

Abstract
In an effort to devise strategies for overcoming bacterial β-lactamases, we studied LN-1-255, a 6-alkylidene-2′-substituted penicillin sulfone inhibitor. By possessing a catecholic functionality that resembles a natural bacterial siderophore, LN-1-255 is unique among β-lactamase inhibitors. LN-1-255 combined with piperacillin was more potent against Escherichia coli DH10B strains bearing blaSHV extended-spectrum and inhibitor-resistant β-lactamases than an equivalent amount of tazobactam and piperacillin. In addition, LN-1-255 significantly enhanced the activity of ceftazidime and cefpirome against extended-spectrum cephalosporin and Sme-1 containing carbapenem-resistant clinical strains. LN-1-255 inhibited SHV-1 and SHV-2 β-lactamases with nm affinity (KI = 110 ± 10 and 100 ± 10 nm, respectively). When LN-1-255 inactivated SHV β-lactamases, a single intermediate was detected by mass spectrometry. The crystal structure of LN-1-255 in complex with SHV-1 was determined at 1.55Å resolution. Interestingly, this novel inhibitor forms a bicyclic aromatic intermediate with its carbonyl oxygen pointing out of the oxyanion hole and forming hydrogen bonds with Lys-234 and Ser-130 in the active site. Electron density for the “tail” of LN-1-255 is less ordered and modeled in two conformations. Both conformations have the LN-1-255 carboxyl group interacting with Arg-244, yet the remaining tails of the two conformations diverge. The observed presence of the bicyclic aromatic intermediate with its carbonyl oxygen positioned outside of the oxyanion hole provides a rationale for the stability of this inhibitory intermediate. The 2′-substituted penicillin sulfone, LN-1-255, is proving to be an important lead compound for novel β-lactamase inhibitor design.
The rapidly increasing number of antibiotic-resistant Gram-negative microorganisms, including the Enterobacteriaceae family and the Pseudomonas, Acinetobacter, and Klebsiella genera, represents a grave threat to human health. The first-line treatments for such infections are β-lactam antibiotics, and the most common mechanism of resistance to such agents is bacterial production of β-lactamases. Regrettably, Enterobacteriaceae resistant to penicillins and extended-spectrum cephalosporins (e.g. ceftazidime, ceftriaxone and cefepime) are continuing to threaten the efficacy of our available β-lactam antibiotics (1–4). Currently, up to 30% of the Enterobacter spp., Escherichia coli, and Klebsiella spp. recovered from patients with infections in hospitals, long-term care facilities, and intensive care units in the United States are resistant to ceftazidime (1, 2, 5–9). Of greatest concern is the emerging number of community-acquired E. coli and Klebsiella spp. that are resistant to these cephalosporins (3, 4, 8–11). This latter group presents a very significant future danger to the current use of β-lactam antibiotics to treat common infections in the ambulatory setting (11).
Resistance to extended-spectrum cephalosporins in E. coli and Klebsiella pneumoniae is typically caused by a plasmid or chromosomal class A bla gene that encodes for extended-spectrum β-lactamases (ESBLs)4 (1, 5, 12, 13). Among the β-lactam class of antibiotics, only carbapenems are recommended for the therapy of infections caused by ESBL-producing bacteria (1, 2, 7). Many ESBL-producing bacteria possess β-lactamases that are susceptible to β-lactamase inhibitors, but the clinical efficacy of β-lactam/β-lactamase inhibitor combinations still remains to be established conclusively (5, 12). β-Lactam/β-lactamase inhibitor combinations offer an extremely attractive approach to combating infections caused by ESBL-producing bacteria. Unfortunately, current commercial β-lactamase inhibitors (clavulanate, sulbactam, and tazobactam) narrowly target class A enzymes. Thus, there is an urgent need for the design and development of more clinically useful β-lactam antibiotic/β-lactamase inhibitor combinations.
Early studies demonstrated that selected 6-alkylidene-2′-substituted penicillin sulfones and 7-alkylidene-3′-substituted cephalosporin sulfones (Fig. 1, A and B) are effective β-lactamase inhibitors (14–17). In an effort to assess the activity and understand the efficacy of these compounds, we investigated one of the most potent of these derivatives, a 6-alkylidene-2′-substituted penicillin sulfone, which has become known as LN-1-255 (18). This inhibitor was designed with a catecholic functionality that could potentially resemble a natural bacterial siderophore, thus enabling LN-1-255 to utilize the iron uptake system to traverse the outer membrane (18).
FIGURE 1.

Chemical structures of the β-lactamase inhibitors. A, general structure of 7-alkylidene-3-substituted cephalosporin sulfones. B, general structure of 6-alkylidene-2′-substituted penicillin sulfones. C, LN-1-255. D, tazobactam sodium.
In this report, LN-1-255 was evaluated as a class A β-lactamase inhibitor and compared with tazobactam against SHV-1, an ESBL variant of SHV-1 (SHV-2), and selected “inhibitor-resistant” SHV β-lactamases. In addition, we tested LN-1-255 as a broad spectrum inhibitor against representative clinical isolates possessing a variety of β-lactamases. To elucidate the mechanism of inactivation, the crystal structure of SHV-1 in complex with LN-1-255 was determined at 1.55 Å resolution. We show that LN-1-255 inactivates SHV-1 by forming a bicyclic aromatic intermediate that resists deacylation.
MATERIALS AND METHODS
Chemical Synthesis—The chemical structure of LN-1-255 and tazobactam are shown in Fig. 1. The synthesis and initial evaluation of LN-1-255 were reported (18) and reviewed (15) by Buynak et al.
Bacterial Strains, Plasmids, and Mutagenesis to Create ESBL Variants of SHV-1—As described previously, blaSHV-1 was first cloned into phagemid vector pBC SK(-) (Stratagene, La Jolla, CA) from a clinical strain of K. pneumoniae 15571 and maintained in E. coli DH10B™ cells (Invitrogen) (19). This E. coli strain with blaSHV-1 was used for minimum inhibitory concentration (MIC) determinations, mutagenesis, and protein purification. The construction of blaSHV-2, blaSHV-5, and other inhibitor-resistant blaSHV enzymes was described previously (20–22).
Antibiotic Susceptibility—The MICs of E. coli DH10B expressing blaSHV β-lactamases were performed in Luria Bertani (LB) agar. MICs were determined using a Steers replicator, which delivered 10 μl of LB broth containing 104 colony-forming units/spot. The inhibitors were screened using piperacillin as the partner β-lactam. In the first set of MICs, both β-lactam/β-lactamase inhibitors were tested using an 8/1 ratio of piperacillin/β-lactamase inhibitor. Piperacillin was obtained from (Sigma), and sodium tazobactam was a kind gift from Wyeth Pharmaceuticals (Pearl River, NY).
Additional susceptibility testing was performed to assess the potency of LN-1-255 with candidate broad spectrum β-lactam antibiotics using a collection of well defined clinical isolates. These isolates represent a worldwide collection of important patient-derived strains that possess contemporary β-lactamases used in screening studies. Against these strains, MICs were performed according to the Clinical Laboratory Standards Institute guidelines. Here, we used microdilution panels with cation-adjusted Mueller Hinton broth. The β-lactam antibiotics and tazobactam employed here were acquired from Sigma. LN-1-255 was used in a fixed (4 μg/ml) ratio with ceftazidime, cefpirome, and meropenem. Tazobactam was tested at a fixed ratio of 4 μg/ml combined with increasing concentrations of piperacillin.
β-Lactamase Purification—The SHV-1 and SHV-2 β-lactamases were harvested from E. coli DH10B according to a previously published method employing preparative isoelectric focusing (pIEF) (20–22). Cell lysis was achieved with the addition of lysozyme and EDTA (Sigma). Crude enzyme preparations were further purified using pIEF. We assessed the purity of these preparations of SHV-1 and SHV-2 using 5% stacking, 12% resolving SDS-PAGE. Gels were stained with Coomassie Brilliant Blue R250 (Fisher, Pittsburgh, PA) to visualize proteins. In certain instances we were required to further purify SHV-1 and SHV-2 β-lactamases using size exclusion chromatography with a Pharmacia ÄKTA purifier system (GE Healthcare). We employed a HiLoad 16/60 Superdex 75 column (GE Healthcare) and eluted with 20 mm phosphate-buffered saline (PBS) (pH 7.4). We determined protein concentrations using Bio-Rad protein assay with bovine serum albumin as a standard.
Kinetic Parameters—Kinetic parameters (Vmax, Km, kcat) for SHV-1 and SHV-2 β-lactamases were determined from initial steady state velocities using an Agilent™ 8453 diode array spectrophotometer. The kinetic determinations were performed at room temperature (23 °C) in 20 mm PBS, pH 7.4. Vmax and Km were obtained with nonlinear least squares fit of the data (Henri-Michaelis-Menten equation) using Enzfitter™ (Biosoft Corp., Ferguson, MO) (20).
We measured the ability of LN-1-255 to inhibit and inactivate SHV-1 and SHV-2. We first determined the dissociation constants for the preacylation complex (KD) according to two published methods (23–26). First, we performed a direct competition assay using 100 μm nitrocefin (BD Biosciences) as the indicator substrate (Δε482 = 17,400 m-1 cm-1), 21 nm SHV-1 or SHV-2, and varying concentration of inhibitor (tazobactam, 0.3–1.2 μm, or LN-1-255, 10–500 nm). After the data were obtained, the KD was “corrected” according to the following equation to account for the affinity of nitrocefin (ncf) for SHV-1 and SHV-2 β-lactamases.
 |
(Eq. 1) |
Next, we also determined the KD by measuring the initial velocity (vi) in the presence of increasing concentrations of LN-1-255 (0–50 nm) or tazobactam (0–500 nm) against the colorimetric substrate nitrocefin (0–100 μm). Care was taken to initiate the reaction with the addition of both nitrocefin and the inhibitor and to limit the initial velocity to the first 5–10 s so that the only inhibition observed would be derived from formation of the noncovalent Henri-Michaelis-Menten complex. Three experimental determinations were made for each unique nitrocefin/inhibitor concentration, and progress curves in the presence of varying concentrations of inhibitor were analyzed by nonlinear regression using a competitive model of inhibition, corrected as above. Each value was presented.
The first-order rate constant for enzyme and inhibitor complex inactivation, kinact, was measured directly by monitoring the reaction time courses in the presence of inhibitor and nitrocefin. A fixed concentration of enzyme (E; 21 nm), 100 μm nitrocefin, and increasing concentrations of tazobactam (0.5–6 μm) or LN-1-255 (50–600 nm) (inactivator (I)) were used in each assay. The kobs values were determined using a nonlinear least squares fit of the data to Equation 2 employing Origin 7.5®.
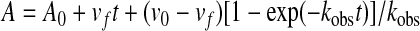 |
(Eq. 2) |
Here, A is absorbance, v0 (expressed in variation of absorbance per unit time) is initial velocity, vf is final velocity, and t is time. Each kobs was plotted versus I and fit to determine kinact and KI (27). The 24-h turnover number, tn (i.e. partitioning of the initial enzyme inhibitor complex between hydrolysis and enzyme inactivation, kcat/kinact, or partition ratio) was determined as reported previously (23, 26, 28).
Ultraviolet Difference (UVD) Spectroscopy—According to a previously published method, UV difference spectra (absorption spectra of LN-1-255 reacted with SHV-1 and SHV-2 β-lactamases in a 1000:1 inhibitor to enzyme (I:E) ratio) were measured from λ = 190–400 nm (29, 30).
Chelation Protocol—Five ml of 1 n HCl (Fisher HealthCare, Pittsburgh, PA) was used to activate 1 g of chelating resin (iminodiacetic acid) (Sigma) to remove ambient iron. HCl was then removed using 10 ml of 20 mm PBS (pH 7.4) after which 5 ml of 1 m LN-1-255 in 20 mm PBS was passed through the column. Flow-through was collected, and LN-1-255 recovery was measured based on inhibitory activity of SHV-1 versus LN-1-255, which was not passed through the column. Recovery was ∼60%. LN-1-255 passed through the chelating resin was then used for mass spectrometry analysis.
Electrospray Ionization (ESI) Mass Spectrometry (MS)—ESI-MS of the intact SHV-1 and SHV-2 β-lactamases inactivated by LN-1-255 was performed on an Applied Biosystems (Framingham, MA) Q-STAR XL quadrupole time-of-flight mass spectrometer equipped with a nanospray source as described previously (26). Experiments were performed by first desalting the reaction mixtures using C18 ZipTips (Millipore, Bedford MA) according to the manufacturer's protocol. The protein sample was then diluted with acetonitrile, 1% formic acid to a concentration of 10 μm. This protein solution was infused at a rate of 0.5 μl/min, and data were collected for 2 min. Spectra were deconvoluted using the Applied Biosystems Analyst program.
Crystallization and Data Collection—Crystals for x-ray diffraction analysis of SHV-1 β-lactamase were obtained as described previously (31, 32). One crystal was soaked for 24 h with 50 mm LN-1-255 in mother liquor containing CYMAL-6 detergent also present in the crystallization condition. The crystal was cryoprotected in 30% 2-methyl-2,4-pentandiol before immersing into liquid nitrogen for data collection. A 1.55 Å data set were collected at the National Synchrotron Light Source X-29 and processed using HKL2000 (33, 34). Refinement was carried out in CNS (crystallography and NMR system software) (35), and model building was done using COOT (36). After initial refinement and model fitting, strong density was observed for a bound bicyclic aromatic LN-1-255 intermediate covalently attached to Ser-70. Topology and parameter files for this LN-1-255 ligand intermediate were obtained using PRO-DRG2 for further refinement in CNS (37). Two conformations were modeled for the LN-1-255 atoms beyond the bicyclic aromatic system with occupancies of 0.6 and 0.4, respectively, as determined using B-factor refinement while varying their relative occupancies and monitoring difference density maps. The final model, refined to an R/Rfree of 16.8/19.7%, contains residues 26–292, 248 water molecules, one complete and one partial CYMAL-6 molecule, and one LN-1-255 intermediate of which a subset of the atoms are in two alternative conformations.
RESULTS
Antibiotic Susceptibility—We compared the in vitro efficacy of piperacillin/LN-1-255 with that of piperacillin/tazobactam (Table 1). Against the blaSHV-1 and blaSHV-2 in E. coli DH10B, susceptibility tests revealed that LN-1-255 was much more potent than an equal amount of tazobactam combined with piperacillin. In addition, we also demonstrated that LN-1-255 combined with piperacillin was very effective at lowering MICs against E. coli containing blaSHV-5 and a series of five different inhibitor-resistant blaSHV enzymes (blaSHV S130G, R244S, M69I, M69L, M69V). This broad effect is extremely notable because piperacillin/tazobactam does not lower MIC significantly when tested against E. coli strains containing inhibitor-resistant SHV β-lactamases.
TABLE 1.
Minimum inhibitory concentrations of Laboratory Isolates According to the Clinical Laboratory Standards Institute, MIC breakpoints for piperacillin and piperacillin/tazobactam are: ≤8 μg/ml, susceptible; 8–16 μg/ml, intermediate; ≥32 μg/ml, resistant.
| Laboratory Isolate | Piperacillin | Piperacillin/tazobactam (8:1 ratio) | Piperacillin/LN-1-255 (8:1 ratio) |
|---|---|---|---|
| μg/ml | μg/ml | μg/ml | |
| E. coli DH10B | 2 | 2/0.25 | 2/0.25 |
| blaSHV-1 | 1024 | 512/64 | 32/4 |
| blaSHV-2 | 512 | 32/4 | 16/2 |
| blaSHV-5 | 1024 | 32/4 | 16/2 |
| blaSHV-S130G | 128 | 128/16 | 64/8 |
| blaSHV R244S | 64 | 32/4 | 32/4 |
| blaSHV-M69I | 256 | 64/8 | 16/2 |
| blaSHV-M69L | 256 | 256/32 | 16/2 |
| blaSHV-M69V | 256 | 256/32 | 16/2 |
Further susceptibility testing was performed using LN-1-255 with ceftazidime, cefpirome, meropenem, and piperacillin. Against this selection of clinical isolates (E. coli and K. pneumoniae), which represents a Gram-negative bacteria harboring contemporary class A and D β-lactamase enzymes (e.g. TEM-10, SHV-5 and -7, OXA-2, and CTX-M-3 β-lactamases), LN-1-255 combined with ceftazidime and cefpirome markedly lowered the MICs (Table 2). Moreover LN-1-255 improved the activity of ceftazidime against E. coli and K. pneumoniae containing the class C β-lactamases CMY-6 and CMY-7. The overall effect seen here was substantial when compared with piperacillin alone and piperacillin/tazobactam. In contrast, improvement was not evident against AmpC cephalosporinase producing Enterobacter cloacae, Citrobacter freundii, and Serratia marcescens.
TABLE 2.
Minimum inhibitory concentrations of clinical isolates According to the Clinical Laboratory Standards Institute, MIC breakpoints for ceftazidime are: ≤8 μg/ml, susceptible; 8–16 μg/ml, intermediate; ≥32 μg/ml, resistant. For meropenem: ≤4 μg/ml, susceptible; 8 μg/ml, intermediate; ≥16 μg/ml, resistant. Standards for cefpirome are not available in the United States. We will regard MICs ≤ 8 μg/ml for cefpirome as susceptible, 8–16 μg/ml as intermediate, and ≥32 μg/ml as resistant.
| Clinical Isolates | Genotype | Ceftazidime | Ceftazidime + LN-1-255 at 4 μg/ml | Cefpirome | Cefpirome + LN-1-255 at 4 μg/ml | Meropenem | Meropenem + LN-1-255 at 4 μg/ml | Piperacillin | Piperacillin + tazobactam at 4 μg/ml |
|---|---|---|---|---|---|---|---|---|---|
| μg/ml | μg/ml | μg/ml | μg/ml | μg/ml | μg/ml | μg/ml | μg/ml | ||
| E. coli | TEM-1 | 0.12 | 0.12 | ≤0.06 | ≤0.06 | ≤0.06 | ≤0.06 | 128 | 2 |
| E. coli | TEM-10 | >128 | 1 | 2 | 0.12 | ≤0.06 | ≤0.06 | >128 | 16 |
| E coli | TEM-12 | 16 | 1 | 2 | 0.25 | ≤0.06 | ≤0.06 | >128 | 4 |
| E. coli | TEM-26 | >128 | 1 | 4 | 0.12 | ≤0.06 | ≤0.06 | >128 | 4 |
| E. coli | SHV-3 | 64 | 2 | 32 | 0.12 | ≤0.06 | ≤0.06 | >128 | >128 |
| E. coli | SHV-5 | >128 | 0.5 | 8 | 0.12 | ≤0.06 | ≤0.06 | >128 | 64 |
| K. pneumoniae | TEM-1, SHV-5 | >128 | 64 | 16 | 8 | ≤0.06 | ≤0.06 | >128 | >128 |
| K. pneumoniae | SHV-7, OXA-2 | 128 | ≤0.06 | 2 | ≤0.06 | ≤0.06 | ≤0.06 | >128 | 4 |
| K. pneumoniae | SHV-18 | 64 | 1 | 2 | 0.5 | ≤0.06 | ≤0.06 | >128 | 16 |
| K. pneumoniae | CTX-M-3 | 32 | 8 | 64 | 16 | ≤0.06 | ≤0.06 | >128 | 64 |
| K. pneumoniae | CTX-M-14 | 16 | 2 | 64 | 16 | ≤0.06 | ≤0.06 | >128 | 32 |
| K. pneumoniae | CTX-M-24 | 2 | 0.12 | 4 | ≤0.06 | ≤0.06 | ≤0.06 | 128 | 2 |
| E. cloacae | AmpC | 64 | 64 | 2 | 1 | ≤0.06 | ≤0.06 | >128 | 128 |
| C. freundii | AmpC | 64 | 64 | 2 | 2 | 0.12 | 0.12 | >128 | >128 |
| S. marcescens | AmpC | 128 | 64 | 1 | 0.12 | ≤0.06 | ≤0.06 | 16 | 8 |
| E. coli | CMY-2 | 128 | 64 | 4 | 2 | 0.12 | 0.12 | >128 | 64 |
| E. coli | CMY-6 | 128 | 2 | >128 | 0.5 | 0.12 | ≤0.06 | >128 | 64 |
| K. pneumoniae | CMY-7 | >128 | 4 | >128 | 32 | ≤0.06 | ≤0.06 | >128 | 128 |
| S. marcescens | Sme-1 | 0.25 | 0.25 | ≤0.06 | 0.12 | 32 | 1 | 32 | 2 |
The median MIC value for meropenem alone and in combination remained unchanged. It is noteworthy that LN-1-255 combined with meropenem readily inhibited bacteria possessing the class A carbapenemase Sme-1, produced by a strain of S. marcescens (MIC decreased from 32 to 1 μg/ml). This represents an important extension of the spectrum of LN-1-255.
Kinetic Parameters—SHV-1 and SHV-2 β-lactamases were purified to homogeneity by pIEF and size exclusion chromatography. Using increasing concentration of nitrocefin as an indicator substrate in a competition reaction with increasing amounts of inhibitor (I), we show that LN-1-255 inhibited SHV-1 and SHV-2 β-lactamases at lower concentrations than tazobactam: LN-1-255, KD = 0.019 ± 0.003 μm and LN-1-255, KD = 0.009 ± 0.002 μm, respectively (Table 3 and supplemental Fig. 1, a–d). The alternative determination of the dissociation constants for the preacylation complex, KD, followed a similar trend (Table 3).
TABLE 3.
Kinetic parameters of inhibition
| Tazobactam | LN-1-255 | |
|---|---|---|
| KDa(μm) | ||
| SHV-1 | 0.333 ± 0.033 | 0.027 ± 0.002 |
| SHV-2 | 0.182 ± 0.018 | 0.015 ± 0.002 |
| KDb(μm) | ||
| SHV-1 | 0.206 ± 0.019 | 0.019 ± 0.003 |
| SHV-2 | 0.041 ± 0.009 | 0.009 ± 0.002 |
| KI (μm) | ||
| SHV-1 | 0.200 ± 0.035 | 0.11 ± 0.01 |
| SHV-2 | 0.030 ± 0.003 | 0.10 ± 0.01 |
| kinact (s–1) | ||
| SHV-1 | 0.14 ± 0.01 | 0.55 ± 0.03 |
| SHV-2 | 0.030 ± 0.003 | 0.71 ± 0.07 |
| kinact/KI (s–1 μm–1) | ||
| SHV-1 | 0.70 ± 0.12 | 5.0 ± 0.7 |
| SHV-2 | 1.00 ± 0.14 | 7.1 ± 1.1 |
| tn | ||
| SHV-1 | 9 | 4 |
| SHV-2 | 9 | 0 |
KD measured using a direct competition reaction
KD measured using progress curves
Comparing kinact determinations, there was a highly significant difference between these two inhibitors. LN-1-255 inactivated SHV-1 and SHV-2 β-lactamases more rapidly than tazobactam (Table 3, supplemental Fig. 1, e–h). Although the KI values reveal that both tazobactam and LN-1-255 are potent at nm concentrations, the kinact/KI ratio explains why LN-1-255 inactivates SHV-1 and SHV-2 more “efficiently” than tazobactam in this test system. In determining the partition ratio, tn, of LN-1-255 for SHV-1 and for SHV-2, we see that there is nearly stoichiometric inactivation: LN-1-255 and SHV-1, kcat/kinact = 4; LN-1-255 and SHV-2, kcat/kinact = 0.
There is a dramatic “real time” inhibition effect, when adding the inhibitor LN-1-255, on the rate of hydrolysis of nitrocefin by SHV-2 β-lactamase: 7.5-fold less LN-1-255 inactivated SHV-2 more rapidly (note arrows in supplemental Fig. 2) when compared with tazobactam. In addition, there is no apparent reactivation.
UVD Spectroscopy—In previous work, UVD spectroscopy has been used to provide insight into the nature of reactive intermediates formed during the inactivation process (28, 30, 38). This approach was useful in detecting a major peak at 280 nm consistent with the formation of the β-aminoacrylate or enamine intermediate in our studies with clavulanate (38).
UVD spectroscopy measured every 12 s using LN-1-255 (I) reacted with SHV-1 or SHV-2 β-lactamase (E)atan I/E ratio of 1000/1 identified intense chromophores at 258 and 307 nm, which formed within 900 s and increased in intensity up to 1 h (Fig. 2, A and B, respectively). We tentatively assign the chromophore at 307 nm to a conjugated aromatic ring (see below).
FIGURE 2.

UVD spectroscopy of SHV-1 (A) and SHV-2 (B) reacted with LN-1-255. Note chromophore formation at 258 and 307 nm.
ESI-MS—To gain insight into the nature of the inactivation products, ESI-MS was performed with a Q-STAR quadrupole time-of-flight mass spectrometer equipped with a nanospray source. The m/z analyses of the SHV-1 and SHV-2 β-lactamases inactivation experiments are summarized in Table 4. The molecular mass of SHV-1 β-lactamase was determined as 28,871 ± 3 Da (Fig. 3A). The mass of SHV-2 was 28,901 ± 3 Da (Fig. 3B). ESI-MS measurements were in agreement with the theoretical masses of SHV-1 and SHV-2 β-lactamases.
TABLE 4.
Mass spectrometry analysis (atomic mass units)
| LN-1-255 | 488 |
| SHV-1 β-lactamase | 28,871 ± 3 |
| SHV-1 β-lactamase + LN-1-255 | 29,359 ± 3 |
| Δ Difference | 488 |
| LN-1-255 | 488 |
| SHV-2 β-lactamase | 28,901 ± 3 |
| SHV-2 β-lactamase + LN-1-255 | 29,389 ± 3 |
| Δ Difference | 488 |
FIGURE 3.

Deconvoluted mass spectrometry of SHV-1 and LN-1-255-inactivating SHV-1 (A) and SHV-2 and LN-1-255-inactivating SHV-2 (B).
Using the partition ratios (kcat/kinact) as a guide, we inactivated SHV-1 and SHV-2 β-lactamases with a 10-fold excess of LN-1-255 (900 s) in 20 mm PBS, pH 7.4, and removed any ambient iron by passage through a chelation column. The deconvoluted spectra for LN-1-255-inactivated SHV-1 and SHV-2 β-lactamases are presented in Fig. 3.
This analysis shows that there is covalent attachment of LN-1-255 to the SHV-1 and SHV-2 β-lactamases. In the time period studied, we did not find evidence of fragmentation of LN-1-255 as seen in the inactivation of TEM-1, CMY-2, SHV-1, R244S, and the S130G variant of SHV-1 with tazobactam and clavulanate (20, 29, 30, 39). Taken together, these mass spectrometry data indicate that there is formation of a stable inactivation product that corresponds to the addition of intact (unfragmented) LN-1-255 to SHV-1 and -2 β-lactamases.
X-ray Crystallographic Refinement and Structure Analysis—The initial unbiased omit electron density map in the active site of SHV-1 revealed strong density of a covalently bound LN-1-255 molecule (Fig. 4 and Table 5). The formation of an LN-1-255 intermediate containing a bicyclic aromatic ring system was evident from the density holes for both the 6- and 5-membered ring depending on the density contour levels. This ring system is pointing toward the solvent yet makes van der Waals interactions with Ala-237 and partially occludes the oxyanion hole. The carbonyl oxygen of this intermediate is therefore not located in the oxyanion hole and is involved in hydrogen bonds with the side chains of Lys-234 and Ser-130 (Fig. 5). The tail of the LN-1-255 intermediate displays weaker electron density and is refined in two alternative conformations. The major conformation of the LN-1-255 tail has its catechol moiety in close proximity to the hydrophobic ring of one of the CYMAL-6 molecules (Fig. 5). This conformation is the only conformation when SHV-1 is soaked with an analog of LN-1-255 that lacks the two hydroxyls of the catechol moiety (data not shown). The second (0.4 occupancy) conformation (Fig. 6) has its catechol moiety sandwiched in between the LN-1-255 bicyclic aromatic ring system and Tyr-105, but its density is also relatively weak.
FIGURE 4.

Electron density of LN-1-255 in the active site of SHV-1 β-lactamase. Unbiased omit Fo - Fc difference density of LN-1-255 covalently bound to Ser-70 contoured at 2.5δ (red) and 1.6δ (green) levels. The LN-1-255 ligand density shows clear density for the bicyclic aromatic double ring moiety, yet the atoms beyond this moiety are modeled in two conformations (gray and black/thin) as the density is more consistent with two conformations (occupancies of 0.6 and 0.4, respectively). The catechol moiety of both conformations display poor electron density, and their positions cannot be accurately modeled. The CYMAL-6 molecules (yellow) are also depicted.
TABLE 5.
Data collection and refinement statistics for SHV-1·LN-1-255 structure
| Data collection | |
| Space group | P212121 |
| Unit cell dimensions (Å) | 49.61, 55.50, 83.50, 90, 90, 90 |
| Wavelength (Å) | 1.1 |
| Resolution (Å) | 50-1.55 (1.61-1.55) |
| Redundancy | 5.3 |
| Data cut-off (σ) | –3.0 (default) |
| Unique reflections | 33,525 |
| 〈I〉/〈σ(I)〉 | 20.8 (2.1) |
| Rmerge (%) | 10.1 (41.0) |
| Completeness (%) | 97.8 (96.3) |
| Refinement | |
| Resolution range (Å) | 50–1.55 (1.57–1.55) |
| Atoms in asymmetric unit | 6,174 |
| R-factor (%) | 16.8 (28.1) |
| R-free (%) | 19.7 (30.7) |
| Root-mean-square deviations from ideality | |
| Bond lengths (Å) | 0.0094 |
| Angles (°) | 1.57 |
| Average temperature factors (Å2) | |
| Protein | 14.9 |
| CYMAL-6 molecules | 26.5 |
| LN-1-255 | 27.7 |
| Waters | 30.7 |
| Ramanchandran plot statistics | |
| Residues in most favored regions | 92.6% |
| Additional allowed regions | 7.4% |
| Generously allowed regions | 0% |
| Disallowed regions | 0% |
FIGURE 5.

Interactions of LN-1-255 in active site of SHV-1 β-lactamase. The bicyclic, aromatic, double ring system forms a planar ring, and its conjugated acyl group attached to Ser-70 is also found to be in the same plane. The carbonyl oxygen atom of this acyl moiety makes hydrogen bonds with Lys-234 and Ser-130. The remaining atoms of the LN-1-255 intermediate are modeled in two conformations (green and purple). Part of one of the CYMAL-6 molecules (yellow) is also depicted.
FIGURE 6.

Superposition of SHV-1 complexes with LN-1-255 and SA-2-13 bound. LN-1-255 is shown in green and purple for the two conformations, SA-2-13 is depicted in black, and the SHV-1 protein atoms are in gray and black (thin lines), respectively. The superposition reveals that binding of LN-1-255 to SHV-1 affects the active site in several positions compared with SA-2-13. These shifts (indicated by red arrows) include the side chain of Ser-130 and the loop region near Val-216, which shifts away to accommodate LN-1-255 binding. In addition, a minor difference is that LN-1-255 keeps residue Asn-170 in the wild-type conformation, whereas SA-2-13 forces Asn-170 to be in two conformations with the second being an outward conformation. Finally, the side chain of Tyr-105 has also somewhat shifted.
DISCUSSION
Major developments in the 6-alkylidenepenam sulfone structural series of β-lactamase inhibitors are shown in Fig. 7. The first example was the 6-benzylidenepenicillin sulfone (Fig. 7A) reported by Foulds et al. (40). This was rapidly followed by the work of Chen et al. (41) demonstrating the superiority of the 6-pyridylmethylidene analog (Fig. 7B). These authors proposed a general inhibitory mechanism (shown in Fig. 8) based on a methanolysis of the inhibitor and isolation of the corresponding methyl ester. Buynak et al. (18) independently varied both the C-6 and C-2 substituents, again demonstrating that the optimal inhibitory substituent at C-6 was the pyridylmethylidene. In addition, they also showed that substantially improved inhibitory activity could be obtained with selected C-2 substituents (Fig. 7C), such as r = O2CCH2Ph. In addition, in vitro activity could be substantially improved by the catecholic functionality of LN-1-255 (18).
FIGURE 7.

Evolution of the 6-alkylidenepenam sulfone structural series of β-lactamase inhibitors. A, 6-benzylidene penicillin sulfone. B, 6-pyridylonethylidene analog. C, addition of C-2 substituent.
FIGURE 8.

Proposed mechanism of enzymatic inhibition by the 6-(pyridylmethylidene)penicillin sulfone series of inhibitors.
Current commercial β-lactamase inhibitors narrowly target the class A enzymes. A clinically useful new β-lactamase inhibitor must have a significantly broader inhibitory spectrum, presumably targeting all serine β-lactamases (classes A, C, and D). To achieve clinical efficacy, the inhibitor should meet specific molecular, microbiological, and physiological requirements. This new inhibitor will first need to be selectively recognized by the bacterial enzyme (i.e. have a high affinity for the active site) and, secondly, form a highly stabilized acyl enzyme. Moreover, the inhibitor will need to demonstrate synergy with an appropriately selected β-lactam antibiotic, thereby significantly lowering MIC values. For Gram-negative microorganisms this will involve (i) rapid penetration (of both inhibitor and antibiotic) through the outer membrane and (ii) resistance to efflux. Lastly, the inhibitor/antibiotic combination will need to display appropriate pharmacokinetic and pharmacodynamic characteristics.
LN-1-255 clearly meets several of the criteria for an effective β-lactamase inhibitor. This compounds satisfies the following standards: (i) efficacy by MIC testing against SHV-1, inhibitor-resistant SHV variants, and ESBLs produced in laboratory and clinical strains; (ii) affinity for the active site of SHV-1 and ESBL variant enzymes; and (iii) stability of the resultant acyl enzyme. It is also clear from kinetics, mass spectrometry, and x-ray crystallography that the dynamics and details of the inactivation process are unique when compared with commercially available inhibitors such as tazobactam.
This work represents the first crystallographic confirmation of Chen's proposed inhibitory mechanism in the penicillin series (41), the individual steps of which are shown in Fig. 8. Regarding the effect of the 2′-substituent, Buynak et al. (18) initially observed a 10–100-fold improvement in the IC50 values of specific 2′-substituted 6-alkylidenepencillins sulfones against representative class A and C enzymes, particularly when utilizing a phenylacetoxy substituent at C-2′. Catecholic analogs were designed next (e.g. LN-1-255) to simultaneously take advantage of this improvement in activity and to facilitate passage through the outer membrane via the iron uptake pathway. Recent data indicate that the improved enzymatic inhibitory activity may be due to tight binding in the initial Michaelis complex (27). As in the case of sulbactam, once the acyl enzyme is formed, the now more highly basic amine nitrogen electrons facilitate the departure of the sulfinate sulfur leading to fragmentation of the dioxothizolidine system and formation of an intermediate imine. Although in the case of sulbactam this imine would either tautomerize to the β-aminoacrylate (enamine) or serve as an electrophile to capture a second nucleophile (e.g. Ser-130), in the case of LN-1-255, the favorable disposition of the basic pyridyl nitrogen atom allows rapid intramolecular capture. Loss of a proton from C-5 results in the formation of the bicyclic 10 π electron aromatic indolizine sytem. The acyl enzyme ester carbonyl is now stabilized not only by the adjacent aromatic system but also by favorable resonance interactions with both nitrogens. Presumably the bulk and planar geometry demanded by the favorable π interactions in this bicylic system result in movement to a different location in the active site, thereby removing the carbonyl oxygen from the oxyanion hole. Additionally, we observed favorable interactions with Tyr-105 in SHV (see below).
Why does the combination of piperacillin and LN-1-255 lower MICs more effectively as compared with piperacillin/tazobactam? The addition of the dihydroxy phenyl catechol moiety may provide LN-1-255 with enhanced bacterial uptake via siderophore channels. This pathway is used by bacteria to incorporate iron that is complexed with bacterially synthesized siderophores (42). This feature can be exploited to improve drug uptake (14, 15). Secondly, the addition of the dihydroxy phenyl catechol moiety may improve affinity, because this moiety has a large hydrophobic side and can stack onto aromatic protein residues that enhance binding by making additional hydrophobic and/or van der Waals interactions.
The impact of the cathecol moiety is further highlighted when LN-1-255 is tested against clinical isolates. LN-1-255 enhances the efficacy of ceftazidime against isolates of E. coli containing TEM-1, TEM-10, TEM-12, and TEM-26 β-lactamases (the latter three are clinically important ESBLs) (Table 2) as well as isolates of K. pneumoniae containing SHV ESBLs, OXA-2, and CTX-M ESBLs. This dramatic increase effectively expands the spectrum of the partner cephem to equal that of a “fourth generation” cephalosporin (cefpirome). When combined with cefpirome, LN-1-255 effectively expands the activity to approximately equal that of meropenem.
The observed bicyclic aromatic LN-1-255 intermediate is remarkably stable as demonstrated by mass spectrometry, UV difference absorption spectroscopy, and x-ray crystallography (Figs. 2, 3, 4, 5, 6). Two unique molecular features of this intermediate that are revealed by crystallography and explain the stability to deacylation. Of primary importance, the aromatic bicyclic moiety and the carbonyl oxygen are part of a large conjugated π system that stabilizes the acyl enzyme complex against nucleophilic attack by the activated water molecule in the active site. Additionally, the carbonyl oxygen of the intermediate is pushed out of the oxyanion hole; this is likely a result of this C=O moiety needing to be planar with the bicyclic aromatic ring system. A reoriented C=O group away from the oxyanion hole decreases the rate of deacylation for two reasons: (a) this moiety is no longer primed, via the backbone NH hydrogen bonds in the oxyanion hole, to be receptive for nucleophilic attack by a water; and (b) the carbonyl carbon atom position is now positioned ∼1.7 Å further away from the deacylation water, a distance likely too great for efficient deacylation by a water molecule. Note that the carbonyl oxygen does not necessarily need to be positioned outside the oxyanion hole to slow down deacylation as observed for a cephem sulfone compound similar to LN-1-255 yielding a bicyclic aromatic intermediate with class C GC-1 β-lactamase (43). An additional difference between the LN-1-255 SHV-1 structure and the cephem sulfone class C structure is that the former intermediate is refined, having a planar bicyclic aromatic ring, in accord with the unbiased omit density (Fig. 4), whereas this aromatic group in the latter compound was not refined in a planar conformation.
It is important to communicate that the crystal structure of LN-1-255 covalently bound to SHV-1 provides inhibitory information, yet does not contribute insights into the initial (Henri-Michaelis) binding complex that likely determines the KI. The complex obtained is that of a late-stage reaction intermediate after substantial molecular rearrangements have occurred (Figs. 4, 5, 6).
Lastly, we emphasize that the binding of LN-1-255 also results in changes in the active site of SHV-1. To illustrate this, we superimposed SHV-1-bound LN-1-255 with our previously determined structure of SHV-1-bound SA-2-13 (32) (Fig. 6). This SA-2-13 bound structure is a high resolution 1.28 Å structure, and SA-2-13 causes little active site changes except for forcing residue Asn-170 into two conformations (one inward wild-type conformation priming the deacylation water and an outward conformation). The superposition reveals that LN-1-255 bound to SHV-1 produces two significant active site shifts. The first is a shift in the Ser-130 side chain position to accommodate the carbonyl oxygen of LN-1-255, which would otherwise yield a ∼2.2 Å steric clash. Second, the (larger) LN-1-255 inhibitor pushes the loop region Val-216 outward to accommodate the bicyclic aromatic double ring system (Fig. 6). These LN-1-255-induced active site shifts are intriguing and could possibly affect the rate at which the intermediate is formed (kinact) and also the rate at which it is deacylated (kcat). In the scheme shown in Fig. 9, we have illustrated the major differences between tazobactam and LN-1-255.
FIGURE 9.

A schematic model of the kinetic differences between LN-1-255 and tazobactam.
In summary, LN-1-255 possesses key features that permit increased cell entry, enhance affinity and acylation efficiency for the active site, and slow deacylation. More widespread MIC testing of LN-1-255 against clinical strains will be required; the affinity of LN-1-255 against other β-lactamases must be studied further. Nevertheless, the discovery of LN-1-255 is an important “first step” toward defining characteristics for rapid cell penetration and optimal structure-activity relationships in the quest for novel and versatile β-lactamase inhibitors.
Supplementary Material
Acknowledgments
We thank Dr. Marion Skalweit Helfand for helpful comments.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 AI062968 (to F. V. D. A.), RO1 AI063517-01 (to R. A. B.), and the Robert A. Welch Foundation (J. D. B.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2.
Footnotes
The abbreviations used are: ESBL, extended spectrum β-lactamase; MIC, minimum inhibitory concentration; pIEF, preparative isoelectric focusing; UVD, ultraviolet difference; ESI-MS, electrospray ionization mass spectrometry; SHV, sulfhydryl reagent variable.
References
- 1.Paterson, D. L., and Bonomo, R. A. (2005) Clin. Microbiol. Rev. 18 657-686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paterson, D. L., Ko, W. C., Von Gottberg, A., Mohapatra, S., Casellas, J. M., Goossens, H., Mulazimoglu, L., Trenholme, G., Klugman, K. P., Bonomo, R. A., Rice, L. B., Wagener, M. M., McCormack, J. G., and Yu, V. L. (2004) Clin. Infect. Dis. 39 31-37 [DOI] [PubMed] [Google Scholar]
- 3.Rodriguez-Bano, J., and Navarro, M. D. (2008) Clin. Microbiol. Infect. 14 Suppl. 1, 104-110 [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez-Bano, J., Navarro, M. D., Romero, L., Muniain, M. A., de Cueto, M., Rios, M. J., Hernandez, J. R., and Pascual, A. (2006) Clin. Infect. Dis. 43 1407-1414 [DOI] [PubMed] [Google Scholar]
- 5.Bradford, P. A. (2001) Clin. Microbiol. Rev. 14 933-951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kotapati, S., Kuti, J. L., Nightingale, C. H., and Nicolau, D. P. (2005) J. Infect. Dis. 51 211-217 [DOI] [PubMed] [Google Scholar]
- 7.Paterson, D. L., Ko, W. C., Von Gottberg, A., Mohapatra, S., Casellas, J. M., Goossens, H., Mulazimoglu, L., Trenholme, G., Klugman, K. P., Bonomo, R. A., Rice, L. B., Wagener, M. M., McCormack, J. G., and Yu, V. L. (2004) Ann. Intern. Med. 140 26-32 [DOI] [PubMed] [Google Scholar]
- 8.Pitout, J. D., Gregson, D. B., Church, D. L., Elsayed, S., and Laupland, K. B. (2005) J. Clin. Microbiol. 43 2844-2849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pitout, J. D., Nordmann, P., Laupland, K. B., and Poirel, L. (2005) J. Antimicrob. Chemother. 56 52-59 [DOI] [PubMed] [Google Scholar]
- 10.Calbo, E., Romani, V., Xercavins, M., Gomez, L., Vidal, C. G., Quintana, S., Vila, J., and Garau, J. (2006) J. Antimicrob. Chemother. 57 780-783 [DOI] [PubMed] [Google Scholar]
- 11.Helfand, M. S., and Bonomo, R. A. (2006) Clin. Infect. Dis. 43 1415-1416 [DOI] [PubMed] [Google Scholar]
- 12.Bush, K., Jacoby, G. A., and Medeiros, A. A. (1995) Antimicrob. Agents Chemother. 39 1211-1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacoby, G. A., and Munoz-Price, L. S. (2005) N. Engl. J. Med. 352 380-391 [DOI] [PubMed] [Google Scholar]
- 14.Beharry, Z., Chen, H., Gadhachanda, V. R., Buynak, J. D., and Palzkill, T. (2004) Biochem. Biophys. Res. Commun. 313 541-545 [DOI] [PubMed] [Google Scholar]
- 15.Buynak, J. D. (2004) Curr. Med Chem. 11 1951-1964 [DOI] [PubMed] [Google Scholar]
- 16.Buynak, J. D., Doppalapudi, V. R., and Adam, G. (2000) Bioorg. Med. Chem. Lett. 10 853-857 [DOI] [PubMed] [Google Scholar]
- 17.Buynak, J. D., Doppalapudi, V. R., Rao, A. S., Nidamarthy, S. D., and Adam, G. (2000) Bioorg. Med. Chem. Lett. 10 847-851 [DOI] [PubMed] [Google Scholar]
- 18.Buynak, J. D., Rao, A. S., Doppalapudi, V. R., Adam, G., Petersen, P. J., and Nidamarthy, S. D. (1999) Bioorg. Med. Chem. Lett. 9 1997-2002 [DOI] [PubMed] [Google Scholar]
- 19.Rice, L. B., Carias, L. L., Hujer, A. M., Bonafede, M., Hutton, R., Hoyen, C., and Bonomo, R. A. (2000) Antimicrob. Agents Chemother. 44 362-367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bethel, C. R., Hujer, A. M., Hujer, K. M., Thomson, J. M., Ruszczycky, M. W., Anderson, V. E., Pusztai-Carey, M., Taracila, M., Helfand, M. S., and Bonomo, R. A. (2006) Antimicrob. Agents Chemother. 50 4124-4131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hujer, A. M., Hujer, K. M., and Bonomo, R. A. (2001) Biochim. Biophys. Acta 1547 37-50 [DOI] [PubMed] [Google Scholar]
- 22.Hujer, A. M., Hujer, K. M., Helfand, M. S., Anderson, V. E., and Bonomo, R. A. (2002) Antimicrob. Agents Chemother. 46 3971-3977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helfand, M. S., Bethel, C. R., Hujer, A. M., Hujer, K. M., Anderson, V. E., and Bonomo, R. A. (2003) J. Biol. Chem. 278 52724-52729 [DOI] [PubMed] [Google Scholar]
- 24.Swaren, P., Golemi, D., Cabantous, S., Bulychev, A., Maveyraud, L., Mobashery, S., and Samama, J. P. (1999) Biochemistry 38 9570-9576 [DOI] [PubMed] [Google Scholar]
- 25.Thomson, J. M., Distler, A. M., and Bonomo, R. A. (2007) Biochemistry 46 11361-11368 [DOI] [PubMed] [Google Scholar]
- 26.Thomson, J. M., Distler, A. M., Prati, F., and Bonomo, R. A. (2006) J. Biol. Chem. 281 26734-26744 [DOI] [PubMed] [Google Scholar]
- 27.Kalp, M., Sheri, A., Buynak, J. D., Bethel, C. R., Bonomo, R. A., and Carey, P. R. (2007) J. Biol. Chem. 282 21588-21591 [DOI] [PubMed] [Google Scholar]
- 28.Bush, K., Macalintal, C., Rasmussen, B. A., Lee, V. J., and Yang, Y. (1993) Antimicrob. Agents Chemother. 37 851-858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pagan-Rodriguez, D., Zhou, X., Simmons, R., Bethel, C. R., Hujer, A. M., Helfand, M. S., Jin, Z., Guo, B., Anderson, V. E., Ng, L. M., and Bonomo, R. A. (2004) J. Biol. Chem. 279 19494-19501 [DOI] [PubMed] [Google Scholar]
- 30.Sulton, D., Pagan-Rodriguez, D., Zhou, X., Liu, Y., Hujer, A. M., Bethel, C. R., Helfand, M. S., Thomson, J. M., Anderson, V. E., Buynak, J. D., Ng, L. M., and Bonomo, R. A. (2005) J. Biol. Chem. 280 35528-35536 [DOI] [PubMed] [Google Scholar]
- 31.Kuzin, A. P., Nukaga, M., Nukaga, Y., Hujer, A., Bonomo, R. A., and Knox, J. R. (2001) Biochemistry 40 1861-1866 [DOI] [PubMed] [Google Scholar]
- 32.Padayatti, P. S., Sheri, A., Totir, M. A., Helfand, M. S., Carey, M. P., Anderson, V. E., Carey, P. R., Bethel, C. R., Bonomo, R. A., Buynak, J. D., and van den Akker, F. (2006) J. Am. Chem. Soc. 128 13235-13242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Minor, W., Tomchick, D., and Otwinowski, Z. (2000) Structure (Lond.) 8 R105-R110 [DOI] [PubMed] [Google Scholar]
- 34.Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276 307-326 [DOI] [PubMed] [Google Scholar]
- 35.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. Sect. D Biol. Crystallogr. 54 905-921 [DOI] [PubMed] [Google Scholar]
- 36.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 37.Schüttelkopf, A. W., and van Aalten, D. M. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 1355-1363 [DOI] [PubMed] [Google Scholar]
- 38.Charnas, R. L., Fisher, J., and Knowles, J. R. (1978) Biochemistry 17 2185-2189 [DOI] [PubMed] [Google Scholar]
- 39.Bonomo, R. A., Liu, J., Chen, Y., Ng, L., Hujer, A. M., and Anderson, V. E. (2001) Biochim. Biophys. Acta 1547 196-205 [DOI] [PubMed] [Google Scholar]
- 40.Foulds, C. D., Kosmirak, M., and Sammes, P. G. (1985) J. Chem. Soc. Perkin Trans. I 964-968
- 41.Chen, Y. L., Chang, C. W., and Hedberg, K. (1986) Tetrahedron Lett. 27 3449-3452 [Google Scholar]
- 42.Chakraborty, R., Storey, E., and van der Helm, D. (2007) Biometals 20 263-274 [DOI] [PubMed] [Google Scholar]
- 43.Crichlow, G. V., Nukaga, M., Doppalapudi, V. R., Buynak, J. D., and Knox, J. R. (2001) Biochemistry 40 6233-6239 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.