

Abstract
Penicillin-binding protein 2 (PBP2) from N. gonorrhoeae is the major molecular target for β-lactam antibiotics used to treat gonococcal infections. PBP2 from penicillin-resistant strains of N. gonorrhoeae harbors an aspartate insertion after position 345 (Asp-345a) and 4-8 additional mutations, but how these alter the architecture of the protein is unknown. We have determined the crystal structure of PBP2 derived from the penicillin-susceptible strain FA19, which shows that the likely effect of Asp-345a is to alter a hydrogen-bonding network involving Asp-346 and the SXN triad at the active site. We have also solved the crystal structure of PBP2 derived from the penicillin-resistant strain FA6140 that contains four mutations near the C terminus of the protein. Although these mutations lower the second order rate of acylation for penicillin by 5-fold relative to wild type, comparison of the two structures shows only minor structural differences, with the positions of the conserved residues in the active site essentially the same in both. Kinetic analyses indicate that two mutations, P551S and F504L, are mainly responsible for the decrease in acylation rate. Melting curves show that the four mutations lower the thermal stability of the enzyme. Overall, these data suggest that the molecular mechanism underlying antibiotic resistance contributed by the four mutations is subtle and involves a small but measurable disordering of residues in the active site region that either restricts the binding of antibiotic or impedes conformational changes that are required for acylation by β-lactam antibiotics.
Neisseria gonorrhoeae, the causative agent of the sexually transmitted infection, gonorrhea, is an obligate human pathogen that primarily colonizes the urogenital tract. For nearly 40 years N. gonorrhoeae was treated with a single dose of penicillin, but in 1987 the prevalence of penicillin-resistant strains necessitated the use of alternative antibiotics (1), primarily fluoroquinolones and third-generation cephalosporins. The rapid emergence and dissemination of fluoroquinolone-resistant N. gonorrhoeae, however, leaves only third-generation cephalosporins (e.g. cefixime and ceftriaxone) as recommended agents (1). Even though these cephalosporins are still effective anti-gonococcal antibiotics, the increasing prevalence of strains with intermediate resistance to these agents suggests that full resistance is not far behind (2-5). These trends signal an urgent need to understand the molecular mechanisms of antibiotic resistance and to devise new treatment options for gonococcal infections.
The molecular targets of β-lactam antibiotics are the penicillin-binding proteins (PBPs),3 a group of enzymes involved in the final stages of peptidoglycan synthesis in bacteria (6-8). PBPs are grouped into three main classes, A, B, and C. Class A and B PBPs are transpeptidases (TPases) that catalyze the formation of peptide cross-links between adjacent glycan strands of peptidoglycan, but are distinguished in that class A PBPs also contain an N-terminal transglycosylase domain that polymerizes glycan strands, whereas class B PBPs possess only TPase activity. Class C PBPs catalyze carboxypeptidase activity (and sometimes endopeptidase activity) in vitro and may function to modulate the degree of cross-linking in peptidoglycan by removing the terminal d-Ala of the peptide.
Four PBPs have been identified in N. gonorrhoeae (9, 10): two high molecular mass transpeptidases, class A PBP1 and class B PBP2, and two low molecular mass class C PBPs, PBPs 3 and 4. Both PBP3 and PBP4, which catalyze carboxypeptidase and endopeptidase activity in vitro (10, 11), can be deleted from N. gonorrhoeae with only minor effects on cell growth and morphology (11), whereas PBPs 1 and 2 are essential for cell viability and are the lethal targets of β-lactam antibiotics (9, 12). Because PBP2 is inhibited at 10-fold lower concentrations of penicillin than PBP1 in penicillin-susceptible strains, PBP2 is the primary killing target of penicillin at its minimum inhibitory concentration (MIC) (9).
Chromosomally mediated penicillin-resistant strains of N. gonorrhoeae arise through the stepwise acquisition of multiple resistance determinants via DNA uptake and homologous recombination (13, 14). These resistance determinants include mutations of PBPs 1 and 2 that lower their susceptibility to β-lactam antibiotics (penA and ponA) (15, 16), mutations in porin IB that reduce entry of antibiotics into the cell (penB) (17, 18), and increased expression of the MtrC-MtrD-MtrE efflux pump that expels antibiotics from the periplasm (mtrR) (19, 20). Together, the mutations in high level penicillin-resistant strains (e.g. FA6140) increase the MIC of penicillin 400-1000-fold relative to a penicillin-susceptible strain (e.g. FA19).
The first step in the development of chromosomally mediated penicillin resistance in N. gonorrhoeae is acquisition of penA, which encodes altered forms of PBP2 (15, 21). Although by itself the penA determinant accounts for a relatively small contribution to penicillin resistance in N. gonorrhoeae, this first step is requisite for the development of high level resistance. Variants of PBP2 from penicillin-resistant strains very often contain an aspartic acid insertion between amino acids 345 and 346 (termed Asp-345a or D-345a) along with 4-8 substitutions near the C terminus of the protein (22, 23). Because PBP2 is an essential enzyme, these mutations must remodel the active site so as to lower the rate of acylation by penicillin without markedly affecting its ability to function as a TPase. This is an intriguing phenomenon because β-lactam antibiotics are structural mimics of the peptide substrate (24) and react with the same serine nucleophile at the active site of the enzyme (25). How PBPs achieve this distinction at the molecular level remains a subject of considerable interest (26-31).
Crystal structures of PBP2x and PBP1a from Streptococcus pneumoniae (in both penicillin-sensitive and penicillin-resistant forms) (27, 31-34) and of PBP2a from methicillin-resistant Staphylococcus aureus (28) have been used to propose mechanisms of penicillin resistance in their respective organisms. However, mechanistic interpretation has been hampered in PBP2x and PBP1a by the large number of mutations between the susceptible and resistant forms (e.g. PBP2x from strain Sp328 contains 83 mutations (27)) and in PBP2a by the lack of a penicillin-susceptible counterpart. PBP2 from N. gonorrhoeae is a more amenable system to understand the molecular basis for penicillin resistance because only a small number of mutations are responsible for conferring antibiotic resistance.
Here, we report the crystal structure of a soluble form of PBP2 derived from a penicillin-susceptible strain (FA19) (35) and of a construct derived from a penicillin-resistant strain (FA6140) (14) of N. gonorrhoeae that contains four mutations near the C terminus of the protein that are associated with penicillin resistance. Even though the kinetic differences between these proteins are significant, there is a surprising lack of structural differences between the two proteins, suggesting a very subtle molecular mechanism of antibiotic resistance. Although a mutant protein containing Asp-345a could not be crystallized, the crystal structure of the native protein suggests that such an insertion will disrupt interactions involving the SXN motif of the enzyme.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification—The sequence encoding the periplasmic domain of PBP2 (residues 44-581) and a four-amino acid spacer (Gly-Ser-Gly-Gly) was fused in-frame to the C-terminal end of maltose-binding protein in the vector pMAL-C2KV/H6 (11). pMAL-C2KV/H6 is similar to pMAL-C2 (New England Biolabs, Beverly, MA) except that it contains the kanamycin resistance gene in place of the β-lactamase gene, a tobacco etch virus protease cleavage site at the junction of the two proteins, and six histidine residues between the second and third amino acids of maltose-binding protein.
The maltose-binding protein-PBP2 fusion protein was purified from 2 liters of Escherichia coli GW6011 (MC4100 lon-146::Tn10) cells harboring the expression plasmid after induction of the cells with 0.3 mm isopropyl-β-d-thiogalactoside for 2 h at 18 °C. Cells were harvested by centrifugation, resuspended in 40 ml of buffer A (20 mm Tris, pH 8.0, 500 mm NaCl, 10% glycerol) containing 2 mm EDTA and 1 mm phenylmethylsulfonyl fluoride, and lysed with an Emulsiflex-C5 disrupter (Avestin, Ottawa, Canada). After centrifugation at 100,000 × g to remove membranes and particulates, ammonium sulfate was added to 40% saturation, and precipitated proteins were isolated by centrifugation at 15,000 × g. The protein pellet was resuspended in buffer A, filtered, and loaded onto a Ni2+-chelating affinity column (GE Healthcare). The column was washed sequentially with 20 column volumes of buffer A and 5 column volumes of buffer A with 15 mm imidazole, and the fusion protein was eluted with a gradient of 15-300 mm imidazole in buffer A. The purified fusion protein was dialyzed at 4 °C for 3 h against buffer A to remove imidazole and simultaneously cleaved with His6-tagged tobacco etch virus protease (50:1 weight ratio). PBP2 was isolated from the digest by a second passage over Ni2+-chelating affinity column. Minor contaminants eluted in the flow-through, whereas PBP2 (>95% purity) eluted in the 15 mm imidazole wash. The purified protein was concentrated to 5-7 mg/ml in buffer A (typical yields were 30 mg protein/2 liters of culture).
Selenomethionine-substituted PBP2 was prepared by expressing the protein in B834(DE3) methionine auxotroph cells (Novagen, San Diego, CA) and growing in M9 minimal media containing all amino acids except that selenomethionine was used in place of methionine. This protein was purified in the same way as for PBP2 except that 4 mm β-mercaptoethanol was added to all buffers.
PBP2 from FA6140 was amplified from genomic DNA with the same gene-specific primers as for wild-type penA and cloned into pMAL-C2KVH6. To create PBP2-6140CT and PBP2-Asp-345a, a silent KpnI site was introduced at codons 366 and 367 in both wild type PBP2 and PBP2-6140 by the QuikChange method (Stratagene, Carlsbad, CA), and the final constructs were created by fragment exchange. All other mutants were created in wild-type PBP2 by the QuikChange method. Purification of these mutant PBP2 variants was accomplished using the same protocol described above for wild-type PBP2.
Kinetic Analysis of PBP2 Variants—The kinetic scheme for the interaction of a PBP with a β-lactam antibiotic is,
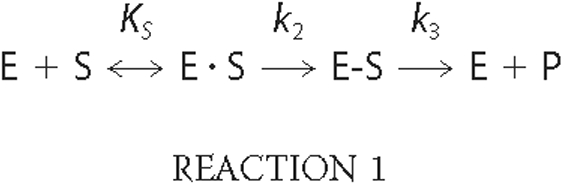
where E·S is the non-covalent Michaelis complex, E-S is the covalent acyl-enzyme complex, and P is the released product. The constant k2/KS, in which KS = (k-1 + k2)/k1 describes the initial rate of formation of covalent β-lactam antibiotic-target conjugates (E-S) at low (subsaturating) concentrations of β-lactam antibiotics (36). The deacylation rate of the penicilloyl-PBP2 complex is very slow and can be ignored. The second order k2/KS rate constants for acylation of PBP2 variants were determined from time courses of formation of acyl-enzyme complexes as described previously (11). In all cases, [14C]penicillin G was present in at least a 10-fold molar excess over PBP2.
Crystallization—Crystals of PBP2 and PBP2-6140CT were obtained by vapor diffusion over wells containing 2.0-2.4 m ammonium sulfate buffered with 100 mm Tris, pH 8.0-8.5. These rectangular-shaped prisms of approximate dimensions 0.5 × 0.1 × 0.3 mm grew after 2-3 weeks of incubation at 21 °C. A cryo-protecting solution was prepared by adding glycerol to the well solution in 2% increments up to a final concentration of 34%. Diffraction analysis revealed that the crystals belong to space group P212121 with cell dimensions a = 57.4, b = 138.6, and c = 228.0 Å. There are two molecules in the asymmetric unit, giving an estimated solvent content of 65% (37). Selenomethionine-containing crystals were prepared in the same manner.
Data Collection and Phasing—Data from selenomethionine-containing crystals were collected at 100 K at beamline 9.2 of the Stanford Synchrotron Radiation Laboratory (Stanford, CA) on a Quantum 4 charge-coupled device detector using the BLU-ICE environment (38). Data at three wavelengths were collected, corresponding to the peak, inflection, and a high energy remote, around the absorption edge of selenium using inverse beam geometry in two 90° segments. These data were processed at a resolution of 2.8 Å using DENZO and SCALEPACK (39) (supplemental Table 1). The structure was solved by SOLVE (40) and RESOLVE (41, 42) using only the first 90° of the peak and remote data sets. Inclusion of the second wedge of these data or of the inflection data set lowered the quality of electron density maps, probably due to radiation damage. Selenomethionine-substituted crystals of PBP2 diffracted slightly better than native crystals, and therefore these crystals were also used to collect a high resolution data set for refinement of the structure. These data were processed using MOSFLM (43) and SCALA (44) (Table 1). Data from crystals of PBP2-6140CT were collected at the SER-CAT ID22 beamline of the Advanced Photon Source on a Mar 300 charge-coupled device detector and processed using HKL2000 (39) (Table 1).
TABLE 1.
X-ray diffraction data and refinement statistics
| PBP 2 wild type | PBP 2-6140CT | |
|---|---|---|
| Data collection | ||
| Resolution range (Å) | 87.0-2.30 (2.36-2.30) | 50.0-2.4 (2.49-2.40) |
| Rmergea (%) | 6.1 (34.9) | 10.1 (52.0) |
| Redundancy | 6.2 (3.7) | 7.1 (4.3) |
| Completeness (%) | 90.3 (52.6) | 98.0 (83.5) |
| <I>/<σI> | 8.8 (2.0) | 29.9 (2.7) |
| Refinement | ||
| Resolution range (Å) | 66.6-2.40 | 48.1-2.40 |
| Completeness in range (%) | 94.6 (70.0) | 97.7 (80.4) |
| Total number of reflections | 68,535 | 72,265 |
| Number of non-hydrogen protein atoms | 6,522 | 6,639 |
| Number of sulfate molecules | 17 | 15 |
| Number of glycerol molecules | 2 | 2 |
| Number of water molecules | 146 | 86 |
| Rfactor (%) | 21.6 | 21.9 |
| Rfree (%) | 23.7 | 25.3 |
| r.m.s.d. from ideal stereochemistry | ||
| Bond lengths (Å) | 0.012 | 0.011 |
| Bond angles (°) | 1.45 | 1.39 |
| B factors | ||
| Mean B factor (main chain) (Å3) | 35.4 | 48.1 |
| r.m.s.d. in main chain B factor (Å3) | 0.48 | 0.44 |
| Mean B factor (side chains and waters) (Å3) | 38.1 | 49.4 |
| r.m.s.d. in side chain B factors (Å3) | 1.42 | 1.18 |
| Ramachandran plot | ||
| Residues in most favored region (%) | 93.0 | 91.7 |
| Residues in additionally allowed regions (%) | 7.0 | 8.3 |
| Residues in generously allowed regions (%) | 0.0 | 0.0 |
| residues in disallowed regions (%) | 0.0 | 0.0 |
| PDB code | 3EQU | 3EQV |
Rmerge = ∑|Ii - Im|/∑Ii, where Ii is the intensity of the measured reflection, and Im is the mean intensity for all observations of that reflection. Numbers within parentheses are for the outer resolution shell of data.
Model Building and Refinement—Electron density maps were displayed using O (45) and used to construct an initial model. This was refined by iterative cycles of automated refinement and manual revisions using O. Initial refinements used XPLOR (46) but later rounds used REFMAC (47). Non-crystallographic symmetry restraints were not used during the refinement. 5% of reflections were set aside for the calculation of Rfree. Solvent molecules were added using ARP/wARP (48). In later rounds TLS parameters were refined, where each domain of the protein was defined as a separate TLS group. The stereochemistry of the model was monitored by PROCHECK (49). The structure of PBP2-6140CT was refined by the same protocol. Refinement statistics of both models are shown in Table 1.
There are two molecules in the asymmetric unit of PBP2, and all common main chain atoms can be superimposed with a root mean square deviation (r.m.s.d.) of 0.70 Å (the same value is 0.75 Å for PBP2-6140CT). The protein is numbered according to the sequence of the protein in vivo. Hence, the first 44 residues, corresponding to the N-terminal tail and transmembrane domains, are not present in the construct. Residues that were not visible in the electron density were excluded from the model. In both molecules of the asymmetric unit, ∼20 residues are absent at the N terminus, such that molecule A starts at Asn-63 and molecule B at Ile-65. Three areas of disorder were evident: a large area of disorder comprising residues 93-163 in molecule A and 92-163 in molecule B, a smaller region comprising 502-512 in molecule A and 504-511 in molecule B, and residues 213-215 that are disordered in molecule B only. Finally, eight residues were excluded from the C terminus in both molecules of the asymmetric unit.
The electron density shows clearly that Ser-465 of both molecules in the asymmetric is phosphorylated and, therefore, has been modeled accordingly. This site is on the surface of the transpeptidase domain at the N-terminal end of α10, on the opposite side of the molecule from the active site. This apparent modification is observed consistently in data sets collected from different crystals of PBP2 (both wild-type and mutant) and using proteins from different preparations. An example of omit |Fo|-|Fc| electron density for one of these (A465) is shown in supplemental Fig. 1. Because PBP2 was expressed in the cytoplasm of E. coli, this phosphorylation may not be physiologically relevant.
Circular Dichroism—Each protein (PBP2, PBP2-6140, and PBP2-6140CT) was diluted to a concentration of 0.1 mg/ml in a buffer containing 10 mm Tris, pH 8.0, 250 mm NaCl, and 10% glycerol. The melting curves for each PBP2 construct were measured an AVIV Biomedical Inc. Model 400 circular dichroism spectrophotometer at 206 nm (corresponding to the minimum in a 260-195-nm wavelength scan). Data were recorded every degree over a temperature range of 25-75 °C. These measurements were repeated six times (with a new sample of protein each time) and then averaged. All spectra were corrected against a control spectrum collected from buffer only, and the mean residual ellipticity was calculated using AVIV software. The melting temperature (Tm) for each construct was calculated as the midpoint of the slope between the mean residual ellipticity values where the protein was folded and unfolded. Mass spectrometry was used to show that the differences observed in the melting curves were not due to different levels of phosphorylation in the three proteins (see the supplemental data).
RESULTS
Structure Description
The crystal structure of a soluble form of PBP2 (residues 42-581) from a penicillin-susceptible strain of N. gonorrhoeae was solved at 2.4 Å by multiwavelength anomalous dispersion methods using selenomethionyl-substituted protein. PBP2 contains two domains, an N-terminal domain and a C-terminal TPase (or penicillin binding) domain that is common to all PBPs (Fig. 1A). The N-terminal domain is an elongated domain ∼45 Å in length featuring several long β-strands and a subdomain of shorter strands and small helices. The subdomain interacts with a protruding loop from the transpeptidase domain in a similar way as observed in other high molecular mass PBP structures (28, 29, 32). Within the N-terminal domain and in both molecules of the crystallographic asymmetric unit, residues 91-163 cannot be located in the electron density map and are presumably disordered. Mass spectrometry confirmed this region was present in the protein construct (data not shown).
FIGURE 1.

The structure of PBP2 from N. gonorrhoeae. A, a ribbon representation of the molecule in which the structure is color-ramped blue-to-red in the N-terminal to C-terminal direction. Elements of secondary structure are labeled according to a nomenclature scheme used previously (55). In this convention, some elements of structure are absent in PBP2, e.g. α3 and α7. In this view, the transpeptidase domain is at the top, and the N-terminal domain (and site of the membrane anchor) is at the bottom. B, the amino acid sequence of wild-type PBP2 from N. gonorrhoeae (74). For the TPase domain, the standard system of nomenclature for secondary structure is used. For the N-terminal domain, the system used for the equivalent domain in PBP2a (28) is adopted. Regions that could not be modeled due to apparent disorder are shown as dashed lines. The conserved motifs are highlighted by boxes. The figure was produced in part by SecSeq (D. E. Brodersen, unpublished software).
The functional role of the N-terminal domain in PBP2 is unknown. However, as has been suggested previously for the C-terminal domain of PBP5 from E. coli (50), such domains may serve to project the catalytic domain away from the cytoplasmic membrane toward the peptidoglycan substrate and/or may mediate protein-protein interactions, which in this case might target PBP2 to the septal region of the gonococcal cell. Such interactions appear to be important for targeting E. coli PBP3 (the homologue of PBP2) to the septum (51-54).
The TPase domain of PBP2 can be superimposed onto the TPase domains of PBP2a from methicillin-resistant S. aureus (28) (r.m.s.d. of 2.0 Å for 221 Cα atoms) and PBP2x from S. pneumoniae (55) (r.m.s.d. of 1.6 Å for 248 Cα atoms) even though it shares only 15 and 21% sequence identity with these domains, respectively. The TPase domain can be divided into two subdomains, an α-subdomain and an α/β-subdomain, with the active site lying in the groove between them. As viewed in Fig. 1A, the α-subdomain comprising helices α2, α4-α6, and α8, lies near the top of the molecule and forms one side of the active site groove (see Fig. 1B for secondary structure assignments). An interesting feature of this domain is a projection of mostly β-ribbon between α2 and α4 that lies at the very top of the molecule. In PBP2 from resistant strains of N. gonorrhoeae, this region contains the aspartic acid insertion found in the majority of penA alleles (22) (see below). The α/β-subdomain, which forms the lower face of the active site, contains a central core of anti-parallel β-strands (β1-β5) packed on both sides by several long helices (α1, α9, α10, and α11). In addition, there is an area of apparent structural disorder between residues 502-512 (inclusive) in molecule A (of the asymmetric unit) and 504-511 in molecule B, located in the β3-β4 loop. Several of the mutations observed in PBP2 variants from penicillin-resistant strains map to this loop (see below).
Active Site
The active site of PBP2 contains three conserved sequence motifs that are observed in nearly all penicillin-interacting enzymes (6) (Fig. 2). The SXXK motif is located at the N-terminal end of helix α2 (shown in yellow in Fig. 2) and contains two residues that are important for catalysis: Ser-310, the serine nucleophile that is acylated by both peptide substrate and β-lactam antibiotics, and Lys-313. The SXN motif, comprising Ser-362, Ser-363, Asn-364, is present on the loop connecting α4 and α5. The KTG motif, comprising Lys-497, Thr-498, and Gly-499, is located on strand β3. Lys-313 is a central player in the dense network of hydrogen bonds in the active site, where it is within hydrogen bonding distance of Ser-310, Asn-364, and the backbone carbonyl of Ser-362. Another potential hydrogen bond is observed between Lys-497 and Ser-362.
FIGURE 2.

Active site of PBP2 from N. gonorrhoeae. Shown are the three conserved motifs with the SXXK motif in yellow, the SXN motif in green, and the KTG motif in orange. Residues are shown in bond format with potential hydrogen bonds shown as dashed lines. Distances are in Å. The fourth loop, colored purple, is the site of the insertion of an aspartate between residues 345 and 346, which is associated with penicillin resistance.
The overall arrangement of the residues in the active site, including the pattern of hydrogen bonding, is highly similar to that seen in other PBPs, commensurate with a common mechanism of catalysis in PBPs. This striking overlap of active site residues between PBPs, however, is hard to reconcile with the vastly different second-order rates of acylation of penicillin measured in these PBPs (from 12 to 99,000 m-1 s-1) (56-58)) and suggests that regions adjacent to the active site, which differ markedly among PBPs, must have a significant impact on the penicillin acylation rate.
Modeling of a β-Lactam Antibiotic Bound in the Active Site of PBP2
Numerous attempts to produce a complex of PBP2 with penicillin G by crystal soaking were unsuccessful; hence, penicillin G was docked into the active site by superimposing the active site motifs of PBP2 with those of a crystal structure of PBP5 (a d-alanine carboxypeptidase) from E. coli in complex with penicillin G.4 The 10 residues corresponding to the three conserved motifs in the active sites of these two PBPs overlapped with an r.m.s.d. of 0.79 Å, further emphasizing the high degree of similarity of the active sites within PBPs (supplemental Fig. 2). One close contact in the initial model, a clash between Tyr-422 and the phenyl ring of penicillin, was relieved by rotation of the phenyl group of the antibiotic. In addition, there is a second potential clash between the carboxylate group of penicillin and Tyr-544, suggesting this residue may hydrogen bond with the carboxylate group in the acyl-enzyme complex. Interestingly, although the orientation of the bound antibiotic is not exactly the same, both of these clashes occur when cefuroxime is docked into the active site of PBP2 in a similar way using the structure of cefuroxime-bound PBP2x (32) (not shown). In PBP2, the clash between Tyr-544 and the penicillin carboxylate could be relieved either by movement of the tyrosine side chain or of the helix (α11) from which it emanates. This is important because α11 harbors mutations in PBP2 from resistant strains that may influence the role of this helix in antibiotic binding.
Kinetics of Interaction of PBP2 Variants with [14C]Penicillin G
PBP2 from the penicillin-resistant chromosomally mediated clinical isolate, FA6140 (MICpen = 4 μg/ml) contains five mutations compared with PBP2 from FA19: that is, an Asp-345a insertion between positions 345 and 346 (D345a) and four mutations clustered near the C terminus (Fig. 3). To determine the structural effect of these mutations on the architecture of PBP2, we attempted to crystallize PBP2 from FA6140 (termed PBP2-6140), but the protein failed to crystallize either in the same conditions used to produce crystals of the wild-type protein or using new screens, suggesting that one or more of the mutations interferes with crystal packing.
FIGURE 3.

The location of mutations within PBP2 associated with penicillin resistance. A, a diagram of the transpeptidase domain of PBP2 showing the three active-site motifs, the site of the Asp-345a insertion, and the four mutations near the C-terminal end of the protein that are present in PBP2-6140. WT, wild type. B, a ribbon representation of the structure of the transpeptidase domain of PBP2 showing the location of the mutations around the active site (containing modeled penicillin in bonds form). Each mutation is marked by a black sphere. The orientation shown is the same as in Fig. 1A.
For this reason we set out to define the distinct contributions of the Asp-345a insertion and the C-terminal mutations to the decrease in penicillin acylation rate in PBP2-6140. Two constructs were produced, one that contained only the D345a insertion (PBP2-D345a) and one that harbored only the four C-terminal mutations (PBP2-6140CT) (Fig. 3). These variants were analyzed for their acylation rate constants (k2/KS) with [14C]penicillin G as described under “Experimental Procedures.” Wild-type PBP2 has a k2/KS value of 63,700 m-1 s-1 for [14C]penicillin G, whereas PBP2-6140 has a k2/KS of 4,000 m-1 s-1, a 16-fold decrease (Table 2). The Asp-345a insertion alone conferred an ∼5-fold decrease in k2/Ks (12,200 m-1 s-1) relative to wild-type PBP2, similar to that previously reported (23, 59). Surprisingly, the four C-terminal mutations conferred an identical 5-fold decrease in k2/Ks (12,400 m-1 s-1) as the Asp-345a insertion (Table 2).
TABLE 2.
k2/Ks values of [14C]penicillin G for PBP 2 variants
| PBP2 variants | k2/Ks | Fold decrease |
|---|---|---|
| m−1s−1 | ||
| WT (n = 14)a | 63,700 ± 5,500 | |
| 6140 (n = 10) | 4,000 ± 2,000 | 15.9 |
| D345a (n = 8) | 12,200 ± 2,400 | 5.2 |
| 6140CT (n = 8) | 12,400 ± 900 | 5.1 |
| F504L (n = 6) | 35,500 ± 4,100 | 1.8 |
| A510V (n = 3) | 50,200 ± 2,700 | 1.3 |
| A516G (n = 5) | 44,700 ± 3,000 | 1.4 |
| P551S (n = 12) | 21,300 ± 800 | 3.0 |
| P551S/F504L (n = 7) | 13,100 ± 700 | 4.9 |
| P551S/A510V (n = 3) | 18,800 ± 60 | 3.4 |
| P551S/A516G (n = 6) | 25,900 ± 2,400 | 2.5 |
The number of determinations of k2/Ks for each construct.
Based on these data, we attempted to crystallize both PBP2-D345a and PBP2-6140CT. Only the latter construct crystallized under the same conditions as wild type, indicating that PBP2-6140 did not crystallize because of the presence of the Asp-345a insertion. The crystal structure of PBP2-6140CT was determined to 2.4 Å resolution (see Table 1 for refinement statistics). The overall fold of PBP2-6140CT is essentially the same as the wild-type enzyme (supplemental Fig. 3), and all common main atoms of both molecules of the asymmetric unit superimpose with an r.m.s.d. of 0.34 Å. The primary change is 10 extra residues that are visible at the N terminus in molecule A of PBP2-6140CT (residues 53-62) compared with wild-type PBP2.
Effects of C-terminal Mutations on PBP2 Structure
Active Site Architecture—In structural terms, the C-terminal mutations must alter the architecture at or around the active site in such a way as to lower the reactivity of the enzyme for β-lactam antibiotics without significantly affecting the ability of the enzyme to catalyze transpeptidation of its natural peptide substrate. The four mutations in PBP2-6140CT cluster in two regions that are both near, but not within, the active site. Superimposition of the active sites of wild-type PBP2 with those of PBP2-6140CT, however, shows little or no difference in the positions of the active site residues (Fig. 4), and any slight differences that do occur are within the expected degree of precision in a crystal structure of this resolution (60).
FIGURE 4.

Superimposition of the active sites of wild-type PBP2 and PBP2-6140CT. For wild type, the backbone of the protein is colored green, and the side chain bonds are yellow, whereas for PBP2-6140CT both of these are colored pink. Note the very close correspondence of the three active site motifs in PBP2 derived from penicillin-susceptible and penicillin-resistant strains of N. gonorrhoeae.
β4 and β3-β4 Loop—The mutations F504L, A510V, and A516G are located on β4 or in the disordered loop that connects this strand to β3 (Figs. 3B and 5A). When molecule A of each structure is compared, there is additional density in PBP2-6140CT, which allows the modeling of three extra residues (Arg-502, Lys-503 and Asn-512) within the loop. In addition, there is a very slight shift in the position of β3 such that it lies closer to the active site in PBP2-6140CT. Two of the three mutations in this area (F504L and A510V) remain disordered in the β3-β4 loop. The A516G mutation is located on β4 immediately adjacent to the KTG motif on β3 but has very little effect on the structure. When molecule B of each asymmetric unit is compared, the differences are less pronounced; the length of the disordered region is the same, and the position of β3 is essentially unchanged.
FIGURE 5.

The effect of mutations associated with penicillin resistance on the structure of PBP2 from N. gonorrhoeae. In this picture, the crystal structures of wild-type PBP2 (green) and PBP2-6140CT (pink) are superimposed. The location of the active site is indicated by the modeled penicillin molecule (gray bonds), and sites of mutations are labeled in red. Elements of secondary structure and selected amino acids are labeled, including those at the active site. A, β3 region, including the β3-β4 disordered loop. B, α11 region.
Helix α11—The other C-terminal mutation, P551S, lies on α11, which is located toward the C terminus of the protein (Fig. 5B). This helix is interesting because in the wild-type enzyme it is kinked at its N-terminal end after two consecutive proline residues at positions 551 and 552, and at its C-terminal end, the side chain of Tyr-544 points directly into the active site (where it may interact with penicillin, as mentioned above). Surprisingly, the presence of serine at position 551 in place of proline in PBP2-6140CT has no discernible effect on the structure of this helix, and the degree of kinking is unchanged in the mutant enzyme. The same is true for both molecules in the asymmetric unit. In addition, there is no localized increase in B factors in this helix compared with the surrounding structure.
One possible explanation for the lack of structural alterations arises by examining the crystal packing interactions in this region. In both molecules A and B of the asymmetric unit of PBP2, the C-terminal end of α11 packs against a three-stranded β sheet of the N-terminal domain of a neighboring molecule. Such an interaction might have reversed any alteration in the structure of α11 present when the protein is in solution rather than when constrained by the crystal lattice. Interestingly, the same crystal contact appears to block the active site of PBP2 and, thus, might explain the failure to introduce β-lactam compounds by crystal-soaking experiments.
To define the individual contributions of the C-terminal mutations to the decrease in penicillin acylation rate, we purified proteins containing each mutation individually and determined their effect on the k2/KS of [14C]penicillin G (Table 2). The A510V, A516G, and F504L mutations by themselves had minor (<2-fold) effects on k2/KS. In contrast, the P551S mutation had a much larger effect on the acylation rate, with a decrease of 3-fold relative to wild-type PBP2. Based on this result, we examined the three other mutations in concert with the P551S mutation. Whereas the A510V or A516G mutation together with P551S did not appreciably change the k2/KS of [14C]penicillin G for PBP2, the combination of P551S and F504L mutations decreased the acylation rate nearly to the level of PBP2 containing all four mutations. These data indicate that essentially all of the effects of the C-terminal mutations are due to F504L and P551S, with the latter mutation being the most important.
Thermal Stabilities of PBP2 Variants
To assess whether the mutations in PBP2 associated with penicillin resistance lower the stability of protein, thermal melting curves were determined for wild-type PBP2, PBP2-6140, and PBP2-6140CT using circular dichroism spectroscopy (Fig. 6). Tm values calculated from these curves were as follows: wild-type PBP2, 54.5 °C; PBP2-6140CT, 52.0 °C; PBP2-6140, 50.0 °C. These data show that both mutant PBPs melt at a lower temperature than wild type, indicating that the mutations associated with penicillin resistance impart a destabilizing effect on the protein fold. Interestingly, the Tm of PBP2-6140CT is intermediate between wild-type PBP2 and PBP2-6140, which correlates with the second order rate constants of acylation.
FIGURE 6.

Thermal stabilities of wild-type and mutant PBP2s. Melting curves were determined using circular dichroism by measuring the mean ellipticity at 206 nm in 1 °C intervals. ▪, wild-type PBP2; ▴, PBP2-6140CT; •, PBP2-6140.
DISCUSSION
In this study, we report the crystal structures and kinetic analysis of PBP2 variants derived from penicillin-susceptible and penicillin-resistant strains of N. gonorrhoeae. These structures are the first of a PBP from N. gonorrhoeae. The five mutations in PBP2 from the penicillin-resistant isolate FA6140 lower the acylation rate of the enzyme for penicillin by 16-fold, which corresponds to a 6-fold increase in MIC (16). They are located close to but not within the active site and can be dissected into two groups, the Asp-345a insertion and the C-terminal mutations.
The most surprising finding from this investigation is that, although the four C-terminal mutations together lower the rate of acylation by ∼5-fold, these amino acid changes have only minor effects on the protein architecture. Both PBP2-6140 and PBP-6140CT have lower thermal stabilities when compared with wild-type PBP2, suggesting the mutations are destabilizing and that they probably increase the overall flexibility of the protein fold. In support of this, the overall B factor of PBP-6140CT is significantly higher than the wild-type enzyme (48 versus 35 Å2), although it should also be noted that this parameter is very specific for a given crystal. The lack of localized increases in B factors around the mutation sites also supports the idea of a global increase in flexibility.
These observations can be explained in two ways; 1) The mutations lead to a slight disordering of the structure that is too small in displacement to observe by x-ray crystallography but which nevertheless has a significant effect on the kinetics, or 2) there is a dynamic equilibrium between conformational states of the enzyme, where one state exhibits higher acylation activity than the other, and that increased flexibility caused by the mutations in PBP2-6140 shifts this equilibrium toward the less active state. The inability to observe these two states by x-ray crystallography is because, in the crystal, both wild-type and mutant PBP2 have been “frozen” in the same conformational state.
The distribution of the mutations associated with penicillin resistance around (but not within) the active site is also consistent with a mechanism of generalized disordering that lowers reactivity with penicillin. A similar pattern of mutations has been observed in several other unrelated systems, suggesting this may be a general phenomenon of resistance. For example, mutations in Drosophila acetylcholinesterase associated with resistance to inhibitors are located in the second shell of residues around the active site (61), and a study of a drug-resistant HIV-1 protease showed that residues that lie outside the active site can be the primary source of resistance (62). In cytochrome P450 14α-sterol demethylases (CYP51) from azole-resistant Candida albicans, mutations appear to prevent key conformational changes but do not contact the inhibitor directly (63).
The increased flexibility in PBP2 could affect binding affinity or acylation. The non-covalent binding affinity of penicillin (KS) could be lowered by hindering the conformational changes required for binding, i.e. it would increase the energetic cost associated with induced fit. Many PBPs undergo conformational changes upon binding β-lactams, including E. coli PBP4 (64), S. pneumoniae PBP1b (65), S. pneumoniae PBP2x (66), and E. coli PBP5.4 The apparent clashes with penicillin in the model of the acylated enzyme suggest that conformational changes are also required in PBP2. Moreover, the P551S mutation, which has the greatest effect on k2/KS, is located at the kink in α11 and might prevent or alter the movement of Tyr-544 (at the N-terminal end of α11) during acylation. The other important mutation, F504L, is on the disordered β3-β4 loop and could influence the position of β3, which does shift slightly in PBP2-6140CT (see Fig. 5A).
Increased flexibility of PBP2 could also lower the rate of acylation by increasing the energetic cost required to form the tetrahedral intermediate for catalysis. Such a state requires very precise geometry to permit catalysis, and increased flexibility in the active site region would make it less likely that such a state is attained. In both mechanisms (reduced binding affinity or acylation), the reactivity of peptide would be less affected than penicillin because both effects can be overcome by a proportionally larger number of interactions between the protein and its substrate. In addition, the impact of lowered affinity for peptide would be lessened by the proximity of PBP2 to peptidoglycan, which increases the effective concentration of its substrate.
The apparent mechanism of penicillin resistance in N. gonorrhoeae, which involves very subtle alterations in PBP2 due to destabilizing mutations, contrasts with investigations in other PBPs. In the case of PBP2a derived from methicillin-resistant S. aureus, it was proposed that the low rate of acylation by β-lactams resulted from a remodeling of the active site involving a twisting of β3 and a shift in α2. These changes, which presumably occurred during the evolution of PBP2a from penicillin-sensitive progenitor, increased the energetic penalty for transitioning from the initial Michaelis-Menten complex to the acyl-enzyme intermediate (28). In support of this hypothesis, mutations in PBP2x from the penicillin-resistant strain of S. pneumoniae sp5259 cause a small shift in the position of β3 (34), and we also observe a slight shift in the same strand in PBP2. In the case of PBP2a, though, the argument was premised upon the observation (at that time) that PBPs do not undergo significant conformational changes upon reacting with β-lactams and that in PBP2a the remodeling meant that conformational changes were now required to form the acyl-enzyme complex. However, several structures of PBPs solved since then show that conformational changes do indeed occur when forming the acyl-enzyme intermediate (see above). Nevertheless, it is interesting to note that two of the four mutations in PBP2 are located in the β3-β4 loop, thus implicating this region as being important for penicillin resistance in more than one PBP.
Many studies have linked penicillin resistance mediated by mutations in PBP2x from S. pneumoniae with significant changes in crystal structures, including disordering of a loop that precedes the SXN motif (27, 33, 34). The contrast between such large alterations in PBP2x and the lack of significant structural differences in PBP2-6140CT is very striking and points to different mechanisms of resistance operating in each organism. One reason for this might be the lack of an outer membrane in Gram-positive bacteria. Thus, alterations in PBPs in S. pneumoniae contribute proportionally more to resistance than in N. gonorrhoeae where other mechanisms, such as alterations in porins and efflux pumps, are also involved. In addition, the subtle changes in PBP2 from N. gonorrhoeae probably have little impact on the reactivity of the enzyme with its peptide substrates, whereas the changes in PBP2x would be expected to alter the reactivity of the enzyme for both peptide substrates and β-lactams. In keeping with this hypothesis, peptidoglycan isolated from penicillin-resistant strains of S. pneumoniae shows a higher degree of peptide branching than that isolated from susceptible strains (67, 68). Although the structure of peptidoglycan from penicillin-resistant strains of N. gonorrhoeae has not been analyzed, only minor differences have been observed in strains of N. meningitidis that have reduced susceptibility to penicillin (69).
Absent from the discussion thus far is the effect on penicillin resistance contributed by the insertion of Asp-345a. Because the aspartate insertion is the one mutation observed most often in penicillin-resistant strains (22), it was considered the most important for lowering reactivity with penicillin, with the other mutations having only a slight (∼2-fold) effect on the protein (23). However, as shown here, the four C-terminal mutations in PBP2 from FA6140 have an identical effect on the second order rate of acylation as the Asp-345a insertion by itself (Table 2). These data suggest that the two types of alterations are mildly antagonistic to one another, as one would expect a nearly 25-fold decrease (the product of the two) if the mutations were independent.
The site of mutation is on a hairpin loop that lies near the SXN motif of the active site (Fig. 7). Interestingly, the equivalent loop in PBP5fm from Enterococcus faecium also contains an insertion mutation (of a serine) that is present in the most penicillin-resistant strains (70, 71). Unfortunately, none of the constructs of PBP2 containing Asp-345a crystallized, leaving the exact structural consequences of this mutation unclear. Examination of this region in the wild-type structure of PBP2, though, shows that the side chain of Asp-346, which is immediately adjacent to the insertion site, forms hydrogen bonds with the side chain hydroxyl of Ser-363 (the X of the SXN motif) and the amide nitrogens of Asn-364 and Val-365. Insertion of an aspartate before Asp-346 may disrupt these interactions and affect enzyme activity by altering the conformation of the SXN motif. Although it might be expected that such an insertion would cause disordering of the hairpin loop, it should be noted that the insertion of an aspartate is the only amino acid that confers penicillin resistance to N. gonorrhoeae (23), arguing in favor of a more specific mechanism to lower acylation by penicillin. One intriguing possibility is that the inserted aspartate replaces the existing aspartate at position 346 and assumes some or all of the hydrogen bonds mediated by that residue. This may explain why only an aspartate can be inserted and not a glutamate, for instance, because the same arrangement may not be possible with the longer side chain. Clearly, the balance between developing penicillin resistance and maintaining transpeptidase activity is very delicate.
FIGURE 7.

The location of the Asp-345a insertion in PBP2 between Arg-345 and Asp-346 in the β2c-β2d loop. PBP2 containing this mutation does not crystallize; hence, only the wild-type structure is shown here. Key interactions that Asp-346 makes with the SXN motif at the active site, are marked with dashed lines, and the distances are shown in Å. In this picture, 3 the backbone of PBP2 is shown as a green ribbon, and selected side chains are colored yellow. The site of the insertion is marked by an arrow.
In conclusion, the molecular mechanism of penicillin resistance in PBP2 contributed by the four mutations toward the C-terminal end of the protein, and in particular P551S and F504L, appears to be very subtle in nature and very different from the large structural changes observed in PBP2x from S. pneumoniae. Although this result might seem unexpected, in hindsight a microscopic rather than macroscopic mechanism of resistance is logical given the strict constraints placed on the evolution of resistance by the necessity to retain activity with peptides while lowering that for β-lactams. By contrast, the effect of Asp-345a is expected to be more significant both in terms of its potential to alter structure and its proximity to an active site motif. Clearly, there remains much to understand, and a key goal now is to view both wild-type and mutant PBP2 in complexes with β-lactam antibiotics. With the recent emergence of gonococcal strains showing intermediate resistance to third-generation cephalosporins caused in part by mutations in PBP2 (2, 5, 72, 73), such investigation is of high priority.
Supplementary Material
Acknowledgments
Use of the Advanced Photon Source was supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract W-31-109-ENG-38. Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the United States Department of Energy, Office of Basic Energy Sciences. The Stanford Synchrotron Radiation Laboratory Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program and the NIGMS. The x-ray crystallography facility used for this work is supported by the Medical University of South Carolina Research Resource Facilities program.
The atomic coordinates and structure factors (codes 3EQU and 3EQV) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported, in whole or in part, by National Institutes of Health Grants GM66861 (to C. D.) and AI36901 (to R. A. N.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1 and S2 and Figs. 1-3.
Footnotes
The abbreviations used are: PBP, penicillin-binding protein; MIC, minimal inhibitory concentration; r.m.s.d., root mean square deviation; TPase, transpeptidase.
G. Nicola, R. A. Nicholas, and C. Davies, unpublished data.
References
- 1.Centers for Disease Control. (2007) MMWR Morb. Mortal Wkly. Rep. 56 332-33617431378 [Google Scholar]
- 2.Ameyama, S., Onodera, S., Takahata, M., Minami, S., Maki, N., Endo, K., Goto, H., Suzuki, H., and Oishi, Y. (2002) Antimicrob. Agents Chemother. 46 3744-3749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang, S. A., Lee, M. V., O'Connor, N., Iverson, C. J., Ohye, R. G., Whiticar, P. M., Hale, J. A., Trees, D. L., Knapp, J. S., Effler, P. V., and Weinstock, H. S. (2003) Clin. Infect. Dis. 37 849-852 [DOI] [PubMed] [Google Scholar]
- 4.Whiley, D. M., Limnios, E. A., Ray, S., Sloots, T. P., and Tapsall, J. W. (2007) Antimicrob. Agents Chemother. 51 3111-3116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindberg, R., Fredlund, H., Nicholas, R., and Unemo, M. (2007) Antimicrob. Agents Chemother. 51 2117-2122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghuysen, J. M. (1991) Annu. Rev. Microbiol. 45 37-67 [DOI] [PubMed] [Google Scholar]
- 7.Ghuysen, J. M. (1994) Trends Microbiol. 2 372-380 [DOI] [PubMed] [Google Scholar]
- 8.Holtje, J. V. (1998) Microbiol. Mol. Biol. Rev. 62 181-203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barbour, A. G. (1981) Antimicrob. Agents Chemother. 19 316-322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stefanova, M. E., Tomberg, J., Davies, C., Nicholas, R. A., and Gutheil, W. G. (2004) Eur. J. Biochem. 271 23-32 [DOI] [PubMed] [Google Scholar]
- 11.Stefanova, M. E., Tomberg, J., Olesky, M., Holtje, J. V., Gutheil, W. G., and Nicholas, R. A. (2003) Biochemistry 42 14614-14625 [DOI] [PubMed] [Google Scholar]
- 12.Ropp, P. A., and Nicholas, R. A. (1997) J. Bacteriol. 179 2783-2787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cannon, J. G., and Sparling, P. F. (1984) Annu. Rev. Microbiol. 38 111-133 [DOI] [PubMed] [Google Scholar]
- 14.Faruki, H., and Sparling, P. F. (1986) Antimicrob. Agents Chemother. 30 856-860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spratt, B. G. (1988) Nature 332 173-176 [DOI] [PubMed] [Google Scholar]
- 16.Ropp, P. A., Hu, M., Olesky, M., and Nicholas, R. A. (2002) Antimicrob. Agents Chemother. 46 769-777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gill, M. J., Simjee, S., Al-Hattawi, K., Robertson, B. D., Easmon, C. S., and Ison, C. A. (1998) Antimicrob. Agents Chemother. 42 2799-2803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olesky, M., Hobbs, M., and Nicholas, R. A. (2002) Antimicrob. Agents Chemother. 46 2811-2820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hagman, K. E., Pan, W., Spratt, B. G., Balthazar, J. T., Judd, R. C., and Shafer, W. M. (1995) Microbiology 141 611-622 [DOI] [PubMed] [Google Scholar]
- 20.Veal, W. L., Nicholas, R. A., and Shafer, W. M. (2002) J. Bacteriol. 184 5619-5624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sparling, P. F., Sarubbi, F. A., Jr., and Blackman, E. (1975) J. Bacteriol. 124 740-749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dowson, C. G., Jephcott, A. E., Gough, K. R., and Spratt, B. G. (1989) Mol. Microbiol. 3 35-41 [DOI] [PubMed] [Google Scholar]
- 23.Brannigan, J. A., Tirodimos, I. A., Zhang, Q. Y., Dowson, C. G., and Spratt, B. G. (1990) Mol. Microbiol. 4 913-919 [DOI] [PubMed] [Google Scholar]
- 24.Tipper, D. J., and Strominger, J. L. (1965) Proc. Natl. Acad. Sci. U. S. A. 54 1133-1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yocum, R. R., Waxman, D. J., Rasmussen, J. R., and Strominger, J. L. (1979) Proc. Natl. Acad. Sci. U. S. A. 76 2730-2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mouz, N., Gordon, E., Di Guilmi, A. M., Petit, I., Petillot, Y., Dupont, Y., Hakenbeck, R., Vernet, T., and Dideberg, O. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 13403-13406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dessen, A., Mouz, N., Gordon, E., Hopkins, J., and Dideberg, O. (2001) J. Biol. Chem. 276 45106-45112 [DOI] [PubMed] [Google Scholar]
- 28.Lim, D., and Strynadka, N. C. (2002) Nat. Struct. Biol. 9 870-876 [DOI] [PubMed] [Google Scholar]
- 29.Contreras-Martel, C., Job, V., Di Guilmi, A. M., Vernet, T., Dideberg, O., and Dessen, A. (2006) J. Mol. Biol. 355 684-696 [DOI] [PubMed] [Google Scholar]
- 30.Carapito, R., Chesnel, L., Vernet, T., and Zapun, A. (2006) J. Biol. Chem. 281 1771-1777 [DOI] [PubMed] [Google Scholar]
- 31.Job, V., Carapito, R., Vernet, T., Dessen, A., and Zapun, A. (2008) J. Biol. Chem. 283 4886-4894 [DOI] [PubMed] [Google Scholar]
- 32.Gordon, E., Mouz, N., Duee, E., and Dideberg, O. (2000) J. Mol. Biol. 299 477-485 [DOI] [PubMed] [Google Scholar]
- 33.Chesnel, L., Pernot, L., Lemaire, D., Champelovier, D., Croize, J., Dideberg, O., Vernet, T., and Zapun, A. (2003) J. Biol. Chem. 278 44448-44456 [DOI] [PubMed] [Google Scholar]
- 34.Pernot, L., Chesnel, L., Le Gouellec, A., Croize, J., Vernet, T., Dideberg, O., and Dessen, A. (2004) J. Biol. Chem. 279 16463-16470 [DOI] [PubMed] [Google Scholar]
- 35.Maness, M. J., and Sparling, P. F. (1973) J. Infect. Dis. 128 321-330 [DOI] [PubMed] [Google Scholar]
- 36.Frere, J. M., Nguyen-Disteche, M., Coyette, J., and Joris, B. (1992) in The Chemistry of β-lactams (Page, M. I., ed) pp. 148-196, Chapman & Hall, Glasgow
- 37.Matthews, B. (1968) J. Mol. Biol. 33 491-497 [DOI] [PubMed] [Google Scholar]
- 38.McPhillips, T. M., McPhillips, S. E., H. J. C., Cohen, A. E., Deacon, A. M., Ellis, P. J., Garman, E., Gonzalez, A., Sauter, N. K., Phizackerley, R. P., Soltis, S. M., and Kuhn, P. (2002) J. Synchrotron Radiat. 9 401-406 [DOI] [PubMed] [Google Scholar]
- 39.Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276 307-326 [DOI] [PubMed] [Google Scholar]
- 40.Terwilliger, T., and Berendzen, J. (1999) Acta Crystallogr. Sect. D 55 849-861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Terwilliger, T. (2000) Acta Crystallogr. D 56 965-972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Terwilliger, T. C. (2002) Acta Cryst. D 58 1937-1940 [DOI] [PubMed] [Google Scholar]
- 43.Leslie, A. G. W. (1992) Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography No. 26
- 44.CCP4 (1994) Acta Crystallogr. Sect. D 50 760-763 [DOI] [PubMed] [Google Scholar]
- 45.Jones, T. A., Zou, J.-Y., Cowan, S. W., and Kjeldgaard, M. (1991) Acta Crystallogr. Sect. A 47 110-119 [DOI] [PubMed] [Google Scholar]
- 46.Brünger, A. T. (1992) X-PLOR, Version 3.1, A System for X-ray Crystallography and NMR, Yale University, New Haven, CT
- 47.Murshudov, G. N., Vagin, A. A., and Dodson, E. J. (1997) Acta Crystallogr. Sect. D 53 240-255 [DOI] [PubMed] [Google Scholar]
- 48.Lamzin, V. S., and Wilson, K. S. (1993) Acta Crystallogr. Sect. D 49 129-147 [DOI] [PubMed] [Google Scholar]
- 49.Laskowski, R. A., MacArthur, M. W., Moss, D. S., and Thornton, J. M. (1993) J. Appl. Crystallogr. 26 283-291 [Google Scholar]
- 50.Davies, C., White, S. W., and Nicholas, R. A. (2001) J. Biol. Chem. 276 616-623 [DOI] [PubMed] [Google Scholar]
- 51.Marrec-Fairley, M., Piette, A., Gallet, X., Brasseur, R., Hara, H., Fraipont, C., Ghuysen, J. M., and Nguyen-Disteche, M. (2000) Mol. Microbiol. 37 1019-1031 [DOI] [PubMed] [Google Scholar]
- 52.Mercer, K. L., and Weiss, D. S. (2002) J. Bacteriol. 184 904-912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morlot, C., Noirclerc-Savoye, M., Zapun, A., Dideberg, O., and Vernet, T. (2004) Mol. Microbiol. 51 1641-1648 [DOI] [PubMed] [Google Scholar]
- 54.Wissel, M. C., and Weiss, D. S. (2004) J. Bacteriol. 186 490-502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pares, S., Mouz, N., Petillot, Y., Hakenbeck, R., and Dideberg, O. (1996) Nat. Struct. Biol. 3 284-289 [DOI] [PubMed] [Google Scholar]
- 56.Mouz, N., Di Guilmi, A. M., Gordon, E., Hakenbeck, R., Dideberg, O., and Vernet, T. (1999) J. Biol. Chem. 274 19175-19180 [DOI] [PubMed] [Google Scholar]
- 57.Graves-Woodward, K., and Pratt, R. F. (1998) Biochem. J. 332 755-761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nicholas, R. A., Krings, S., Tomberg, J., Nicola, G., and Davies, C. (2003) J. Biol. Chem. 278 52826-52833 [DOI] [PubMed] [Google Scholar]
- 59.Schultz, D. E., Spratt, B. G., and Nicholas, R. A. (1991) Protein Expression Purif. 2 339-349 [DOI] [PubMed] [Google Scholar]
- 60.Cruickshank, D. W. (1999) Acta. Crystallogr. D Biol. Crystallogr. 55 583-601 [DOI] [PubMed] [Google Scholar]
- 61.Harel, M., Kryger, G., Rosenberry, T. L., Mallender, W. D., Lewis, T., Fletcher, R. J., Guss, J. M., Silman, I., and Sussman, J. L. (2000) Protein Sci. 9 1063-1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Muzammil, S., Ross, P., and Freire, E. (2003) Biochemistry 42 631-638 [DOI] [PubMed] [Google Scholar]
- 63.Podust, L. M., Poulos, T. L., and Waterman, M. R. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 3068-3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kishida, H., Unzai, S., Roper, D. I., Lloyd, A., Park, S. Y., and Tame, J. R. (2006) Biochemistry 45 783-792 [DOI] [PubMed] [Google Scholar]
- 65.Macheboeuf, P., Di Guilmi, A. M., Job, V., Vernet, T., Dideberg, O., and Dessen, A. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 577-582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yamada, M., Watanabe, T., Baba, N., Takeuchi, Y., Ohsawa, F., and Gomi, S. (2008) Antimicrob. Agents Chemother. 52 2053-2060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Garcia-Bustos, J., and Tomasz, A. (1990) Proc. Natl. Acad. Sci. U. S. A. 87 5415-5419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Severin, A., Figueiredo, A. M., and Tomasz, A. (1996) J. Bacteriol. 178 1788-1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Antignac, A., Boneca, I. G., Rousselle, J. C., Namane, A., Carlier, J. P., Vazquez, J. A., Fox, A., Alonso, J. M., and Taha, M. K. (2003) J. Biol. Chem. 278 31529-31535 [DOI] [PubMed] [Google Scholar]
- 70.Rybkine, T., Mainardi, J. L., Sougakoff, W., Collatz, E., and Gutmann, L. (1998) J. Infect. Dis. 178 159-163 [DOI] [PubMed] [Google Scholar]
- 71.Sauvage, E., Kerff, F., Fonze, E., Herman, R., Schoot, B., Marquette, J. P., Taburet, Y., Prevost, D., Dumas, J., Leonard, G., Stefanic, P., Coyette, J., and Charlier, P. (2002) Cell. Mol. Life Sci. 59 1223-1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ito, M., Deguchi, T., Mizutani, K. S., Yasuda, M., Yokoi, S., Ito, S., Takahashi, Y., Ishihara, S., Kawamura, Y., and Ezaki, T. (2005) Antimicrob. Agents Chemother. 49 137-143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takahata, S., Senju, N., Osaki, Y., Yoshida, T., and Ida, T. (2006) Antimicrob. Agents Chemother. 50 3638-3645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Spratt, B. G., and Cromie, K. D. (1988) Rev. Infect. Dis. 10 699-711 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.