPherson A. Eur J Biochem. 1990;189:1–23. doi: 10.1111/j.1432-1033.1990.tb15454.x. [DOI] [PubMed] [Google Scholar]
- 22.Matthews B W. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 23.Terwilliger T C, Eisenberg D. Acta Crystallogr A. 1983;39:813–817. [Google Scholar]
- 24.Furey W. PHASES Manual. Pittsburgh: Veterans Administration Medical Center; 1995. [Google Scholar]
- 25.Wang B C. Methods Enzymol. 1985;115:90–112. doi: 10.1016/0076-6879(85)15009-3. [DOI] [PubMed] [Google Scholar]
- 26.Roussel, A. & Cambillau, C. (1989) turbo-frodo Manual (Silicon Graphics, Mountain View, CA), Version 4.2.
- 27.Read R J. Acta Crystallogr A. 1986;42:140–149. [Google Scholar]
- 28.Brunger, A. T. (1992) x-plor: User’s Guide (Yale Univ., New Haven, CT), Version 3.1.
- 29.Carson, M. (1996) ribbons (Univ. of Alabama, Birmingham, AL), Version 2.0.
- 30.Crane B R, Siegel L M, Getzoff E D. Science. 1995;270:59–67. doi: 10.1126/science.270.5233.59. [DOI] [PubMed] [Google Scholar]
- 31.Smith W W, Burnett R M, Darling D G, Ludwig M L. J Mol Biol. 1977;117:195–225. doi: 10.1016/0022-2836(77)90031-6. [DOI] [PubMed] [Google Scholar]
- 32.Finocchiaro G, Ito M, Ikeda Y, Tanaka K. J Biol Chem. 1988;263:15773–15780. [PubMed] [Google Scholar]
- 33.Bedzyk L A, Escudero K W, Gill R E, Griffin K J, Frerman F E. J Biol Chem. 1993;268:20211–20217. [PubMed] [Google Scholar]
- 34.Boynton Z L, Bennett G N, Rudolph R B. J Bacteriol. 1996;178:3015–3024. doi: 10.1128/jb.178.11.3015-3024.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arigoni F, Kaminski P A, Hennecke H, Elmerich C. Mol Gen Genet. 1991;25:514–520. doi: 10.1007/BF00261695. [DOI] [PubMed] [Google Scholar]
- 36.Gubler M, Zurcher T, Hennecke H. Mol Microbiol. 1989;3:141–148. doi: 10.1111/j.1365-2958.1989.tb01803.x. [DOI] [PubMed] [Google Scholar]
- 37.Earl C D, Ronson C W, Ausubel F M. J Bacteriol. 1987;169:1127–1136. doi: 10.1128/jb.169.3.1127-1136.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Colombo I, Finocchiaro G, Garavaglia B, Garbuglio N, Yamaguchi S X, Frerman F E, Berra B, DiDonato S. Hum Mol Genet. 1994;3:429–435. doi: 10.1093/hmg/3.3.429. [DOI] [PubMed] [Google Scholar]
- 39.Yamaguchi S, Orii T, Maeda K, Oshima M, Hashimoto T. J Inherited Metab Dis. 1990;13:783–786. doi: 10.1007/BF01799588. [DOI] [PubMed] [Google Scholar]
- 40.Indo Y, Glassberg R, Yokota I, Tanaka K. Am J Hum Genet. 1991;49:575–580. [PMC free article] [PubMed] [Google Scholar]
- 41.Freneaux E, Sheffield V C, Molin L, Shires A, Rhead W J. J Clin Invest. 1992;90:1679–1686. doi: 10.1172/JCI116040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Byrne M P, Manuel R L, Lowe L G, Stites W E. Biochemistry. 1995;34:13949–13960. doi: 10.1021/bi00042a029. [DOI] [PubMed] [Google Scholar]
- 43.Mizzer J P, Thorpe C. Biochemistry. 1981;20:4965–4970. doi: 10.1021/bi00520a024. [DOI] [PubMed] [Google Scholar]
- 44.Kim J-J P, Wang M, Paschke R. Proc Natl Acad Sci USA. 1993;90:7523–7527. doi: 10.1073/pnas.90.16.7523. [DOI] [PMC free article] [PubMed] [Google Scholar]