

Abstract
Entry of the bacterial pathogen Listeria monocytogenes into host epithelial cells is critical for infection and virulence. One major pathway for Listeria entry involves binding of the bacterial protein Internalin B to the host receptor tyrosine kinase Met (hepatocyte growth factor receptor). Activation of Met and downstream signaling cascades is critical for Listeria entry. Internalin B is composed of several structural domains including an N-terminal leucine-rich repeat that is sufficient for binding Met and stimulating downstream signal transduction. Internalin B is monomeric, whereas the leucine-rich repeat is dimeric when expressed as an isolated fragment. The different quaternary states of Internalin B and the leucine-rich repeat suggest that these two Met ligands might cause distinct biological effects. Here we demonstrate that Internalin B and the leucine-rich repeat fragment exhibit agonist properties that differentially influence Met down-regulation in lysosomes. Specifically, Met stability is increased in response to the leucine-rich repeat fragment compared with Internalin B. Interestingly, Internalin B and the leucine-rich repeat stimulate equivalent rates of clathrin-mediated Met internalization. However, the leucine-rich repeat is defective in promoting lysosomal down-regulation of Met and instead enhances receptor recycling to the cell surface. In addition, the leucine-rich repeat causes prolonged Met activation (phosphorylation) and increased cell motility compared with Internalin B. Taken together, our findings indicate that individual domains of Internalin B differentially regulate Met trafficking. The ability of the leucine-rich repeat fragment to promote Met recycling could account for the increased cell motility induced by this ligand.
The Listeria monocytogenes surface protein Internalin B (InlB)2 is a 630-amino acid protein critical for bacterial invasion into a broad range of host cells including endothelial cells, hepatocytes, and epithelial cell lines such as Vero and HeLa cells (1–4). In addition to the bacterial-bound form, soluble InlB is detected in bacterial supernatants (5, 6) and is active in promoting Vero cell infection by a Listeria mutant lacking InlB (7). The host receptor for InlB is Met, a receptor tyrosine kinase for the endogenous ligand hepatocyte growth factor (HGF) (8). Tight regulation of Met signaling elicits multiple cellular responses critical for mammalian development including proper cellular growth, survival, and migration (for review, see Ref. 9). In adult tissues, Met signaling is intrinsic for organ homeostasis and tissue remodeling (10–12).
InlB shares many of the agonist activities of HGF including increased cell proliferation, epithelial cell motility, and membrane ruffling (5, 8, 13). Recent structural studies show that HGF and InlB directly bind to discrete sites on the extracellular, sema domain of Met (14–16), consistent with early biochemical studies showing that InlB and HGF do not compete for receptor occupancy (8, 14). Despite these differences, HGF and InlB activate similar signaling cascades downstream of Met autophosphorylation including pathways involving Grb2, Gab1, phosphatidylinositol 3-kinase and MAP kinase (MAPK) (2, 5, 7, 17, 18). We previously reported that like HGF, InlB induces Met endocytosis in a process requiring clathrin-heavy chain, the clathrin adaptor epidermal growth factor phosphorylation substrate 15 (Eps15), Grb2, and the E3 ubiquitin ligase Cbl (18, 19). After internalization, InlB- and HGF-activated Met are targeted for lysosomal degradation. Met degradation is dependent on phosphatidylinositol 3-kinase activity and hepatocyte receptor substrate (Hrs) (18), a protein that interacts with ubiquitinated cargo and is important for endosomal sorting (20–23).
InlB is a modular protein consisting of a N-terminal cap followed by a 213-amino acid leucine-rich repeat (LRR), an interrepeat domain, and a C-terminal region containing “GW” modules that anchor InlB non-covalently to the bacterial cell wall (24, 25). InlB binds Met in a 1:1 stoichiometry primarily through the concave surface of the LRR region, although a second contact involving the inter-repeat region of InlB strengthens this interaction (14). Biochemical studies confirm that a fragment containing only the N-cap and LRR domains (i.e. the LRR fragment) comprises the minimal region for binding and inducing Met phosphorylation (7, 8).
Surprisingly, the LRR fragment used in studies on Met activation (7, 17) has a different quaternary structure than InlB. Whereas full-length InlB is monomeric, the isolated LRR domain is a disulfide-linked dimer. LRR dimerization results from a cysteine residue in InlB that is normally unavailable for disulfide formation. Truncation of InlB to generate the LRR fragment results in the surface exposure of this cysteine residue located near the C terminus of the LRR fragment. The different quaternary structures of monomeric InlB and the dimeric LRR fragment raise the possibility that these two Met ligands could exhibit some differences in biological activity.
Although it is unclear whether the LRR fragment is produced physiologically during Listeria infection, we are interested in using InlB and its derivatives as tools to examine aspects of Met-mediated signal transduction and trafficking. Our structure/function analysis of InlB identified the LRR fragment with agonist properties reminiscent of InlB, with the exception that LRR promotes Met stability. Down-regulation plays a key role in regulating the temporal activity of signaling receptors, and mutations or other conditions that interfere with down-regulation are thought to contribute to the development of several cancers (26, 27). “Down-regulation” refers to ligand-induced depletion of a surface receptor, typically resulting from receptor internalization and degradation in a lysosomal compartment (28). We found that compared with InlB, the LRR fragment is defective in promoting Met down-regulation. LRR and InlB induce Met internalization at similar rates, indicating that the differences in receptor degradation caused by these two ligands are due to alterations in intracellular trafficking. Compared with InlB, the LRR fragment is defective in targeting Met to the degradative pathway and, instead, promotes receptor recycling to the plasma membrane. InlB and LRR are comparable in their abilities to induce initial Met activation (i.e. Met tyrosine phosphorylation); however, receptor phosphorylation in response to the LRR fragment is more sustained than that induced by InlB. The ability of LRR to prolong Met phosphorylation raised the possibility that the truncated ligand might be more potent than InlB for inducing one or more Met-mediated biological activities. Consistent with this idea, we show that the LRR fragment was more effective than InlB in promoting epithelial cell motility. Collectively, our results indicate that individual domains of InlB differentially regulate Met trafficking and impact Met-mediated biological activity.
EXPERIMENTAL PROCEDURES
Reagents, Antibodies, and Plasmids—All general reagents were obtained from Fisher or Sigma-Aldrich unless indicated otherwise. The following antibodies were purchased as indicated: transferrin receptor (Invitrogen), anti-actin (Sigma-Aldrich), anti-human HGF receptor (R & D Systems Inc., Minneapolis, MN), clathrin heavy chain (CHC), caveolin, EEA1 (BD Biosciences), Met C-12, and Met C-28 (Santa Cruz Biotechnologies Inc, Santa Cruz, CA), phospho-Met Tyr-1234, Tyr-1235 (Upstate Biotechnology, Billerica, MA), phospho-Met Tyr-1003 (ABR-Affinity BioReagents, Golden, CO), peroxidase-conjugated goat anti-mouse immunoglobulin G (IgG), and goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). Plasmids encoding N-terminal His-tagged LRR or LRR-C242A construct in pET28a were kindly provided by Dr. Partho Ghosh (University of California, San Diego, CA) and have been described elsewhere (17, 18, 25). Recombinant human HGF was purchased from PeproTech Inc, Rocky Hill, NJ, and Alexa594-labeled-transferrin (Tfn) and Alexa594-labeled dextran was obtained from Invitrogen.
Cell Line and Cell Culture—Human cervical epithelial adenocarcinoma (HeLa) and Madin-Darby canine kidney (MDCK) cells were grown in complete media (Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 1× penicillin/streptomycin) at 37 °C with 5% CO2. The H10 cell line derived from kidney epithelial cells from embryos of Met null –/– mice (29) was kindly provided by Dr. Lloyd Cantley (Yale University School of Medicine). H10 derivatives stably expressing wild type (WT) or mutant Met alleles (KinD) (19) were grown in the Dulbecco's modified Eagle's medium/F-12 (1:1) media supplemented with 10% fetal bovine serum, 5 μg/ml puromycin at 37 °C with 5% CO2. Cells were routinely starved in media lacking sera (incomplete media) overnight at 37 °C before use unless specified otherwise. For studies using cycloheximide, cells were pretreated with 10 μg/ml cycloheximide at 37 °C for 30–60 min to prevent de novo synthesis of Met during receptor stimulation as indicated in the appropriate figure legends.
Ligand Purification and Labeling, Confocal Microscopy—The purification and labeling of InlB and its derivatives has been described in detail elsewhere (18). Gel filtration chromatography was used to separate dimeric LRR from LRR monomers. Briefly, 2–2.5 mg of LRR in 120 μl was injected (10 μl/run) onto a Superdex 75 (GE Healthcare) gel filtration column at a flow rate of 1 ml/min in 500 mm NaCl, 50 mm HEPES, pH 7.4, and the separation of LRR dimers from monomers was monitored by size and SDS/PAGE. For confocal microscopy, serum-starved cells grown on coverslips were co incubated in incomplete media containing 5.0 μg/ml Alexa594-labeled Tfn or 5.0 μg/ml Alexa594-labeled dextran with 2 nm Alexa488-labeled InlB or 2 or 10 nm Alexa488-labeled LRR or LRR-C242A. The cells were rapidly cooled and analyzed for ligand uptake as previously published (18, 19). All images were taken using identical acquisition parameters, and the amount of internalized LRR or LRR-C242A is expressed as a percentage of internalized InlB. Colocalization analyses were preformed as previously described (30) on cells stained for endogenous EEA1 or CD63. Routinely, a minimum of 10 cell areas was analyzed using MetaMorph software v7.1.3.0 (Molecular Devices, Union City, CA), and the data were normalized to the percentage of co-localization with EEA1 at the 0-min chase time.
Flow Cytometry-based Recycling Assay—Serum-starved HeLa cells grown on 10-cm plates were incubated with 10 μg/ml cycloheximide for 1 h at 37 °C to inhibit protein synthesis. The cells were then stimulated with 2 nm InlB or 10 nm LRR for 15 min at 37 °C to induce Met internalization. Residual surface Met was cleaved by incubation with 0.25% trypsin (Invitrogen) for 30 min on ice. Cells were collected by centrifugation and washed in PBS containing 2% fetal bovine serum (FACS buffer) before resuspension in serum-free media containing 10 μg/ml cycloheximide. Cells were incubated at 37 °C for 0, 5, 10, or 15 min to allow Met trafficking. After the appropriate chase time cells were shifted to ice to prevent further endocytic trafficking of Met and were resuspended to a concentration of 1 × 107 cells/ml in FACS buffer. To determine the level of Met present on the cell surface, 2 × 105 cells were aliquoted into triplicate wells of a 96-well plate. Cells were incubated with 10 μg/ml goat anti-HGF receptor antibody for 30 min on ice followed by 20 min of incubation with donkey anti-goat phycoerythrin-conjugated secondary antibody. After staining, cells were fixed with 2% formaldehyde (Ted Pella Inc., Redding, CA) in FACS buffer for 20 min at room temperature. Residual formaldehyde was removed by washing in FACS buffer before measurement of the mean yellow fluorescent signal for 20,000 events by a BD FACSArray. Experiments were performed in triplicate, and the values were expressed as the level of Met present on the cell surface after each chase time. Data were normalized to the level of Met present on the cell surface after 0 min of chase, so that the surface level of Met at time 0 was 1.
Small Interfering RNA (siRNA) Transfection Studies—siRNA depletion experiments were performed using commercially available control, Grb2, and Met (Dharmacon, Chicago, IL) or CHC (Santa Cruz Biotechnologies Inc., Santa Cruz, CA) siRNAs. Cells were cultured for 1 day before transfection and transfected using empirically determined concentrations of siRNA using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's specifications. Typically cells were treated with 40–60 pmol of siRNA with 1–1.5 μl of Lipofectamine 2000, respectively, to routinely accomplish a 95% decrease in protein levels as determined by Western analysis. All experiments were routinely performed 72 h after siRNA transfection.
Cell Surface Enzyme-linked Immunosorbent Assay—Duplicate sets of HeLa cells were plated in 4-well tissue culture plates (Nunc A/S, Rochester, NY) at a concentration of 2 × 105/well. The following day sera-starved cells were stimulated with 2 nm InlB, 2 nm LRR, 10 nm LRR, or 2 nm LRR-C242A in incomplete media for 2–8 min at 37 °C. The cells were rapidly cooled on ice to inhibit endocytosis, washed three times with ice-cold PBS, and then fixed with 4% formaldehyde (Ted Pella Inc., Redding, CA) in PBS for 10 min at room temperature. The cells were incubated with primary anti-human HGF receptor antibody in PBS containing 10% horse serum for 1 h at room temperature, and unbound antibody was removed using three ice-cold PBS washes, then incubated with the appropriate horseradish peroxidase-conjugated secondary antibody. For measuring total receptor, one duplicate plate of cells was first fixed in paraformaldehyde and then permeabilized with 0.2% Triton X-100 for 5 min before the addition of the primary antibody. One well of cells lacking primary antibody was used as an additional negative control. To detect antibody-bound protein complexes, cells were incubated with 1-Step-ABTS reagent (Thermo Scientific, Rockford, IL) for 30 min at room temperature, and the colorimetric reaction was terminated by the addition of 1% SDS. The absorbance at 405 nm was measured using a VERSAmax microplate reader and SOFTmax® PRO 3.0 software (Molecular Devices, Sunnyvale, CA). Experiments were performed in triplicate, and values were calculated as the ratio of surface Met (X) to total receptor (Y) at each time point (tx), normalized to the control value at time zero (t0). For the kinetic studies, values representing the relative amount of internalized Met were calculated using the formula ((X/Y)t0 – (X/Y)tx)/tx.
Western Blot Analysis—Western analysis was performed using ECL plus (GE Healthcare), and the resulting digitized blots were quantified using MetaMorph software as previously described (19).
Wound Healing and Three-dimensional Tubulogenesis Assays—Cell migration was analyzed using an in vitro wound-healing model (31). Briefly, confluent monolayers of MDCK or HeLa cells were cultured in 6-well plates and serum-starved for 48 or 24 h, respectively, in media supplemented with 0.2% fetal bovine serum before each experiment. MDCK cells were wounded mechanically by scraping with a 200-μl pipette tip (3 separate wounds per well). Immediately after wounding, the cells were incubated in media containing ligand. At the indicated time points after wounding, wound areas were imaged with a NIKON TE2000 multifunction inverted microscope. Multiple positioning marks were made to ensure that identical areas were imaged between time points. The relative gap distance from three independent experiments was measured in pixels using MetaMorph software. MDCK tubulogenesis assays were performed in three-dimensional matrix gels as previously described (32). Briefly, MDCK cells were trypsinized and suspended at 3 × 104 cells/ml in a neutralized rat tail collagen type I (BD Biosciences) solution composed of 3.5 mg/ml glutamine, 2.35 mg/ml NaHCO3, 1× minimum Eagle's medium, 20 mm Hepes (pH 7.6), and 2 mg/ml collagen I. 150 μl of the cell-collagen I mixture was added to each well of 8-well culture slides. After the collagen gel solidified, the cells were incubated in complete media, which was changed every other day. Cysts routinely formed at 7–8 days under these conditions. For inducing tubulogenesis, cysts were incubated in complete media containing 2 nm InlB, 2 nm HGF, or 10 nm LRR and renewed every 24 h. One group of cells incubated in growth media lacking ligand served as a control. Phase contrast imaging was preformed using a NIKON TE2000 multifunction inverted microscope equipped with a 20× objective.
RESULTS
The LRR Fragment Is Unable to Target Met to the Degradative Pathway—We previously reported using InlB as a tool to understand the molecular mechanisms of Met endocytosis and intracellular trafficking (18, 19). In particular, we were interested in identifying the minimal region of InlB required for Met internalization and degradation. We focused on the LRR fragment of InlB (herein referred to as LRR) as it has been shown to bind Met, induce receptor tyrosine phosphorylation, and activate downstream signaling cascades (7, 8, 33). InlB and LRR were expressed and purified as recombinant His-tagged proteins and used to examine Met endocytosis and degradation. Non-reducing SDS-PAGE shows that, consistent with earlier reports (7, 8), InlB purifies as a monomeric species, whereas LRR is composed of monomers and disulfide-linked dimers when purified under non-reducing conditions (supplemental Fig S1A). LRR dimers convert to inactive monomers in the presence of reducing agents. Conversely, an LRR mutant incapable of forming disulfide-linked homodimers due to a mutation in the sole cysteine residue in LRR (C242A) purifies as a monomeric species under non-reducing conditions (supplemental Fig S1A). To confirm that LRR dimers but not LRR monomers are functionally active, the LRR preparation was subjected to sizing chromatography. Fractions enriched in LRR monomers or dimers were isolated, and their ability to induce Met phosphorylation at tyrosine 1234 and 1235 in the Met kinase domain was examined. For these studies we used HeLa cells, an established cell model for examining the mechanisms for receptor endocytosis and degradation. HeLa cells express high levels of Met on their surface and have a well documented response to Met signaling (34–36). Western analysis using site-specific phosphotyrosine antibodies confirmed tyrosine phosphorylation at positions 1234 and 1235 in response to dimeric LRR (supplemental Fig. S1, B and C). Met activation was not detected in response to monomeric LRR or the mutant LRR-C242A, consistent with a previous study (7).
We next examined the fate of internalized Met in response to InlB or LRR treatment. Two different LRR concentrations (2 or 10 nm) were used, as discrepancies in the level of Met activation by LRR have been reported (7, 8, 14) possibly due to differences in ligand preparations. HeLa cells pretreated with 10 μg/ml cycloheximide (30 min) were incubated in media lacking ligand or with LRR or InlB for 10, 30, 60, or 120 min in the continued presence of cycloheximide (Fig. 1, A and B). Cycloheximide was included to prevent new protein synthesis that would otherwise contribute to an underestimation of Met degradation. The level of Met and activated receptor (as assessed by tyrosine phosphorylation of residues 1234 and 1235) decreased in response to InlB over the time course of the experiment (Fig. 1B). Quantification of the Western bands revealed that levels of Met and phospho-Met 60–120 min after treatment with InlB were negligible, consistent with ligand-induced receptor internalization and degradation. The apparent increase in Met levels at 5 min was not observed in repeats of the experiment and is not statistically significant. Interestingly, treatment with 10 nm LRR, and to a lesser extent, 2 nm LRR caused a delay in Met degradation. Whereas total Met was reduced to 32.6% that of its original levels in cells treated with InlB at 60 min, the steady state levels of Met in response to 2 nm LRR and 10 nm LRR only decreased to 64.8 and 86.8% that of starting levels, respectively (Fig. 1C). These results indicate that LRR is less effective than InlB in promoting Met degradation. In addition, Met phosphorylation at tyrosine 1234 and 1235 was sustained up to 120 min in cells treated with LRR, whereas phospho-Met was undetectable after 30 min in response to full-length InlB (Fig. 1). Hence, the decrease in Met degradation observed in response to LRR correlates with prolonged Met activation.
FIGURE 1.

Met degradation is delayed in response to LRR. A, structures of full-length InlB and its N-terminal LRR fragment and the mutant C242A. Numbers represent amino acids. B, HeLa cells were incubated in medium containing 10 μg/ml of cycloheximide (37 °C/30 min) before stimulation with cycloheximide-containing media supplemented with 2 nm InlB, 2 nm LRR, 10 nm LRR, or medium lacking ligand (0 min) for the indicated times. The resulting cell lysates were examined by Western analysis for total Met (Met), phosphorylated Met (p-Met, Y1234, Y1235), and actin. C, mean Met protein levels ± S.E. were quantified using MetaMorph software, and Met band density was normalized to actin density in the corresponding lane. Representative data are shown for 2–3 separate experiments.
The LRR Fragment and InlB Induce Comparable Met Activation—We previously reported that Met kinase activity was required for receptor endocytosis and, hence, degradation in response to InlB (19). The differences we observed in LRR-versus InlB-induced Met degradation could be the result of compromised Met activation. Therefore, we examined Met phosphorylation (i.e. activation) over a range of ligand concentrations. Western analysis of lysates prepared from HeLa cells treated with 2 nm InlB using site-specific phosphotyrosine antibodies detected Met phosphorylation at tyrosine 1003, 1234, and 1235 (Fig. 2, A and B). Conversely, a comparable level of Met phosphorylation in response to the LRR fragment was only observed in cells treated with higher concentrations (10 nm) of this ligand (Fig. 2, A and B). As expected, no Met activation was detected in control cells stimulated with LRR-C242A. Contrary to our results, a previous study reported a 500-fold difference in the potency of the LRR fragment for activating Met (7). The explanation for this discrepancy remains unclear but may reflect differences in ligand preparations and/or cells lines used in the respective studies. Regardless, our results and those reported by Banerjee et al. (7) indicate that, when used at comparable concentrations, the LRR fragment is a less potent Met agonist than full-length InlB.
FIGURE 2.

Met activation in response to the LRR fragment of InlB is dose-dependent. A, HeLa cells were treated with medium containing InlB, LRR, the mutant LRR-C242A, or lacking ligand (Con) as indicated for 5 min at 37 °C. The resulting cell lysates were examined by Western analysis using antibodies for total Met (Met), key phosphotyrosines in the cytosolic region of Met (Y1234, Y1235, or Y1003), or actin. B, mean Met protein levels ± S.E. were quantified using MetaMorph software, and Met band density was normalized to actin density in the corresponding lane. Representative data are shown for 2–3 separate experiments.
LRR Induces Normal Met Internalization—InlB induces Met
endocytosis through clathrin-coated pits in a Grb2-dependent manner
(18,
19). The delay in Met
degradation observed in response to LRR could be the result of altered
kinetics for Met internalization and/or receptor endocytosis via a
clathrin-independent route. The rate of Met internalization in response to
InlB and/or LRR was examined using a cell surface enzyme-linked immunosorbent
assay to quantify the loss of surface Met
(Fig. 3). Duplicate sets of
HeLa cells were treated with media containing InlB or LRR for increasing times
at 37 °C. The cells were then rapidly cooled, and the relative amount of
immunoreactive receptor remaining on the cell surface of one set of plates was
measured using an antibody specific to the extracellular domain of Met. The
second set of time-matched plates was permeabilized and then incubated with
anti-Met antibody to measure the amount of total (cell surface plus
intracellular) receptor. The ratio of surface to total Met was plotted against
time as an indication of the relative amount of endocytosed Met, and the
specific internalization rate constant (Ke) for each
ligand was calculated as a linear regression coefficient
(18). To correlate the level
of Met phosphorylation with the internalization rate, we used two different
concentrations of LRR in these studies, 2 and 10 nm LRR. As shown
in Fig. 3A, treatment
with 2 nm InlB caused rapid Met uptake from the cell surface
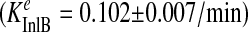 ,
consistent with our previous studies using T47D/Met cells
(18). A similar rate of
receptor Met endocytosis was observed in HeLa cells treated with 10
nm LRR
,
consistent with our previous studies using T47D/Met cells
(18). A similar rate of
receptor Met endocytosis was observed in HeLa cells treated with 10
nm LRR
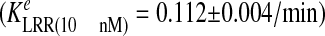 ,
conditions that induce comparable levels of Met phosphorylation as 2
nm InlB. However, Met internalization was slower in response to 2
nm LRR
,
conditions that induce comparable levels of Met phosphorylation as 2
nm InlB. However, Met internalization was slower in response to 2
nm LRR
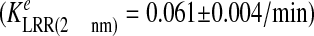 ,
which induced lower levels of receptor phosphorylation than the same
concentration of full-length InlB. We confirmed our findings using confocal
microscopy (Fig. 3, B and
C) with Alexa488-labeled LRR and
Alexa488-labeled InlB to quantify Met internalization.
Serum-starved HeLa cells were incubated with Alexa488-labeled InlB
(2 nm), Alexa488-labeled LRR (2 nm or 10
nm), or Alexa488-labeled mutant LRR-C242A (2
nm) for 10 min at 37 °C. Under these conditions comparable
levels of internalized ligand were detected in HeLa cells treated with 10
nm Alexa488-labeled LRR or 2 nm
Alexa488-labeled InlB, whereas decreased ligand levels were
detected in cells treated with 2 nm Alexa488-labeled
LRR. As expected, negligible internalized ligand was detected in cells treated
with Alexa488-labeled LRR-C242A. Thus, InlB and LRR are equally
effective at triggering Met endocytosis under conditions that induce
comparable Met phosphorylation. Using established populations of cell lines
immortalized from the kidneys of Met null (–/–) mice that stably
express comparable levels of wild-type Met (WT-Met) or the kinase-deficient
mutant K1110A (KinD-Met), we confirmed that Met kinase activity is required
for receptor uptake in response to LRR. The cells were incubated in media
containing InlB or LRR at 37 °C for increasing times, and the relative
amount of internalized WT-Met versus KinD-Met was examined using the
surface enzyme-linked immunosorbent assay (supplemental Fig. S2). As expected,
treatment with InlB and LRR causes WT-Met endocytosis. Conversely, higher cell
surface levels of KinD-Met were detected at each time point, indicating the
need for Met kinase activity in LRR-stimulated receptor endocytosis,
consistent with our earlier studies using InlB
(19).
,
which induced lower levels of receptor phosphorylation than the same
concentration of full-length InlB. We confirmed our findings using confocal
microscopy (Fig. 3, B and
C) with Alexa488-labeled LRR and
Alexa488-labeled InlB to quantify Met internalization.
Serum-starved HeLa cells were incubated with Alexa488-labeled InlB
(2 nm), Alexa488-labeled LRR (2 nm or 10
nm), or Alexa488-labeled mutant LRR-C242A (2
nm) for 10 min at 37 °C. Under these conditions comparable
levels of internalized ligand were detected in HeLa cells treated with 10
nm Alexa488-labeled LRR or 2 nm
Alexa488-labeled InlB, whereas decreased ligand levels were
detected in cells treated with 2 nm Alexa488-labeled
LRR. As expected, negligible internalized ligand was detected in cells treated
with Alexa488-labeled LRR-C242A. Thus, InlB and LRR are equally
effective at triggering Met endocytosis under conditions that induce
comparable Met phosphorylation. Using established populations of cell lines
immortalized from the kidneys of Met null (–/–) mice that stably
express comparable levels of wild-type Met (WT-Met) or the kinase-deficient
mutant K1110A (KinD-Met), we confirmed that Met kinase activity is required
for receptor uptake in response to LRR. The cells were incubated in media
containing InlB or LRR at 37 °C for increasing times, and the relative
amount of internalized WT-Met versus KinD-Met was examined using the
surface enzyme-linked immunosorbent assay (supplemental Fig. S2). As expected,
treatment with InlB and LRR causes WT-Met endocytosis. Conversely, higher cell
surface levels of KinD-Met were detected at each time point, indicating the
need for Met kinase activity in LRR-stimulated receptor endocytosis,
consistent with our earlier studies using InlB
(19).
FIGURE 3.

LRR induces rapid internalization of Met. A, HeLa cells were treated for 2, 4, and 8 min in medium with InlB, LRR, or without ligand (0 min) at 37 °C. The cells were rapidly cooled to 4 °C, and the level of residual surface Met was determined using a cell enzyme-linked immunosorbent assay for Met. The ratio of surface to total Met was plotted against time as an indication of the relative amount of internalized Met (see “Experimental Procedures”), and the specific internalization rate constant (Ke) for each ligand was calculated as a linear regression coefficient ± S.E. Data are representative of three independent experiments. B, HeLa cells were incubated in media containing Alexa488-labeled InlB, LRR, or LRR-C242A at 37 °C for 10 min and analyzed by confocal microscopy. Representative images of endocytosed InlB, LRR (closed arrows), and low levels of LRR-C242A (open arrows) are shown. Scale, 10 μm. C, the relative amount of endocytosed ligand was quantified using Metamorph, and the data are normalized with respect to internalized InlB ± S.E. from 2–3 experiments (*, p < 0.01 ANOVA).
InlB and LRR Induce Clathrin-mediated Met Endocytosis—We used RNA interference to examine whether Met internalization in response to LRR is clathrin-mediated and Grb2-dependent. HeLa cells were transfected with siRNAs targeting CHC or Grb2 (Fig. 4). Under these conditions, near complete depletion of CHC was achieved. As expected, levels of Met, transferrin receptor, and EEA1, markers for clathrin-mediated endocytosis and early endosomes, respectively (37–39), and caveolin 1, a component of caveolae that has been implicated in cell motility, lipid trafficking and epidermal growth factor (EGF) receptor signaling (38, 40–43) remained unaffected by treatment with siRNA targeting CHC (Fig. 4A). We reasoned that if LRR promoted clathrin-independent Met endocytosis, then receptor uptake would not be altered in cells depleted of endogenous CHC. To test this idea CHC-depleted cells were co-incubated with Alexa594-transferrin (Alexa-Tfn) and Alexa488-labeled InlB (Alexa-InlB) or Alexa488-labeled LRR (Alexa-LRR) for 10 min at 37 °C using ligand concentrations that induce comparable levels of Met phosphorylation. Ligand uptake was measured using confocal microscopy. CHC-siRNA abrogated Met endocytosis in response to InlB and its derivative LRR but did not affect receptor internalization in cells transfected with control siRNA. Uptake of the fluid phase marker Alexa594-labeled dextran 10,000 was unaffected (supplemental Fig. S3), whereas Alexa-Tfn internalization was reduced under these conditions (Fig. 4A). Collectively, these findings are consistent with a specific block in clathrin-mediated endocytosis. Similarly, siRNA-mediated depletion of Grb2 blocked Met uptake in response to LRR and InlB. The requirement for Grb2 was specific for Met endocytosis, as clathrin-mediated uptake of Alexa-Tfn was unaffected under these conditions (Fig. 4B). Thus, like InlB, LRR triggers Met internalization through clathrin-coated pits in a Grb2-dependent manner.
FIGURE 4.

Internalization of Met in response to LRR is clathrin- and Grb2-dependent. A, left panel, lysates from untransfected HeLa cells (Un) or cells transfected with control (Con) or CHC siRNA were examined by Western analysis using antibodies for CHC, transferrin receptor, Met, EEA1, and caveolin. Right panel, duplicate sets of control or CHC-depleted HeLa cells were co-treated for 10 min at 37 °C with Alexa594-Tfn and Alexa488-InlB, or Alexa488-LRR, and the relative amount of internalized ligand was analyzed by confocal microscopy. Values represent the mean fluorescence intensity ±S.E. from 2–3 experiments and are expressed as a percentage of control values. B, left panel, Western analysis was performed as indicated to confirm siRNA-mediated depletion of Grb2 in HeLa cells. Right Panel, the relative amounts of internalized Tfn, InlB, and LRR in a 10-min/37 °C pulse in HeLa cells transfected with control (Con) or Grb2 siRNAs were quantified by confocal microscopy. The average values of 2–3 experiments are expressed as a percentage of control values ± S.E. (*, p < 0.01, ANOVA).
Internalized Met Recycles in Response to LRR—Our data indicate no difference in the internalization properties of Met in response to InlB or LRR, suggesting that post-endocytic mechanisms contribute to the prolonged stability and signaling of Met in response to LRR. After InlB-induced Met internalization into early endosomes, the receptor is normally sorted for degradation in the lysosome (18). The delay in Met degradation we observed after treatment with the LRR fragment could be the result of a slower rate of transit through early endosomes and/or enhanced receptor recycling to the cell surface. Therefore, we used confocal microscopy to examine the distribution of internalized Met. Alexa-LRR and Alexa-InlB were employed as indicators for receptor co-localization with endogenous EEA1 and CD63, specific markers for early endosomes and multivesicular bodies (MVB), respectively (44, 45). HeLa cells were allowed to internalize Alexa-LRR or Alexa-InlB into early endosomes at 37 °C for 15 min. After internalization, the cells were washed and then chased at 37 °C for 0, 30, or 60 min in media lacking ligand to promote Met trafficking from early endosomes. The cells were then fixed and co-stained for endogenous EEA1 or CD63 and examined using confocal microscopy (supplemental Fig. S4). Increased chase times resulted in decreased Alexa-InlB co-localization with EEA1 with a concomitant increase in ligand co-staining with CD63, consistent with Met trafficking from early endosomes to MVBs (Fig. 5A). The lower levels of Alexa-InlB that co-localized with CD63 at 60 min (49%) may be the result of InlB-Met complex dissociation or lysosomal degradation under these chase conditions. In contrast to Alexa-InlB-treated cells, high levels of Alexa-LRR co-localized with EEA1 at the 30- and 60-min chase times (Fig. 5B). The decreased extent of Alexa-LRR co-localization with CD63 at 30 min is consistent with a delay in Met trafficking from early endosomes to MVBs.
FIGURE 5.

Met transit from early endosomes to the MVB is delayed in response to LRR. HeLa cells were allowed to internalize Alexa488-labeled InlB or LRR for 15 min at 37 °C and then chased at 37 °C in the absence of ligand for increasing times to promote ligand transport from early endosomes to the MVB. Cells were fixed and stained for endogenous EEA1 or CD63, and the relative amount of internalized ligand that co-localizes with these proteins was quantified using confocal microscopy. Values are expressed as the percentage of co-localization ± S.E. normalized to an area from three independent experiments (*, p < 0.01, ANOVA).
To further examine the fate of InlB- versus LRR-activated Met, we developed a flow cytometry assay to quantify the recovery of internalized Met in the plasma membrane due to receptor recycling. Cycloheximide-treated HeLa cells were incubated in media lacking or containing ligand (2 nm InlB or 10 nm LRR) for 15 min at 37 °C to promote Met internalization into early endosomes. The cells were shifted to 4 °C to halt receptor trafficking and then incubated with ice-cold 0.25% trypsin to remove residual surface-associated receptor. After trypsin inactivation, the cells were shifted to 37 °C in the presence of cycloheximide for increasing times to promote receptor trafficking. Flow cytometry was used to examine the reappearance of internalized receptor on the cell surface using an antibody specific for the ectodomain of Met. Under these conditions receptor recycling was undetectable in cells treated with InlB or untreated control cells (Fig. 6), consistent with a previous study showing that InlB treatment results in the lysosomal degradation of Met (18). Conversely, increasing levels of surface Met were readily detected in LRR-treated cells during the chase conditions, consistent with the recycling of internalized Met back to the plasma membrane. These results suggest that receptor recycling to the cell surface is likely one of the causes of increased Met stability in response to LRR.
FIGURE 6.

Internalized Met recycles in response to LRR. Cycloheximide-treated HeLa cells were incubated in media lacking (Con) or containing ligand (2 nm InlB or 10 nm LRR) for 15 min at 37 °C to internalize Met. The cells were shifted to 4 °C, incubated in ice-cold trypsin to digest residual surface receptor, and then shifted to 37 °C in cycloheximide-containing media as indicated. Recycled receptor was labeled with anti-Met antibody and quantified by flow cytometry. Results represented the mean fluorescent intensities normalized to control cells under each experimental condition from triplicate experiments. Bars represent the means for data across all experiments with S.E. (*, p < 0.01, ANOVA).
Cell Motility Increases in Response to LRR—Our observation that Met signaling in response to 10 nm LRR is sustained (refer to Fig. 1B) suggested that LRR might promote cellular responses resulting from Met signaling such as cell motility and/or branching morphogenesis. MDCK cells are an epithelial cell line that readily undergoes colony dispersal, increased cell motility, and branching morphogenesis in response to Met signaling (8, 46, 47). Thus, we tested the ability of MDCK cells to form tubules in semisolid collagen in response to InlB or LRR. As shown in supplemental Fig. S5, MDCK cells grown in three-dimensional collagen gels form simple cystic structures in the absence of ligand. Treatment of the control cysts with HGF for 7 days induced marked branching tubulogenesis into the surrounding gel. Similarly, treatment for 7 days with InlB or LRR resulted in the formation of long, thick tubules that were indistinguishable from those induced by HGF.
We next examined MDCK cell motility in response to various Met ligands. Serum-starved MDCK cells were incubated in media without ligand or containing HGF, InlB, wild type, or mutant LRR for 8 h, and the scattering response was examined using phase contrast microscopy. Under these conditions, HGF and InlB were indistinguishable in their ability to induce cell scattering. Conversely, LRR treatment stimulated MDCK scattering in a dose-dependent manner (Fig. 7A). No scattering was observed in the absence of ligand or after treatment with the inactive LRR mutant C242A.
FIGURE 7.

LRR promotes cell motility. A, colonies of serum-starved MDCK cells were incubated for 8 h in medium lacking ligand (Con) or supplemented with HGF (2 nm), InlB (2 nm), LRR (2 nm or 10 nm), or the mutant LRR-C242A (2 nm), and the scattering response was examined by phase-contrast microscopy. Bar, 20 μm. The results are representative of three independent experiments. B, confluent monolayers of serum-starved MDCK were wounded with a pipette tip (n = 3/well) and immediately incubated in media without (Con) or with ligand (2 nm InlB, 10 nm LRR). The cells were imaged at identical points along the wound at 0 h or after incubation at 37 °C for 6 h. Representative images from three independent experiments are shown. C, the relative wound gap was measured in pixels using Metamorph software, and the data are normalized to control cells. Bars represent the mean ± S.E. Significant differences between LRR- and InlB-treated cells with control cells were detected (*, p < 0.01, ANOVA) and between LRR and InlB-treated cells (**, p < 0.01, ANOVA).
We quantified the effect of LRR and InlB on MDCK motility using scratch assays. In these studies cells were grown to confluency and serum-starved for 48 h in media containing 0.1% sera to ensure that cell migration rather than cell growth was measured. Linear scratches were induced using a pipette tip, and the width of the wound was measured in pixels. The cells were then incubated in medium with 0.2% serum without or with 2 nm InlB or 10 nm LRR for up to 6 h, at which time the width of the denuded area was measured. As shown in Fig. 7, B and C, cell motility increased in response to InlB and, to a greater extent LRR, relative to control cells incubated in media lacking ligand. The wound width was reduced to 45.99 ± 1.7 and 28.56 ± 1.5% that of the original size in response to InlB and LRR respectively (Fig. 7). In control cells the wound decreased to 92.7 ± 2.25% that of its original width at 6 h. To confirm that the increase in cell motility induced by LRR was not cell type-specific, we performed scratch assays using HeLa cells. As shown in supplemental Fig. S6, HeLa cell motility in response to LRR was higher than that induced by full-length InlB, consistent with our motility studies using MDCK cells. Thus, increased Met stability in response to LRR correlates with increased motility of HeLa and MDCK cells.
DISCUSSION
Here we performed a structure/function analysis of the Listeria protein InlB. A key finding from our studies is that InlB and its N-terminal LRR fragment differentially regulate the endocytic trafficking of Met. Notably, our results indicate that in contrast to InlB, LRR attenuates Met degradation and prolongs Met phosphorylation, correlating with increased cell motility. The effect of LRR on Met degradation is specific as InlB and LRR induce comparable rates of clathrin-mediated Met internalization. Moreover, Met recycles back to the cell surface in response to LRR but not InlB. These results suggest that alterations in Met degradation contribute at least in part to differences in the potencies of InlB and LRR with respect to receptor signaling.
How do InlB and LRR differ in their efficiency in inducing Met degradation? One possible explanation is related to the relative binding affinity of InlB versus LRR for Met. In the case of the EGF receptor, EGF but not transforming growth factor-α induces efficient receptor degradation. EGF remains tightly associated with the EGF receptor after receptor internalization, whereas transforming growth factor-α rapidly dissociates from the receptor within the acidic environment of early endosomes, resulting in receptor recycling to the cell surface (48). Our studies show a requirement for higher concentrations of LRR compared with InlB to achieve comparable levels of Met autophosphorylation. These findings suggest that differences in the binding affinities of InlB and LRR for Met likely exist. However, LRR appears to remain associated with Met during transit through early endosomes and MVBs. Like Alexa-InlB, internalized Alexa-LRR is readily detected in EEA1- and CD63-positive endocytic compartments. Although we cannot exclude the possibility that ligand affinity for Met is altered during transit through endocytic compartments, our studies support the conclusion that binding of LRR or InlB is not adversely disrupted by early endosomal pH.
An alternative explanation involves the different quaternary states of InlB and LRR. Receptor dimerization is the proposed mechanism for Met activation, as bivalent antibodies directed against the ectodomain of Met induce receptor phosphorylation and trigger several downstream biological responses, including cell motility, proliferation, invasion, tubulogenesis, and angiogenesis (49). InlB is a monomeric protein and could activate Met through a mechanism similar to that observed with EGF. EGF induces a conformational change in its receptor, resulting in the unmasking of a cryptic dimerization motif in the receptor extracellular domain (for review, see Refs. 50 and 51). In this way binding of EGF converts the EGF receptor from a dimerization-inhibited state to a dimerization-competent state. The key to this transition involves the initial formation of an asymmetric EGF-EGF receptor dimer (52), in which one of the EGF-bound monomers relieves autoinhibition from the other receptor monomer resulting in an active conformation. By analogy, InlB binding to Met might induce a conformational change that promotes receptor dimerization and, hence, receptor activation. In contrast to InlB, LRR is a dimer in solution. Therefore, LRR may activate Met simply by clustering or cross-linking two receptors. This would suggest that additional regions absent from LRR (e.g. inter-repeat or GW domains) could be required to induce a conformational change sufficient for maximal Met activation.
A recent model described by Niemann et al. (14) may provide an alternative explanation for the different agonist properties of full-length InlB and its LRR fragment. Although InlB is a monomer in solution, the authors propose that soluble InlB might be clustered and form multimers upon interacting with heparan sulfate proteoglycans on the surface of host cells via the GW domains of InlB. In this model InlB and LRR each act as multimers capable of clustering Met. The structural data of Niemann et al. (14) suggests that the binding of InlB to Met induces a change in the orientation of the sema and Ig1 domains in Met. The authors propose that the inter-repeat region in InlB is needed for this re-orientation, which they refer to as “clamping.” According to this model, the LRR fragment can bind and cluster Met but cannot induce the structural change. Perhaps the inability of the LRR fragment to clamp the receptor-ligand complex results in an activated receptor with altered signaling activity compared with the situation obtained with InlB binding.
Conformational changes induced by the binding of InlB versus LRR likely alter the ability of kinase-active Met to engage components of the endocytic machinery and/or attenuate downstream signaling molecules important for receptor degradation. Recent studies suggest a strong correlation between Cbl-mediated Met ubiquitination and receptor down-regulation. In addition to functioning as an adaptor protein for Met (53), Cbl also serves as an E3 ubiquitin ligase (54–57) to ubiquitinate target proteins including Met (56, 57). Although Cbl E3 ubiquitin ligase activity regulates Met endocytosis, Cbl-mediated Met ubiquitination is not required for receptor internalization per se but is needed for efficient receptor down-regulation (19, 28). Consistent with these findings, a somatic intronic mutation identified in lung cancer results in a Met receptor mutant that shows decreased Cbl binding, increased Met stability, and tumorigenesis (58). Similarly, an artificial mutation introduced into the juxtamembrane region of Met (Y1003F) which inhibits recruitment of Cbl increases Met stability and promotes cell proliferation in response to HGF (28). However, it is unclear from these studies whether the respective Met mutants recycle to the cell surface or remain on intracellular membranes. In our hands internalized Met does not normally recycle back to the cell surface in response to HGF.3 Although the mechanistic basis of LRR- versus InlB-induced changes in Met trafficking remain unclear, it is tempting to speculate that Cbl activation and binding or Cbl-mediated ubiquitination of Met may be attenuated in LRR-treated cells, leading to receptor recycling and, hence, increased Met stability and signaling. Consistent with this scenario, LRR treatment promotes motility of MDCK and HeLa. Because lysosomal sorting is a saturable process (59), additional factors downstream of Cbl may also be rate-limiting for Met degradation in LRR-treated cells.
In conclusion, we report the first evidence that Met alters its trafficking pattern in response to a truncated derivative of InlB, the LRR fragment. Altered Met trafficking results in increased receptor stability and recycling to the cell surface, correlating with prolonged Met signaling and cell motility. These studies support the contention that alterations in Met endocytic trafficking can augment receptor signaling and, hence, cellular responses. Additional studies will be required to determine how InlB and its fragment LRR alter the ability of kinase-active Met to interact with critical components of the endocytic machinery important for receptor sorting and degradation.
Supplementary Material
Acknowledgments
We thank members of the Elferink laboratory for helpful comments.
This work was supported, in whole or in part, by National Institutes of Health Grants CA-112605 and CA-119075 (to L. A. E.). This work was also supported by National Science Foundation Grant IBN-343739 (to L. A. E.) and Canadian Institutes of Health Research Grant MT-15497 (to K. I.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S6.
Footnotes
The abbreviations used are: InlB, Internalin B; LRR, leucine-rich repeat; CHC, clathrin heavy chain; EEA1, early endosomal antigen; EGF, epidermal growth factor; HGF, hepatocyte growth factor; Met, hepatocyte growth factor receptor; LRR, leucine-rich repeat; MVB, multivesicular body; Tfn, transferrin; ANOVA, analysis of variance; MDCK, Madin-Darby canine kidney; WT, wild type; PBS, phosphate-buffered saline; FACS, fluorescence-activated cell sorter; siRNA, small Interfering RNA.
K. Hill and L. A. Elferink, personal communication.
References
- 1.Dramsi, S., Biswas, I., Maguin, E., Braun, L., Mastroeni, P., and Cossart, P. (1995) Mol. Microbiol. 16 251–261 [DOI] [PubMed] [Google Scholar]
- 2.Ireton, K., Payrastre, B., Chap, H., Ogawa, W., Sakaue, H., Kasuga, M., and Cossart, P. (1996) Science 274 780–782 [DOI] [PubMed] [Google Scholar]
- 3.Greiffenberg, L., Goebel, W., Kim, K. S., Weiglein, I., Bubert, A., Engelbrecht, F., Stins, M., and Kuhn, M. (1998) Infect. Immun. 66 5260–5267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parida, S. K., Domann, E., Rohde, M., Muller, S., Darji, A., Hain, T., Wehland, J., and Chakraborty, T. (1998) Mol. Microbiol. 28 81–93 [DOI] [PubMed] [Google Scholar]
- 5.Ireton, K., Payrastre, B., and Cossart, P. (1999) J. Biol. Chem. 274 17025–17032 [DOI] [PubMed] [Google Scholar]
- 6.Jonquieres, R., Bierne, H., Fiedler, F., Gounon, P., and Cossart, P. (1999) Mol. Microbiol. 34 902–914 [DOI] [PubMed] [Google Scholar]
- 7.Banerjee, M., Copp, J., Vuga, D., Marino, M., Chapman, T., van der Geer, P., and Ghosh, P. (2004) Mol. Microbiol. 52 257–271 [DOI] [PubMed] [Google Scholar]
- 8.Shen, Y., Naujokas, M., Park, M., and Ireton, K. (2000) Cell 103 501–510 [DOI] [PubMed] [Google Scholar]
- 9.Birchmeier, C., Birchmeier, W., Gherardi, E., and Vande Woude, G. F. (2003) Nat. Rev. Mol. Cell Biol. 4 915–925 [DOI] [PubMed] [Google Scholar]
- 10.Takami, T., Kaposi-Novak, P., Uchida, K., Gomez-Quiroz, L. E., Conner, E. A., Factor, V. M., and Thorgeirsson, S. S. (2007) Cancer Res. 67 9844–9851 [DOI] [PubMed] [Google Scholar]
- 11.Huh, C. G., Factor, V. M., Sanchez, A., Uchida, K., Conner, E. A., and Thorgeirsson, S. S. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 4477–4482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borowiak, M., Garratt, A. N., Wustefeld, T., Strehle, M., Trautwein, C., and Birchmeier, C. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 10608–10613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bierne, H., and Cossart, P. (2002) J. Cell Sci. 115 3357–3367 [DOI] [PubMed] [Google Scholar]
- 14.Niemann, H. H., Jager, V., Butler, P. J., van den Heuvel, J., Schmidt, S., Ferraris, D., Gherardi, E., and Heinz, D. W. (2007) Cell 130 235–246 [DOI] [PubMed] [Google Scholar]
- 15.Niemann, H. H., Petoukhov, M. V., Hartlein, M., Moulin, M., Gherardi, E., Timmins, P., Heinz, D. W., and Svergun, D. I. (2008) J. Mol. Biol. 377 489–500 [DOI] [PubMed] [Google Scholar]
- 16.Stamos, J., Lazarus, R. A., Yao, X., Kirchhofer, D., and Wiesmann, C. (2004) EMBO J. 23 2325–2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Copp, J., Marino, M., Banerjee, M., Ghosh, P., and van der Geer, P. (2003) J. Biol. Chem. 278 7783–7789 [DOI] [PubMed] [Google Scholar]
- 18.Li, N., Xiang, G. S., Dokainish, H., Ireton, K., and Elferink, L. A. (2005) Traffic 6 459–473 [DOI] [PubMed] [Google Scholar]
- 19.Li, N., Lorinczi, M., Ireton, K., and Elferink, L. A. (2007) J. Biol. Chem. 282 16764–16775 [DOI] [PubMed] [Google Scholar]
- 20.Raiborg, C., Bache, K. G., Gillooly, D. J., Madshus, I. H., Stang, E., and Stenmark, H. (2002) Nat. Cell Biol. 4 394–398 [DOI] [PubMed] [Google Scholar]
- 21.Bache, K. G., Raiborg, C., Mehlum, A., and Stenmark, H. (2003) J. Biol. Chem. 278 12513–12521 [DOI] [PubMed] [Google Scholar]
- 22.Katzmann, D. J., Stefan, C. J., Babst, M., and Emr, S. D. (2003) J. Cell Biol. 162 413–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Odorizzi, G., Katzmann, D. J., Babst, M., Audhya, A., and Emr, S. D. (2003) J. Cell Sci. 116 1893–1903 [DOI] [PubMed] [Google Scholar]
- 24.Lecuit, M., Ohayon, H., Braun, L., Mengaud, J., and Cossart, P. (1997) Infect. Immun. 65 5309–5319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Braun, L., Nato, F., Payrastre, B., Mazie, J. C., and Cossart, P. (1999) Mol. Microbiol. 34 10–23 [DOI] [PubMed] [Google Scholar]
- 26.Peschard, P., and Park, M. (2003) Cancer Cell 3 519–523 [DOI] [PubMed] [Google Scholar]
- 27.Bache, K. G., Slagsvold, T., and Stenmark, H. (2004) EMBO J. 23 2707–2712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abella, J. V., Peschard, P., Naujokas, M. A., Lin, T., Saucier, C., Urbe, S., and Park, M. (2005) Mol. Cell. Biol. 25 9632–9645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kjelsberg, C., Sakurai, H., Spokes, K., Birchmeier, C., Drummond, I., Nigam, S., and Cantley, L. G. (1997) Am. J. Physiol. 272 F222–F228 [DOI] [PubMed] [Google Scholar]
- 30.Strick, D. J., and Elferink, L. A. (2005) Mol. Biol. Cell 16 5699–5709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Santos, M. F., McCormack, S. A., Guo, Z., Okolicany, J., Zheng, Y., Johnson, L. R., and Tigyi, G. (1997) J. Clin. Investig. 100 216–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O'Brien, L. E., Yu, W., Tang, K., Jou, T. S., Zegers, M. M., and Mostov, K. E. (2006) Methods Enzymol. 406 676–691 [DOI] [PubMed] [Google Scholar]
- 33.Jonquieres, R., Pizarro-Cerda, J., and Cossart, P. (2001) Mol. Microbiol. 42 955–965 [DOI] [PubMed] [Google Scholar]
- 34.Kermorgant, S., Zicha, D., and Parker, P. J. (2003) J. Biol. Chem. 278 28921–28929 [DOI] [PubMed] [Google Scholar]
- 35.Hammond, D. E., Carter, S., McCullough, J., Urbe, S., Vande Woude, G., and Clague, M. J. (2003) Mol. Biol. Cell 14 1346–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hammond, D. E., Urbe, S., Vande Woude, G. F., and Clague, M. J. (2001) Oncogene 20 2761–2770 [DOI] [PubMed] [Google Scholar]
- 37.Klausner, R. D., Van Renswoude, J., Ashwell, G., Kempf, C., Schechter, A. N., Dean, A., and Bridges, K. R. (1983) J. Biol. Chem. 258 4715–4724 [PubMed] [Google Scholar]
- 38.Hopkins, C. R., and Trowbridge, I. S. (1983) J. Cell Biol. 97 508–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simonsen, A., Lippe, R., Christoforidis, S., Gaullier, J. M., Brech, A., Callaghan, J., Toh, B. H., Murphy, C., Zerial, M., and Stenmark, H. (1998) Nature 394 494–498 [DOI] [PubMed] [Google Scholar]
- 40.Mineo, C., Gill, G. N., and Anderson, R. G. (1999) J. Biol. Chem. 274 30636–30643 [DOI] [PubMed] [Google Scholar]
- 41.Schlegel, A., and Lisanti, M. P. (2001) Cytokine Growth Factor Rev. 12 41–51 [DOI] [PubMed] [Google Scholar]
- 42.Hassan, G. S., Williams, T. M., Frank, P. G., and Lisanti, M. P. (2006) Am. J. Physiol. Heart Circ. Physiol. 290 2393–2401 [DOI] [PubMed] [Google Scholar]
- 43.Navarro, A., Anand-Apte, B., and Parat, M. O. (2004) FASEB J. 18 1801–1811 [DOI] [PubMed] [Google Scholar]
- 44.Kobayashi, T., Vischer, U. M., Rosnoblet, C., Lebrand, C., Lindsay, M., Parton, R. G., Kruithof, E. K., and Gruenberg, J. (2000) Mol. Biol. Cell 11 1829–1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patki, V., Virbasius, J., Lane, W. S., Toh, B. H., Shpetner, H. S., and Corvera, S. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 7326–7330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gherardi, E., Gray, J., Stoker, M., Perryman, M., and Furlong, R. (1989) Proc. Natl. Acad. Sci. U. S. A. 86 5844–5848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Royal, I., and Park, M. (1995) J. Biol. Chem. 270 27780–27787 [DOI] [PubMed] [Google Scholar]
- 48.Ebner, R., and Derynck, R. (1991) Cell Regul. 2 599–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prat, M., Crepaldi, T., Pennacchietti, S., Bussolino, F., and Comoglio, P. M. (1998) J. Cell Sci. 111 237–247 [DOI] [PubMed] [Google Scholar]
- 50.Hubbard, S. R. (2006) Cell 125 1029–1031 [DOI] [PubMed] [Google Scholar]
- 51.Schlessinger, J. (2002) Cell 110 669–672 [DOI] [PubMed] [Google Scholar]
- 52.Zhang, X., Gureasko, J., Shen, K., Cole, P. A., and Kuriyan, J. (2006) Cell 125 1137–1149 [DOI] [PubMed] [Google Scholar]
- 53.Petrelli, A., Gilestro, G. F., Lanzardo, S., Comoglio, P. M., Migone, N., and Giordano, S. (2002) Nature 416 187–190 [DOI] [PubMed] [Google Scholar]
- 54.Joazeiro, C. A., Wing, S. S., Huang, H., Leverson, J. D., Hunter, T., and Liu, Y. C. (1999) Science 286 309–312 [DOI] [PubMed] [Google Scholar]
- 55.Levkowitz, G., Waterman, H., Ettenberg, S. A., Katz, M., Tsygankov, A. Y., Alroy, I., Lavi, S., Iwai, K., Reiss, Y., Ciechanover, A., Lipkowitz, S., and Yarden, Y. (1999) Mol. Cell 4 1029–1040 [DOI] [PubMed] [Google Scholar]
- 56.Peschard, P., Ishiyama, N., Lin, T., Lipkowitz, S., and Park, M. (2004) J. Biol. Chem. 279 29565–29571 [DOI] [PubMed] [Google Scholar]
- 57.Peschard, P., Fournier, T. M., Lamorte, L., Naujokas, M. A., Band, H., Langdon, W. Y., and Park, M. (2001) Mol. Cell 8 995–1004 [DOI] [PubMed] [Google Scholar]
- 58.Kong-Beltran, M., Seshagiri, S., Zha, J., Zhu, W., Bhawe, K., Mendoza, N., Holcomb, T., Pujara, K., Stinson, J., Fu, L., Severin, C., Rangell, L., Schwall, R., Amler, L., Wickramasinghe, D., and Yauch, R. (2006) Cancer Res. 66 283–289 [DOI] [PubMed] [Google Scholar]
- 59.French, A. R., Sudlow, G. P., Wiley, H. S., and Lauffenburger, D. A. (1994) J. Biol. Chem. 269 15749–15755 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.