

Abstract
Carbon dioxide is fundamental to the physiology of all organisms. There is considerable interest in the precise molecular mechanisms that organisms use to directly sense CO2. Here we demonstrate that a mammalian recombinant G-protein-activated adenylyl cyclase and the related Rv1625c adenylyl cyclase of Mycobacterium tuberculosis are specifically stimulated by CO2. Stimulation occurred at physiological concentrations of CO2 through increased kcat. CO2 increased the affinity of enzyme for metal co-factor, but contact with metal was not necessary as CO2 interacted directly with apoenzyme. CO2 stimulated the activity of both G-protein-regulated adenylyl cyclases and Rv1625c in vivo. Activation of G-protein regulated adenylyl cyclases by CO2 gave a corresponding increase in cAMP-response element-binding protein (CREB) phosphorylation. Comparison of the responses of the G-protein regulated adenylyl cyclases and the molecularly, and biochemically distinct mammalian soluble adenylyl cyclase revealed that whereas G-protein-regulated enzymes are responsive to CO2, the soluble adenylyl cyclase is responsive to both CO2 and bicarbonate ion. We have, thus, identified a signaling enzyme by which eukaryotes can directly detect and respond to fluctuating CO2.
Inorganic carbon
(Ci)3 is
central to prokaryotic and eukaryotic physiology. The predominant biologically
active forms of Ci are CO2 and
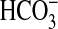 and their relative contributions
to the total Ci pool are pH-dependent. Biological roles for
CO2 and
and their relative contributions
to the total Ci pool are pH-dependent. Biological roles for
CO2 and 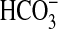 include
photosynthetic carbon fixation
(1), pH homeostasis
(2), carbon metabolism
(3), activation of virulence in
pathogenic organisms (4), sperm
maturation (5), and as an
alarmone in Drosophila
(6,
7).
include
photosynthetic carbon fixation
(1), pH homeostasis
(2), carbon metabolism
(3), activation of virulence in
pathogenic organisms (4), sperm
maturation (5), and as an
alarmone in Drosophila
(6,
7).
Given its importance in biology, the identification of CO2 responsive signaling pathways is key to understanding how organisms cope with fluctuating CO2. Two seven transmembrane receptors, Gr21a and Gr63a, have been shown to confer CO2 responsiveness in Drosophila neurons (6, 7). Guanylyl cyclase D expressing olfactory neurons also mediate sensitivity to CO2 in mice (8). A role for cGMP-activated channels in CO2 sensing has been observed in CO2 avoidance behavior in Caenorhabditis (9, 10). Despite these impressive advances, no eukaryotic signaling enzymes unequivocally demonstrated to respond to CO2 have been identified.
The mammalian soluble adenylyl cyclase (sAC) synthesizes the second
messenger 3′,5′-cAMP and is directly stimulated by
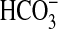 (11–13).
Stimulation of sAC by
(11–13).
Stimulation of sAC by 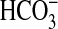 has an
unequivocal role in sperm maturation
(5,
14–16).
sAC is a member of the Class III family of adenylyl cyclases (ACs), a family
that also includes the G-protein-regulated ACs and many examples from
prokaryotic genomes (17,
18). The Class III ACs can be
divided into four subclasses (a–d) based upon polymorphisms within the
active site (19). sAC is a
member of Class IIIb, a subclass characterized partly by replacement of a
substrate binding Asp with Thr. The Class IIIa ACs include the mammalian
G-protein-stimulated ACs and numerous prokaryotic examples. These have been
previously assumed to be non-responsive to Ci
(12).
has an
unequivocal role in sperm maturation
(5,
14–16).
sAC is a member of the Class III family of adenylyl cyclases (ACs), a family
that also includes the G-protein-regulated ACs and many examples from
prokaryotic genomes (17,
18). The Class III ACs can be
divided into four subclasses (a–d) based upon polymorphisms within the
active site (19). sAC is a
member of Class IIIb, a subclass characterized partly by replacement of a
substrate binding Asp with Thr. The Class IIIa ACs include the mammalian
G-protein-stimulated ACs and numerous prokaryotic examples. These have been
previously assumed to be non-responsive to Ci
(12).
All prokaryotic Class IIIb ACs examined to date respond to Ci
including enzymes from organisms as diverse as Anabaena PCC 7120,
Mycobacterium tuberculosis, Stigmatella aurantiaca, and
Chloroflexus aurantiacus
(20,
21). Two Class IIIb ACs,
Slr1991 of Synechocystis PCC 6803 and CyaB1 of Anabaena PCC
7120, have been proven to respond to CO2 and not
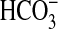 , giving rise to the idea of AC as
a true gas-sensing molecule
(22,
23). The finding that Class
IIIb ACs respond to CO2 and not
, giving rise to the idea of AC as
a true gas-sensing molecule
(22,
23). The finding that Class
IIIb ACs respond to CO2 and not
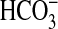 necessitates an examination of the
assumption that G-protein-regulated ACs and related prokaryotic enzymes do not
respond to Ci.
necessitates an examination of the
assumption that G-protein-regulated ACs and related prokaryotic enzymes do not
respond to Ci.
Here we demonstrate, contrary to previous work, that a recombinant
G-protein-regulated AC and the Class IIIa Rv1625c AC of M.
tuberculosis H37Rv show a pH-dependent response to Ci due to
specific stimulation by CO2 at physiologically relevant
concentrations. CO2 interacted directly with the apoprotein and
modulated the activity of both the prokaryotic enzyme and G-protein-regulated
AC in vivo. Finally, we contrasted the responses of sAC- and
G-protein-regulated ACs to different species of Ci and propose that
the mammalian cAMP signaling pathway is able to discriminate between
CO2 and 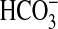 in
vivo.
in
vivo.
EXPERIMENTAL PROCEDURES
Recombinant Proteins—Rv1625c204–443 wild type and mutant proteins, Slr1991120–337 wild type and mutant proteins, recombinant protein corresponding to amino acids 1–469 of human sAC (truncated splice variant (13); sACT), recombinant protein corresponding to the first catalytic domain (amino acids 263–476; 7C1) of human AC type 7, and recombinant protein corresponding to the second catalytic domain (amino acids 821–1090; 2C2) of rat AC type 2 were expressed and purified as previously described (22, 24–27). A mixture of 7C1 with an excess of 2C2 (7C1·2C2) represents a catalytically active G-protein responsive AC without the transmembrane domains of the native molecule. Recombinant protein representing the short splice variant of bovine Gsα was purified and activated with GTPγS·Mg2+ as previously described (28). Single amino acid mutations were introduced by site-directed mutagenesis using appropriate primers and the appropriate wild type construct as template. Double amino acid mutations were introduced by site-directed mutagenesis using appropriate primers and the appropriate single amino acid mutant construct as template. All constructs were confirmed by double-stranded sequencing. Mutagenic primer sequences are provided in Table S1. Plasmids encoding Rv1625c204–443 K296A and D256A mutagenic proteins were a kind gift of Joachim Schultz (25).
Adenylyl Cyclase Assays—AC assays were performed at 37 °C (Rv1625c204–443) or30 °C (7C1·2C2) in a final volume of 100 μl and contained 50 mm buffer, 2 mm [2,8-3H]cAMP (150 Bq), and [α-32P]ATP (25 kBq) if not stated otherwise (29). Protein concentrations were adjusted to maintain substrate utilization at <10%. The following buffers were used at pH 6.5 (Mes), pH 7.0–7.5 (Mops), and pH 8.0–8.5 (Tris-hydrochloride). Enzyme, buffer, and substrate were prepared at the appropriate pH. CO2 was quantified by titration against NaOH. Assay pH was stable over a period of at least 40 min. For dose-response experiments, NaHCO3 was added to the assay, and the CO2 concentration was calculated using the Henderson-Hasselbalch equation, and the total salt concentration was adjusted with NaCl. All errors correspond to the S.E. If absent, errors were smaller than the symbol used to depict the data point.
Adenylyl Cyclase Assays at Ci Disequilibrium—For
Ci disequilibrium assays, dissolved CO2 was prepared by
bubbling into double-distilled H2O at 0 °C to saturation and
quantified by titration against NaOH. NaHCO3 and NaCl were prepared
in double-distilled H2O at 0 °C. CO2,
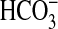 , or Cl– were
subsequently added to the assay at 0 °C simultaneous with substrate to the
required concentration. Buffer and substrate for assays were prepared at the
appropriate pH and temperature for the experiment. pH changes in assays were
monitored using a pH electrode (Biotrode; Hamilton) connected to a computer
with a PC card (Orion Sensorlink). The pH was measured in a time-driven
acquisition mode in assays identical to those used for biochemistry. All pH
measurements were accurate to ± 0.02 pH units (manufacturers
specifications). All errors correspond to the S.E.
, or Cl– were
subsequently added to the assay at 0 °C simultaneous with substrate to the
required concentration. Buffer and substrate for assays were prepared at the
appropriate pH and temperature for the experiment. pH changes in assays were
monitored using a pH electrode (Biotrode; Hamilton) connected to a computer
with a PC card (Orion Sensorlink). The pH was measured in a time-driven
acquisition mode in assays identical to those used for biochemistry. All pH
measurements were accurate to ± 0.02 pH units (manufacturers
specifications). All errors correspond to the S.E.
CO2 Activation of AC in Vivo—pCTXLacZ, a plasmid with lacZ expression driven from a cAMP-responsive promoter, and pQE30-Rv1625c204–443 (25) were transformed into Escherichia coli M15 (pREP4). Cells were grown in Luria broth with 100 μg ml–1 ampicillin, 50 μgml–1 kanamycin, and 5 μgml–1 tetracycline at 30 °C until an A600 of 0.6. Rv1625c204–443 protein production was induced with 30 μm isopropyl 1-thio-β-d-galactopyranoside for 3 h. Cells were pelleted at 4000 × g for 10 min and resuspended in Luria broth containing 50 mm Tris, pH 7.1. Cell suspensions were bubbled with either 10% (v/v) CO2 in air or in air for 30 min at 30 °C. Cells were disrupted with 0.1 mg of sodium deoxycholate and 1% (v/v) toluene and mixed for 10 min at 30 °C. The lysate was made up to 50 mm sodium phosphate, pH 7.0, 0.5 mm ortho-nitrophenol-β-d-galactopyranoside and incubated for 15 min at 30 °C. Reactions were stopped with 2 m sodium carbonate, and absorbance was read at 420 nm. A standard curve was generated using 0–250 μm ortho-nitrophenol.
CO2 Binding Assays—1 ml of 50% (v/v) Sephadex G50 in 50 mm Mes, pH 6.5 (bed volume 0.5 ml), was pre-spun at 1500 × g for 30 s. A freshly prepared binding reaction of 23 nmol of protein, 30 mm NaH14CO3, pH 6.5, and 50 mm Mes, pH 6.5, (total volume 50 μl) was immediately added and centrifuged at 1500 × g for 30 s, and the flow-through collected into 50 μl of 2 m NaOH. Scintillation counting was used to measure 14C counts in the flow-through.
Measurement of Intracellular pH—HEK 293T cells attached to a 24-mm diameter glass coverslip were loaded with the pH-sensitive fluorescent dye 2′,7′-bis(carboxyethyl)-5(6)-carboxyfluorescein (BCECF) through exposure to 1 μm BCECF-AM (an acetoxymethyl ester derivative) for 30 min. Intracellular pH was measured by exciting a small patch of cells at 490 and 440 nm using a microspectroflourometric system and measuring emission at 535 nm. pHi was calibrated using the high potassium nigericin method (30).
cAMP Accumulation in Vivo—HEK 293T cells were cultured in 12-well plates and labeled overnight with 1.5 μCi of [3H]adenine at 80–90% confluence. Cells were washed with phosphate-buffered saline solution and incubated at the required CO2 concentration in preincubation media (10 mm HEPES-NaOH, 117 mm NaCl, 4.5 mm KCl, 1 mm MgCl2, 11 mm glucose, 10 mm sucrose, and 2.5 mm CaCl2) containing 1 mm isobutylmethylxanthine. Preincubation mixes were pre-gassed with the desired CO2 concentration and adjusted to pH 7.0. The assay was initiated after 30 min by the addition of agonist and incubated at the required CO2 concentration. Assays were stopped with 5% (w/v) trichloroacetic acid containing 1 mm ATP and 1 mm cAMP. Products were quantified by twin column chromatography (29). For immunoblotting, samples were harvested after treatment as above except in the absence of [3H]adenine and isobutylmethylxanthine. Immunoblotting was performed using standard methodologies with anti-phospho-CREB (Ser133) and anti-α-tubulin as load control.
RESULTS AND DISCUSSION
The M. tuberculosis H37Rv genome contains at least 15 putative ACs and one cAMP phosphodiesterase, suggesting an important role for cAMP in the physiology of Mycobacterium (31–34). cAMP is implicated in the pathogenesis of mycobacteria, and CO2 has been suggested as a signal to enable Mycobacterium to avoid phagosomal acidification (35, 36). The Rv1625c gene of M. tuberculosis encodes an enzyme consisting of six putative transmembrane helices and a single Class IIIa AC catalytic domain (25, 37). The predicted topology, therefore, resembles one-half of a mammalian G-protein-regulated AC enzyme. A further similarity arises in the active site where six key catalytic residues distributed among the two catalytic domains of the G-protein-regulated ACs are present in Rv1625c to generate a homodimeric enzyme with two active sites (Fig. 1a).
FIGURE 1.

Rv1625c is stimulated by CO2. a, alignment of
the catalytic domains of Rv1625c, human AC type 7 C1 domain, rat AC type 2 C2
domain, Slr1991 of Synechocystis, and CyaB1 of Anabaena.
Numbers denote the amino acid sequence number. Arrows indicate
conserved metal binding aspartate residues. The triangle indicates
the substrate binding lysine residue, and the circle is the polymorphic D/T of
Class IIIa/b ACs. b, ratio of the specific activities of
Rv1625c204–443 when assayed in the presence of 30
mm total Ci or NaCl at various pH values (1.8
μm protein, 200 μm Mn2+-ATP, n
= 8). The inset shows the percentage of total Ci made up
by CO2 and 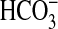 over the pH
range tested. The figure shows specific activity in the presence of 20
mm NaCl (triangles, right-hand axis) and relative
stimulation with Ci (squares, left hand axis). c, cAMP
produced by Rv1625c204–443 under conditions of Ci
disequilibrium (36 μm Rv1625c204–443, 0 °C,
10 s, 20 mm CO2, 20 mm NaHCO3, 20
mm NaCl, 100 mm Mes, pH 6.5, 200 μm
Mn2+-ATP, n = 20; *, p < 0.05). The
inset shows a representative control experiment demonstrating that
the pH was identical in all assays (circles, NaCl;
triangles, NaHCO3; squares,CO2;
arrow, assay start point). d,
Rv1625c204–443 specific activity (n = 6) was plotted
against increasing CO2. The assay mixture contained 433
nm protein and 200 μm Mn2+-ATP, pH 6.5.
The total salt concentration was adjusted to 30 mm for all data
points.
over the pH
range tested. The figure shows specific activity in the presence of 20
mm NaCl (triangles, right-hand axis) and relative
stimulation with Ci (squares, left hand axis). c, cAMP
produced by Rv1625c204–443 under conditions of Ci
disequilibrium (36 μm Rv1625c204–443, 0 °C,
10 s, 20 mm CO2, 20 mm NaHCO3, 20
mm NaCl, 100 mm Mes, pH 6.5, 200 μm
Mn2+-ATP, n = 20; *, p < 0.05). The
inset shows a representative control experiment demonstrating that
the pH was identical in all assays (circles, NaCl;
triangles, NaHCO3; squares,CO2;
arrow, assay start point). d,
Rv1625c204–443 specific activity (n = 6) was plotted
against increasing CO2. The assay mixture contained 433
nm protein and 200 μm Mn2+-ATP, pH 6.5.
The total salt concentration was adjusted to 30 mm for all data
points.
The Class IIIa Rv1625c AC was reported to be insensitive to Ci
under experimental conditions where
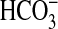 was the predominant form of
Ci. We expressed the AC domain of Rv1625c as a recombinant protein
(Rv1625c204–443) and investigated the response of enzyme to
constant Ci at varying pH (Fig.
1b). Relative stimulation
(Ci:Cl–) varied from less than 1 at pH 8.5 (0.1
mm CO2, 19.6 mm
was the predominant form of
Ci. We expressed the AC domain of Rv1625c as a recombinant protein
(Rv1625c204–443) and investigated the response of enzyme to
constant Ci at varying pH (Fig.
1b). Relative stimulation
(Ci:Cl–) varied from less than 1 at pH 8.5 (0.1
mm CO2, 19.6 mm
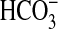 , 0.3 mm
CO2–3) to 6.3 at pH 6.5 (7.7 mm
CO2, 12.3 mm
, 0.3 mm
CO2–3) to 6.3 at pH 6.5 (7.7 mm
CO2, 12.3 mm
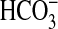 ). Stimulation of Rv1625c specific
activity was most evident below pH 7.5, explaining a failure to previously
observe a stimulation with Ci
(20). A requirement for low pH
to observe a response to Ci is consistent with a role for
CO2 as the activating species but may also be due to the altered
protonation status of Rv1625c204–443 limiting the ability of
the enzyme to respond to
). Stimulation of Rv1625c specific
activity was most evident below pH 7.5, explaining a failure to previously
observe a stimulation with Ci
(20). A requirement for low pH
to observe a response to Ci is consistent with a role for
CO2 as the activating species but may also be due to the altered
protonation status of Rv1625c204–443 limiting the ability of
the enzyme to respond to 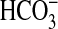 at higher
pH. We, therefore, assayed Rv1625c204–443 under conditions of
Ci disequilibrium to determine whether CO2 or
at higher
pH. We, therefore, assayed Rv1625c204–443 under conditions of
Ci disequilibrium to determine whether CO2 or
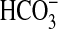 is the activating species
(22,
38). AC assays performed under
conditions of Ci disequilibrium exploit the fact that acquisition
of the
is the activating species
(22,
38). AC assays performed under
conditions of Ci disequilibrium exploit the fact that acquisition
of the 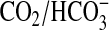 equilibrium is significantly slowed at low temperature. We defined conditions
for assaying AC under conditions of disequilibrium by following the
acquisition of the
equilibrium is significantly slowed at low temperature. We defined conditions
for assaying AC under conditions of disequilibrium by following the
acquisition of the
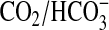 equilibrium through measuring the pH of a weakly buffered (5 mm)
Mes solution on the addition of 20 mm CO2 or
NaHCO3 in the presence or absence of carbonic anhydrase at 0 °C
(data not shown).4 In
this manner we defined conditions for assaying AC under conditions of
disequilibrium using 20 mm CO2 or
equilibrium through measuring the pH of a weakly buffered (5 mm)
Mes solution on the addition of 20 mm CO2 or
NaHCO3 in the presence or absence of carbonic anhydrase at 0 °C
(data not shown).4 In
this manner we defined conditions for assaying AC under conditions of
disequilibrium using 20 mm CO2 or
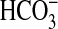 as a 10-s assay period at 0 °C
after the addition of Ci. Under these conditions, Ci is
predominantly in the form added to the assay (CO2 or
as a 10-s assay period at 0 °C
after the addition of Ci. Under these conditions, Ci is
predominantly in the form added to the assay (CO2 or
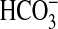 ) and has not significantly
advanced toward the equilibrium determined by assay pH (clamped with 100
mm Mes in the actual AC assays). Control experiments demonstrated
that under the conditions used for the assay final pH was equivalent when
either CO2,
) and has not significantly
advanced toward the equilibrium determined by assay pH (clamped with 100
mm Mes in the actual AC assays). Control experiments demonstrated
that under the conditions used for the assay final pH was equivalent when
either CO2, 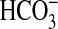 , or
Cl– were added, demonstrating that any observed stimulation
was due to addition of Ci and not a change in assay pH
(Fig. 1c;
inset). Ci disequilibrium assays proved that
Rv1625c204–443 responded to CO2 and not
, or
Cl– were added, demonstrating that any observed stimulation
was due to addition of Ci and not a change in assay pH
(Fig. 1c;
inset). Ci disequilibrium assays proved that
Rv1625c204–443 responded to CO2 and not
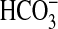 (Fig. 1c). This
demonstrates that a Class IIIa AC is able to respond to Ci and
confirms that the response is to CO2, as with Class IIIb ACs.
(Fig. 1c). This
demonstrates that a Class IIIa AC is able to respond to Ci and
confirms that the response is to CO2, as with Class IIIb ACs.
Given the similarity in response to CO2 seen in Rv1625c204–443 and Class IIIb ACs, we examined the kinetic parameters for Rv1625c and compared them to the Class IIIb ACs (Table 1). CO2 stimulated Rv1625c204–443 specific activity through an increase in kcat, similar to findings with Class IIIb ACs, supporting the idea that the two subclasses share a similar mechanism of response to CO2 (20, 22). A dose-response curve with increasing Ci revealed a 5-fold stimulation at 11.6 mm CO2 (Fig. 1d). Concentrations over 12 mm caused a gradual decrease in specific activity from this peak, making an EC50 impossible to unambiguously calculate. Stimulation was significant to a 95% confidence interval at 1.9 mm CO2.
TABLE 1.
Kinetic parameters for Rv1625c204–443 and 7C1·2C2
433 nM Rv1625c204–443 (n = 12) or 1.1 μm 7C1 and 5.8 μm 2C2 (n = 9) were assayed at pH 6.5 in the presence of 20 mm total salt (7.7 mm CO2).
|
Parameter
|
Rv1625204-443
|
7C1·2C2
|
||
|---|---|---|---|---|
| Cl- | CO2 | Cl- | CO2 | |
| Vmax (nmol of cAMP mg-1 min-1) | 30.4 ± 0.8 | 76.0 ± 2.8 | 44.9 ± 2.8 | 76.4 ± 4.1 |
| Km[ATP] (mm) (S.D.) | 0.54 ± 0.02 | 1.72 ± 0.09 | 1.89 ± 0.25 | 2.04 ± 0.23 |
| kcat (s-1) | 5.9 | 14.6 | 6.4 | 10.7 |
Given the clear relationship between Rv1625c and the Class IIIb ACs with respect to the kinetics of activation in response to CO2, we investigated the activation mechanism. Mutation of a key substrate determining lysine (Lys-646) in the Class IIIb CyaB1 AC of Anabaena ablated the response of the enzyme to CO2 (20). We generated recombinant protein for the corresponding mutation in Rv1625c (K296A) and assessed its response to CO2. Surprisingly, Rv1625c204–443 K296A retained responsiveness to CO2.4 This finding was not unique to Rv1625c as the corresponding mutation in the Class IIIb Slr1991 AC of Synechocystis (K177A) was also responsive to CO2.4 It is plausible that the substrate determining lysine is not actually a direct site of action for CO2, and we sought evidence for an alternative binding site. Ci has been proposed to help recruit the second metal ion to the active site of the Class IIIb CyaC AC of Spirulina platensis (39). Assay of Rv1625c204–443 at varying Mn2+ concentrations revealed that CO2 increased the slope of the dose response (6.6) compared with NaCl (3.0), indicating an increase in cooperativity between binding sites (Fig. 2a). On the basis of their findings in CyaC, Steegborn et al. (39) suggested that Ci interacted directly with an active site metal ion. Given our findings on Mn2+ recruitment for Rv1625c, we further investigated this hypothesis. Attempts to identify the metal co-factor as a site of CO2 interaction through enzyme assay proved uninformative, and we, therefore, developed an alternative methodology.
FIGURE 2.

CO2 binds Rv1625c in vitro and activates in vivo. a, Rv1625c204–443-specific activity (n = 6) was plotted against increasing Mn2+. The assay mixture contained 1.8 μm protein and 200 μm Mn2+-ATP, pH 6.5, and 20 mm NaCl (triangles) or 20 mm NaHCO3 (7.7 mm CO2, squares). b, recovered CO2 from a binding assay in the presence of Rv1625c204–443, bovine serum albumin (BSA), or buffer alone. c, recovered CO2 from a binding assay in the presence of Slr1991120–337 wild type (wt), Slr1991120–337 D137A D181A (Δmetal), BSA, or buffer alone. d, cAMP-dependent lacZ activity in E. coli under control (vector) conditions or in the presence of Rv1625c204–443 in samples treated with air or 10% (v/v) CO2 in air (n = 9; *, p < 0.05). The y axis denotes the concentration of ortho-nitrophenol (ONP) in the lacZ assays performed.
Radiolabeled CO2 bound to protein has been previously recovered after mixing and rapid gel filtration (40). We, therefore, performed a binding analysis to examine the requirements for CO2 binding to enzyme. CO2 bound Rv1625204–443 with no requirement for metal or substrate (Fig. 2b). Identical results were obtained for the Class IIIb ACs Slr1991 and CyaB1.4 Control proteins including bovine serum albumin and an alternative hexahistidine-tagged protein4 showed recovery indistinguishable from buffer alone, indicating an absence of any specific CO2 binding. These data would appear to eliminate a requirement for metal in the active site for CO2 binding, but it is possible that metal co-purified with protein and remained bound to enzyme. We, therefore, investigated CO2 binding in a mutant protein in which both metal binding aspartate residues were mutated to alanine (39, 41). The low yield of protein for Rv1625c204–443 D256A/D300A made this experiment impossible for Rv1625c. We, therefore, performed the equivalent experiment in the mutant protein Slr1991120–337 D137A/D181A (Fig. 2c). This confirmed that CO2 binding occurred in the absence of metal despite the fact that the protein was catalytically inactive.4 At physiological pH and CO2 concentrations, only N-terminal α-amino groups and lysine side chain ε-amino groups are likely to be sufficiently dissociated to react with CO2 (42). This is borne out in crystal structures in which carbamates are formed at lysine side chain ε-amino groups (e.g. Refs. 43–45). It is also possible that changes in the local environment may permit arginine to participate in a CO2 binding site (46). Our findings indicate that the hypothesis that Ci interacts with active site metal is incorrect and that future mechanistic studies should be directed toward sites within the apoprotein.
No prokaryotic ACs have been demonstrated to respond in vivo to
increases in
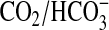 . This
is of obvious importance if prokaryotic ACs are to be posited as sensors of
CO2. A demonstration that Rv1625c is responsive to CO2
in vivo is problematic as the numerous ACs in Mycobacterium
make specific effects on Rv1625c impossible to distinguish. We, therefore,
monitored the activity of Rv1625c expressed in E. coli using a cAMP
responsive lacZ reporter construct as a suitable alternative. Using
cAMP-driven expression of lacZ as readout, we observed a consistent increase
in Rv1625c204–443 activity at elevated CO2
(Fig. 2d). LacZ
produced due to endogenous cAMP was not responsive to CO2
(Fig. 2d,
Vector). As transcription of the E. coli cya gene (the Class
I E. coli AC) is down-regulated by cAMP, expression of
Rv1625c204–443 likely reduced endogenous cAMP production and
eliminated the possibility that our observations were due to the endogenous
Cya AC (47). This demonstrates
that a prokaryotic AC can be stimulated by CO2 in an intact
bacterium and, thus, fulfils a key criterion for AC as a functional
CO2 sensor in bacteria.
. This
is of obvious importance if prokaryotic ACs are to be posited as sensors of
CO2. A demonstration that Rv1625c is responsive to CO2
in vivo is problematic as the numerous ACs in Mycobacterium
make specific effects on Rv1625c impossible to distinguish. We, therefore,
monitored the activity of Rv1625c expressed in E. coli using a cAMP
responsive lacZ reporter construct as a suitable alternative. Using
cAMP-driven expression of lacZ as readout, we observed a consistent increase
in Rv1625c204–443 activity at elevated CO2
(Fig. 2d). LacZ
produced due to endogenous cAMP was not responsive to CO2
(Fig. 2d,
Vector). As transcription of the E. coli cya gene (the Class
I E. coli AC) is down-regulated by cAMP, expression of
Rv1625c204–443 likely reduced endogenous cAMP production and
eliminated the possibility that our observations were due to the endogenous
Cya AC (47). This demonstrates
that a prokaryotic AC can be stimulated by CO2 in an intact
bacterium and, thus, fulfils a key criterion for AC as a functional
CO2 sensor in bacteria.
Building on our findings with Rv1625c, we investigated CO2 as a
stimulating ligand for a related mammalian G-protein regulated AC, an
experiment of some importance as CO2-stimulated signaling enzymes
are not known in eukaryotes (7C1·2C2;
Fig. 1a). We
investigated the response of 7C1·2C2 to 20
mm total Ci over the pH range 6.5–8.5
(Fig. 3a). Similar to
Rv1625c, optimal stimulation of 7C1·2C2 by
Ci occurred at low pH, suggesting a direct response to
CO2. Assay under conditions of Ci disequilibrium proved
7C1·2C2 was responsive to CO2 but not
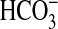 (Fig. 3b). A dose
response with increasing CO2 revealed a maximum 2–3-fold
stimulation at 7 mm CO2
(Fig. 3c). Specific
activity decreased rather than plateaued at higher CO2
concentrations; therefore, an E.C.50 was impossible to calculate.
Stimulation was significant to a 95% confidence interval at 1.7 mm
CO2. Relative stimulation of 7C1·2C2
by CO2 was similar when forskolin and/or Gαs were
used to activate the enzyme, and CO2 did not affect the affinity of
7C1·2C2 for Gαs4.
CO2 increased the affinity of 7C1·2C2
for its metal co-factor (Fig.
3d), indicating a common mechanism of activation with
Rv1625c supported by kinetic analysis
(Table 1).
(Fig. 3b). A dose
response with increasing CO2 revealed a maximum 2–3-fold
stimulation at 7 mm CO2
(Fig. 3c). Specific
activity decreased rather than plateaued at higher CO2
concentrations; therefore, an E.C.50 was impossible to calculate.
Stimulation was significant to a 95% confidence interval at 1.7 mm
CO2. Relative stimulation of 7C1·2C2
by CO2 was similar when forskolin and/or Gαs were
used to activate the enzyme, and CO2 did not affect the affinity of
7C1·2C2 for Gαs4.
CO2 increased the affinity of 7C1·2C2
for its metal co-factor (Fig.
3d), indicating a common mechanism of activation with
Rv1625c supported by kinetic analysis
(Table 1).
FIGURE 3.

Stimulation of a G-protein regulated AC by CO2in vitro. a, ratio of the specific activities of 1.1 μm 7C1 and 5.8 μm 2C2 when assayed in the presence of 20 mm total Ci or NaCl at various pH values (500 μm Mg2+-ATP, 7 μm Gαs, n = 6). The figure shows specific activity in the presence of 20 mm NaCl (triangles; right-hand axis) and relative stimulation with Ci (squares, left-hand axis). b, cAMP produced by 7C1·2C2 under conditions of Ci disequilibrium (20 μm 7C1, 3.2 μm 2C2, 0 °C, 10 s, 20 mm CO2/NaHCO3/NaCl, 100 mm Mes, pH 6.5, 1 mm Mg2+-ATP, 100 μm forskolin, n = 6, *, p < 0.05). Control experiments demonstrated that the pH was identical in all assays. c, 7C1·2C2 specific activity (n = 9) was plotted against increasing CO2 at pH 6.5. The assay mixture contained 1.1 μm 7C1, 5.8 μm 2C2, 7 μm Gαs, and 500 μm Mg2+-ATP. The total salt concentration was adjusted to 30 mm for all data points. d, 7C1·2C2 specific activity (n = 6) was plotted against increasing Mg2+. The assay mixture contained 1.1 μm 7C1, 5.8 μm 2C2, 7 μm Gαs, and 500 μm Mg2+-ATP, pH 6.5, and 20 mm NaCl (triangles) or 20 mm NaHCO3 (7.7 mm CO2; squares).
As sAC is proposed but not proven to respond to
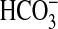 , we investigated any overlap in
specificity for Ci between sAC and the G-protein regulated ACs.
sACT relative stimulation (Ci:Cl–)
varied from 2.0 at pH 8.5 to 3.1 at pH 6.5
(Fig. 4a). The result
at pH 8.5 is consistent with a role for
, we investigated any overlap in
specificity for Ci between sAC and the G-protein regulated ACs.
sACT relative stimulation (Ci:Cl–)
varied from 2.0 at pH 8.5 to 3.1 at pH 6.5
(Fig. 4a). The result
at pH 8.5 is consistent with a role for
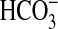 as an activating ligand; however,
the slight increase in -fold stimulation as pH is lowered suggests a response
to CO2. Under conditions of Ci disequilibrium, both
CO2 and
as an activating ligand; however,
the slight increase in -fold stimulation as pH is lowered suggests a response
to CO2. Under conditions of Ci disequilibrium, both
CO2 and 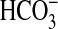 stimulated
sACT (Fig.
4b).
stimulated
sACT (Fig.
4b).
FIGURE 4.

sAC is activated by CO2 and
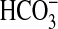 . a, 300 ng of
sACT was assayed at 30 °C for 30 min with 0.8 mm
ATP, 5 mm MgCl2, and 5 mm CaCl2
and either 20 mm total Ci or NaCl (n = 4). The figure
shows specific activity in the presence of 20 mm NaCl
(triangles; right-hand axis) and relative stimulation with
Ci (squares; left-hand axis). b, 5 μg of
sACT was assayed at 0 °C for 10 s at pH 6.4 with 0.8
mm ATP, 5 mm MgCl2, and 5 mm
CaCl2 with either 20 mm CO2,
NaHCO3, or NaCl (n = 15; *, p < 0.05).
. a, 300 ng of
sACT was assayed at 30 °C for 30 min with 0.8 mm
ATP, 5 mm MgCl2, and 5 mm CaCl2
and either 20 mm total Ci or NaCl (n = 4). The figure
shows specific activity in the presence of 20 mm NaCl
(triangles; right-hand axis) and relative stimulation with
Ci (squares; left-hand axis). b, 5 μg of
sACT was assayed at 0 °C for 10 s at pH 6.4 with 0.8
mm ATP, 5 mm MgCl2, and 5 mm
CaCl2 with either 20 mm CO2,
NaHCO3, or NaCl (n = 15; *, p < 0.05).
We next investigated whether CO2 stimulated G-protein activated
cAMP signaling in vivo. As
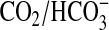 is a
potent biological buffer, we defined conditions under which changes in
internal pH (pHi) were minimized on changing
CO2. Moving from a lower to a higher CO2 concentration
gave a transient cellular acidification and vice versa
(Fig. 5a). Assays
were, therefore, performed after allowing pH homeostasis to occur, although it
is pertinent to note that G-protein-regulated ACs have been demonstrated to be
offered some protection from changes in pHi through the
action of Na+/H+ antiporters
(48). Stimulation of
G-protein-activated ACs with the β-adrenergic receptor agonist
isoproterenol gave an increase in cAMP accumulation in 5% (v/v) CO2
in air versus air (0.03% CO2; atmospheric concentrations
of CO2 in solution are ∼0.015 μm, although the
true cellular concentration is likely to be higher due to the continual
production of metabolic CO2)
(Fig. 5b). The
magnitude of this response was similar to that observed when sAC was
challenged with Ci in vivo
(12,
13). No further stimulation
was observed at 10% (v/v) CO2 as it is likely that full
CO2 activation in a precise physiological setting requires an
associated carbonic anhydrase to maintain CO2 flux
(49,
50). Similar results were
obtained when cAMP production was stimulated with forskolin, indicating that
the stimulating effect of CO2 does not occur upstream of AC
(Fig. 5c). The lack of
stimulation by CO2 in the absence of agonist for
G-protein-regulated AC confirmed that sAC was not the source of cAMP. The
inclusion of the anti-sAC inhibitor KH7 confirmed this finding
(Fig. 5c). We assessed
downstream activation of cAMP signaling by immunoblotting using an antibody
against the phosphorylated form of the cAMP-dependent protein kinase target
protein cAMP-response
element-binding protein (CREB). A small but
significant and independently repeatable increase in phosphorylation on serine
133 of CREB in the presence of agonist was observed at 5% (v/v) CO2
compared with 0.03% (v/v) CO2
(Fig. 5d).
is a
potent biological buffer, we defined conditions under which changes in
internal pH (pHi) were minimized on changing
CO2. Moving from a lower to a higher CO2 concentration
gave a transient cellular acidification and vice versa
(Fig. 5a). Assays
were, therefore, performed after allowing pH homeostasis to occur, although it
is pertinent to note that G-protein-regulated ACs have been demonstrated to be
offered some protection from changes in pHi through the
action of Na+/H+ antiporters
(48). Stimulation of
G-protein-activated ACs with the β-adrenergic receptor agonist
isoproterenol gave an increase in cAMP accumulation in 5% (v/v) CO2
in air versus air (0.03% CO2; atmospheric concentrations
of CO2 in solution are ∼0.015 μm, although the
true cellular concentration is likely to be higher due to the continual
production of metabolic CO2)
(Fig. 5b). The
magnitude of this response was similar to that observed when sAC was
challenged with Ci in vivo
(12,
13). No further stimulation
was observed at 10% (v/v) CO2 as it is likely that full
CO2 activation in a precise physiological setting requires an
associated carbonic anhydrase to maintain CO2 flux
(49,
50). Similar results were
obtained when cAMP production was stimulated with forskolin, indicating that
the stimulating effect of CO2 does not occur upstream of AC
(Fig. 5c). The lack of
stimulation by CO2 in the absence of agonist for
G-protein-regulated AC confirmed that sAC was not the source of cAMP. The
inclusion of the anti-sAC inhibitor KH7 confirmed this finding
(Fig. 5c). We assessed
downstream activation of cAMP signaling by immunoblotting using an antibody
against the phosphorylated form of the cAMP-dependent protein kinase target
protein cAMP-response
element-binding protein (CREB). A small but
significant and independently repeatable increase in phosphorylation on serine
133 of CREB in the presence of agonist was observed at 5% (v/v) CO2
compared with 0.03% (v/v) CO2
(Fig. 5d).
FIGURE 5.

Stimulation of a G-protein regulated AC by CO2in vivo. a, monitoring of HEK 293T cell pHi in response to changing CO2. b, percentage conversion of ATP into cAMP in HEK 293T cells exposed to varying CO2 under basal conditions (empty bars) or in the presence of 50 nm isoproterenol (filled bars)(n = 12). c, percentage conversion of ATP into cAMP in HEK 293T cells exposed to air (0.03% CO2; open bars) or 5% (v/v) CO2 (filled bars) with 5 μm forskolin and 1 μm KH7 (n = 12; *, p < 0.05). d, lower panel shows immunoblot of HEK 293T cell material after treatment with and without isoproterenol at varying CO2. The upper panel shows the ratio of phospho-CREB:α-tubulin bands from the quantified bands.
Our findings demonstrate that the G-protein-activated ACs are specifically CO2-activated signaling enzymes, and this is supported by similar data in a related prokaryotic enzyme. It is possible that our findings are specific only for the 7C1·2C2 protein used in this study, as any amino acid residue(s) required for CO2 binding may not be conserved among G-protein-regulated ACs in general. We hypothesize, however, that CO2 regulation will be a general feature of most if not all G-protein-activated AC isoforms. A diverse range of Class IIIa, -b, and -d ACs have been demonstrated to respond to Ci. Given the extent of sequence diversity between these AC subclasses, it is unlikely that the relatively closely related G-protein-regulated ACs of Class IIIa will differ significantly in their responses to CO2, but an examination of individual isoforms will be required to formally prove this.
Importantly, our findings overturn previous assumptions about these enzymes
as non-responsive to Ci. Furthermore, we demonstrate that
CO2 interacts directly with apoprotein to stimulate metal
recruitment and not through metal contact as previously proposed. We
demonstrate that sAC is not the sole Ci-sensitive AC in mammals as
thought and that sAC and G-protein-regulated ACs show differential sensitivity
to Ci species with G-protein-regulated ACs responsive to
CO2 and sAC responsive to
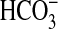 and CO2. Not only is
the cAMP signaling pathway in its entirety, therefore, able to act as a
sensing system for Ci, but different aspects of this pathway are
able to discriminate between CO2 and
and CO2. Not only is
the cAMP signaling pathway in its entirety, therefore, able to act as a
sensing system for Ci, but different aspects of this pathway are
able to discriminate between CO2 and
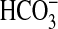 . An interesting facet of this
differential regulation is that sAC detection of Ci may be entirely
independent of intracellular pH, whereas the G-protein-responsive AC signaling
in response to Ci may occur predominantly under conditions of
pathophysiology, e.g. severe respiratory acidosis or alkalosis. Some
tissues are, however, exposed to large variations in pCO2 and have
G-protein-activated cAMP signaling central to their physiology. The duodenum
is exposed to a pCO2 of up to 400 mm Hg, and the cAMP activated
cystic fibrosis transmembrane conductance regulator is key to
. An interesting facet of this
differential regulation is that sAC detection of Ci may be entirely
independent of intracellular pH, whereas the G-protein-responsive AC signaling
in response to Ci may occur predominantly under conditions of
pathophysiology, e.g. severe respiratory acidosis or alkalosis. Some
tissues are, however, exposed to large variations in pCO2 and have
G-protein-activated cAMP signaling central to their physiology. The duodenum
is exposed to a pCO2 of up to 400 mm Hg, and the cAMP activated
cystic fibrosis transmembrane conductance regulator is key to
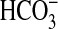 secretion in this tissue
(51). Acidification of the
epididymal lumen with an associated low
secretion in this tissue
(51). Acidification of the
epididymal lumen with an associated low
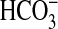 concentration is essential to
maintain stored spermatozoa in a quiescent state
(52). It might be envisaged
that these conditions would be sufficient to maintain sAC in an inactive
state, but our data demonstrating that sAC is able to respond to
CO2 suggest that this cannot be the sole mechanism for keeping sAC
activity switched off. A potential specific role for CO2 signaling
through G-protein-regulated ACs is evident in respiratory alkalosis. A key
marker of this systemic hypocapnia is a blunted phosphaturic response to cAMP
signaling through parathyroid hormone and may be explained by reduced
activation of AC in response to a lowered pCO2
(53). Further to this,
CO2 is known to regulate cAMP concentrations in the carotid body, a
peripheral chemosensor, independent of pH
(54). Although a role for sAC
versus G-protein-regulated ACs in this tissue remains to be
investigated, the clear role for adenosine-mediated cAMP production in the
carotid body is supportive of the latter
(55).
concentration is essential to
maintain stored spermatozoa in a quiescent state
(52). It might be envisaged
that these conditions would be sufficient to maintain sAC in an inactive
state, but our data demonstrating that sAC is able to respond to
CO2 suggest that this cannot be the sole mechanism for keeping sAC
activity switched off. A potential specific role for CO2 signaling
through G-protein-regulated ACs is evident in respiratory alkalosis. A key
marker of this systemic hypocapnia is a blunted phosphaturic response to cAMP
signaling through parathyroid hormone and may be explained by reduced
activation of AC in response to a lowered pCO2
(53). Further to this,
CO2 is known to regulate cAMP concentrations in the carotid body, a
peripheral chemosensor, independent of pH
(54). Although a role for sAC
versus G-protein-regulated ACs in this tissue remains to be
investigated, the clear role for adenosine-mediated cAMP production in the
carotid body is supportive of the latter
(55).
As is the case with sAC, G-protein-regulated ACs respond to CO2 with activation of downstream signaling molecules. Changes in cAMP concentrations and CREB activation are unlikely to be an artifact of the cell culture conditions used or the composition of the assay buffer as the results obtained are clearly corroborated by data using recombinant protein. Although the percentage activation of CREB detailed in this study is far below the increase in cAMP concentrations observed, it is important to consider that HEK 293T cells may not be representative of cAMP signaling systems that are responsive to CO2 in the organism (for example, see the hypothesized roles for cAMP signaling through CO2 discussed). Despite these drawbacks, the system used here has successfully demonstrated that CO2-regulated changes in hormone-activated cAMP concentrations can activate CREB phosphorylation. This is an important proof of principle even if the activation is not functional within the context of the HEK 293T cells used in this study. In vivo activation of AC is also conserved in a prokaryotic counterpart of the mammalian Class IIIa enzyme. Future research from these key findings should assess the role of G-protein-regulated ACs in response to fluctuating CO2 as discussed. The cAMP signaling pathway, therefore, represents a novel signaling pathway able to directly respond to CO2 in eukaryotes.
Supplementary Material
Acknowledgments
We thank Roger Sunahara, Matthew Wolfgang, Joachim Schultz, Lonny Levin, and Jochen Buck for the kind gift of reagents used in this work and Lonny Levin for comments on the manuscript.
Author's Choice—Final version full access.
This work was supported by the Wellcome Trust, the Leverhulme Trust, and the Biotechnology and Biological Sciences Research Council. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1.
Footnotes
The abbreviations used are: Ci, inorganic carbon; AC, adenylyl cyclase; sAC, soluble AC; CREB, cAMP-response element-binding protein; Mes, 2-[N-morpholino]ethanesulfonic acid; Mops, 3-[N-morpholino]propanesulfonic acid; GTPγS, guanosine 5′-3-O-(thio)triphosphate.
P. D. Townsend, P. M. Holliday, D. R. W. Hodgson, and M. J. Cann, unpublished observations.
References
- 1.Falkowski, P. G., and Raven, J. A. (2007) Aquatic Photosynthesis, 2nd Ed., pp. 156–200, Princeton University Press, Princeton
- 2.Roos, A., and Boron, W. F. (1981) Physiol. Rev. 61 296–434 [DOI] [PubMed] [Google Scholar]
- 3.Smith, K. S., and Ferry, J. G. (2000) FEMS Microbiol. Rev, 24 335–366 [DOI] [PubMed] [Google Scholar]
- 4.Bahn, Y. S., and Muhlschlegel, F. A. (2006) Curr. Opin. Microbiol. 9 572–578 [DOI] [PubMed] [Google Scholar]
- 5.Esposito, G., Jaiswal, B. S., Xie, F., Krajnc-Franken, M. A., Robben, T. J., Strik, A. M., Kuil, C., Philipsen, R. L., van Duin, M., Conti, M., and Gossen, J. A. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 2993–2998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones, W. D., Cayirlioglu, P., Kadow, I. G., and Vosshall, L. B. (2007) Nature 445 86–90 [DOI] [PubMed] [Google Scholar]
- 7.Kwon, J. Y., Dahanukar, A., Weiss, L. A., and Carlson, J. R. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 3574–3578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu, J., Zhong, C., Ding, C., Chi, Q. Y., Walz, A., Mombaerts, P., Matsunami, H., and Luo, M. M. (2007) Science 317 953–957 [DOI] [PubMed] [Google Scholar]
- 9.Bretscher, A. J., Busch, K. E., and de Bono, M. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 8044–8049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hallem, E. A., and Sternberg, P. W. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 8038–8043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buck, J., Sinclair, M. L., Schapal, L., Cann, M. J., and Levin, L. R. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 79–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen, Y., Cann, M. J., Litvin, T. N., Iourgenko, V., Sinclair, M. L., Levin, L. R., and Buck, J. (2000) Science 289 625–628 [DOI] [PubMed] [Google Scholar]
- 13.Jaiswal, B. S., and Conti, M. (2001) J. Biol. Chem. 276 31698–31708 [DOI] [PubMed] [Google Scholar]
- 14.Hess, K. C., Jones, B. H., Marquez, B., Chen, Y., Ord, T. S., Kamenetsky, M., Miyamoto, C., Zippin, J. H., Kopf, G. S., Suarez, S. S., Levin, L. R., Williams, C. J., Buck, J., and Moss, S. B. (2005) Dev. Cell 9 249–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie, F., and Conti, M. (2004) Dev. Biol. 265 196–206 [DOI] [PubMed] [Google Scholar]
- 16.Xie, F., Garcia, M. A., Carlson, A. E., Schuh, S. M., Babcock, D. F., Jaiswal, B. S., Gossen, J. A., Esposito, G., van Duin, M., and Conti, M. (2006) Dev. Biol. 296 353–362 [DOI] [PubMed] [Google Scholar]
- 17.Baker, D. A., and Kelly, J. M. (2004) Mol. Microbiol. 52 1229–1242 [DOI] [PubMed] [Google Scholar]
- 18.Sunahara, R. K., and Taussig, R. (2002) Mol. Interv. 2 168–184 [DOI] [PubMed] [Google Scholar]
- 19.Linder, J. U., and Schultz, J. E. (2003) Cell. Signal. 15 1081–1089 [DOI] [PubMed] [Google Scholar]
- 20.Cann, M. J., Hammer, A., Zhou, J., and Kanacher, T. (2003) J. Biol. Chem. 278 35033–35038 [DOI] [PubMed] [Google Scholar]
- 21.Kobayashi, M., Buck, J., and Levin, L. R. (2004) Dev. Genes Evol. 214 503–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hammer, A., Hodgson, D. R., and Cann, M. J. (2006) Biochem. J. 396 215–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raven, J. A. (2006) Biochem. J. 396 e5–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dessauer, C. W., and Gilman, A. G. (1996) J. Biol. Chem. 271 16967–16974 [DOI] [PubMed] [Google Scholar]
- 25.Guo, Y. L., Seebacher, T., Kurz, U., Linder, J. U., and Schultz, J. E. (2001) EMBO J. 20 3667–3675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Litvin, T. N., Kamenetsky, M., Zarifyan, A., Buck, J., and Levin, L. R. (2003) J. Biol. Chem. 278 15922–15926 [DOI] [PubMed] [Google Scholar]
- 27.Yan, S. Z., and Tang, W. J. (2002) Methods Enzymol. 345 231–241 [DOI] [PubMed] [Google Scholar]
- 28.Lee, E., Linder, M. E., and Gilman, A. G. (1994) Methods Enzymol. 237 146–164 [DOI] [PubMed] [Google Scholar]
- 29.Salomon, Y., Londos, C., and Rodbell, M. (1974) Anal. Biochem. 58 541–548 [DOI] [PubMed] [Google Scholar]
- 30.Hegyi, P., Rakonczay, Z., Gray, M. A., and Argent, B. E. (2004) Pancreas 28 427–434 [DOI] [PubMed] [Google Scholar]
- 31.Shenoy, A. R., Capuder, M., Draskovic, P., Lamba, D., Visweswariah, S. S., and Podobnik, M. (2007) J. Mol. Biol. 365 211–225 [DOI] [PubMed] [Google Scholar]
- 32.Shenoy, A. R., Sreenath, N., Podobnik, M., Kovacevic, M., and Visweswariah, S. S. (2005) Biochemistry 44 15695–15704 [DOI] [PubMed] [Google Scholar]
- 33.Shenoy, A. R., and Visweswariah, S. S. (2006) Trends Microbiol. 14 543–550 [DOI] [PubMed] [Google Scholar]
- 34.Shenoy, A. R., and Visweswariah, S. S. (2006) FEBS Lett. 580 3344–3352 [DOI] [PubMed] [Google Scholar]
- 35.Lowrie, D. B., Aber, V. R., and Jackett, P. S. (1979) J. Gen Microbiol. 110 431–441 [DOI] [PubMed] [Google Scholar]
- 36.Lowrie, D. B., Jackett, P. S., and Ratcliffe, N. A. (1975) Nature 254 600–602 [DOI] [PubMed] [Google Scholar]
- 37.Reddy, S. K., Kamireddi, M., Dhanireddy, K., Young, L., Davis, A., and Reddy, P. T. (2001) J. Biol. Chem. 276 35141–35149 [DOI] [PubMed] [Google Scholar]
- 38.Cooper, T. G., and Filmer, D. (1969) J. Biol. Chem. 244 1081–1083 [PubMed] [Google Scholar]
- 39.Steegborn, C., Litvin, T. N., Levin, L. R., Buck, J., and Wu, H. (2005) Nat. Struct. Mol. Biol. 12 32–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lorimer, G. H. (1979) J. Biol. Chem. 254 5599–5601 [PubMed] [Google Scholar]
- 41.Tesmer, J. J., Sunahara, R. K., Johnson, R. A., Gosselin, G., Gilman, A. G., and Sprang, S. R. (1999) Science 285 756–760 [DOI] [PubMed] [Google Scholar]
- 42.Lorimer, G. H. (1983) Trends Biochem. Sci. 8 65–68 [Google Scholar]
- 43.Golemi, D., Maveyraud, L., Vakulenko, S., Samama, J. P., and Mobashery, S. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 14280–14285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hall, P. R., Zheng, R., Antony, L., Pusztai-Carey, M., Carey, P. R., and Yee, V. C. (2004) EMBO J. 23 3621–3631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morollo, A. A., Petsko, G. A., and Ringe, D. (1999) Biochemistry 38 3293–3301 [DOI] [PubMed] [Google Scholar]
- 46.Cotelesage, J. J. H., Puttick, J., Goldie, H., Rajabi, B., Novakovski, B., and Delbaere, L. T. J. (2007) Int. J. Biochem. Cell Biol. 39 1204–1210 [DOI] [PubMed] [Google Scholar]
- 47.Inada, T., Takahashi, H., Mizuno, T., and Aiba, H. (1996) Mol. Gen. Genet. 253 198–204 [DOI] [PubMed] [Google Scholar]
- 48.Willoughby, D., Masada, N., Crossthwaite, A. J., Ciruela, A., and Cooper, D. M. (2005) J. Biol. Chem. 280 30864–30872 [DOI] [PubMed] [Google Scholar]
- 49.Klengel, T., Liang, W. J., Chaloupka, J., Ruoff, C., Schroppel, K., Naglik, J. R., Eckert, S. E., Mogensen, E. G., Haynes, K., Tuite, M. F., Levin, L. R., Buck, J., and Muhlschlegel, F. A. (2005) Curr. Biol. 15 2021–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Missner, A., Kugler, P., Saparov, S. M., Sommer, K., Matthai, J. C., Zeidel, M. L., and Pohl, P. (2008) J. Biol. Chem. 283 25340–25347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clarke, L. L., and Harline, M. C. (1998) Gastroenterology 114 94 [Google Scholar]
- 52.Pastor-Soler, N., Beaulieu, V., Litvin, T. N., Da Silva, N., Chen, Y., Brown, D., Buck, J., Levin, L. R., Breton, S., Sun, X. C., Zhai, C. B., Cui, M., and Bonanno, J. A. (2003) J. Biol. Chem. 278 49523–49529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoppe, A., Metler, M., Berndt, T. J., Knox, F. G., and Angielski, S. (1982) Am. J. Physiol. 243 F471–F475 [DOI] [PubMed] [Google Scholar]
- 54.Summers, B. A., Overholt, J. L., and Prabhakar, N. R. (2002) J. Neurophysiol. 88 604–612 [DOI] [PubMed] [Google Scholar]
- 55.Chen, J., Dinger, B., and Fidone, S. J. (1997) J. Appl. Physiol. 82 1771–1775 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.