

Summary
Particle-induced peri-prosthetic osteolysis is the major cause for orthopedic implant failure. This failure is mediated mainly by the action of osteoclasts, the principal cells responsible for bone resorption and osteolysis. Therapeutic interventions to alleviate osteolysis have been focused on understanding and targeting mechanisms of osteoclastogenesis. The nuclear transcription factor NFAT is an essential terminal differentiation factor of osteoclastogenesis. This transcription factor is known to cooperate with c-jun/AP-1 in mediating RANKL-induced osteoclastogenesis. We have previously determined that RANKL is an essential cytokine mediator of particle-induced osteoclastogenesis, and that PMMA particles activate JNK and c-jun/AP-1 in bone marrow macrophages (osteoclast precursors). In the current study, we investigated the effect of PMMA particles on the NFAT signaling pathway in osteoclast precursor cells. Our findings point out that PMMA particles stimulate nuclear translocation of NFAT2 in wild type osteoclast precursors which is associated with increased osteoclastogenesis. More importantly, induction of osteoclastogenesis was selectively blocked in a dose-dependent fashion by the calcineurin inhibitors, Cyclosporine-A and FK506. Further, this activation was also blocked in a time-dependent fashion by the NFAT inhibitor VIVIT. Finally, we provide novel evidence that PMMA particles induce binding of NFAT2 and AP-1 proteins. Thus our findings demonstrate that activation of the NFAT pathway in conjunction with MAP kinases is essential for basal and PMMA-stimulated osteoclastogenesis.
Keywords: NFAT, osteoclasts, PMMA, JNK
Introduction
Understanding the mechanisms underlying inflammatory osteolysis is crucial for the design of appropriate therapies. Particle-induced peri-prosthetic osteolysis is the major cause for orthopedic implant failure [1, 2]. Wear particulates, such as polymethylmethacrylate (PMMA), polyethylene (PE) and metals provoke a chronic inflammatory response at the prosthetic-bone interface [3]. The inflammatory response is evident by recruitment of macrophages/phagocytes that ingest debris and this response is further cultivated by secretion of pro-inflammatory cytokines, which in turn lead to recruitment and activation of osteoclasts [4].
The role of elevated levels of cytokines and their contribution to inflammatory osteolysis in response to particulate debris has been well established in vitro and in vivo [2, 5, 6]. As predicted, cytokines that dominate the inflammatory response feed into the osteolytic process aiding differentiation and activation of osteoclast precursors and mature osteoclasts. The presence of osteoclastogenic cytokines, primarily RANK ligand and TNF which are secreted by osteoblasts and activated T lymphocytes, has been documented in healthy and diseased tissues [7–9]. For example, RANKL has been detected in sites of bone erosion, supporting the theme that inflammatory sites attract osteoclasts and their precursors [5, 10, 11].
The biologic response of accessory cells to the inflammatory microenvironment involves secretion and elevation of pro-inflammatory mediators in the circulation [5, 10, 11]. These events further heighten the inflammatory response and favor conditions of tissue breakdown and bone erosion. The latter event is primarily mediated by bone resorbing osteoclasts. Osteoclasts are multi-nucleated cells formed by fusion monocytes/macrophages in response to RANK ligand [12, 13]. Differentiation and activation of these cells is further enhanced by circulating pro-inflammatory factors such as TNF, bacterial endotoxins, interleukin-1, E2 prostaglandins, and more [14, 15]. Osteoclastogenesis entails activation of key signaling pathways in the precursor cells in response to RANKL and pro-osteoclastogenic stimuli. Determination of this differentiation process is under the aegis of the transcription factors NF-κB, NFATc1 (also termed NFAT2) and their products [15–17].
In this regard, our recent work has shown that the transcription factor NF-κB is an essential mediator of both inflammatory and osteolytic responses [18–21]. Indeed, application of a dominant-negative form of the NF-κB inhibitory protein, IκB, which retains NF-κB in the cytoplasm, was sufficient to block osteoclast formation and activity. Moreover, blocking activation of the upstream IKK complex that is responsible for phosphorylation of IκB and subsequent activation of NF-κB, using a small peptide that perturbs assembly of the IKK complex and attenuates NF-κB activation [22], was sufficient to inhibit particle-induced osteolysis in vitro and in vivo [23].
Similar to NF-κB, the transcription factor NFAT2 (also known as NFATc1) is critical for osteoclast commitment [16]. Recent studies have shown that NFAT2 expression is dependent upon RANKL-stimulation of TRAF6 and c-Fos pathways [16, 17]. Further, activation of NFAT2 is sustained via the calcium oscillation-dependent calcineurin pathway [24]. NFAT transcription factors can act as transcriptional activators or inhibitors based on the cellular context. For example, binding to AP1 components activates lymphocyte machinery, whereas association with HDAC results with negative regulation of gene activity.
Our previous studies have shown that PMMA particles activate key signaling pathways including NF-κB and MAPK cascades and cooperate with RANKL-induced signaling in osteolysis [2, 19, 23, 25]. In the current study, we investigated the role of NFAT2 as a fundamental osteoclast intracellular signaling mechanism which maybe activated by PMMA particles. Our findings point out that expression of the transcription factor NFAT2 is regulated by PMMA particles. NFAT2 translocation to nuclei of osteoclast precursors is evident after one hour and maximal within 24 hours of exposure to PMMA particles. More importantly, our data provide evidence that inhibitors of the NFAT2 activation pathway inhibit PMMA-induced osteoclastogenesis. Finally, we show for the first time that PMMA particles induce NFAT2 binding to c-Jun and c-Fos.
Materials and Methods
Reagents
Recombinant TNF-α and M-CSF were purchased from R&D industries (Minneapolis, MN). Soluble RANKL was from PEPROTECH (Rocky Hill, NJ). Cyclosporine-A (CsA), FK506 and NFAT inhibitor VIVIT were from Calbiochem (San Diego, CA). Chemicals are from Sigma (St. Louis, MO) unless otherwise indicated.
Polymethylmethacrylate particles
Spherical PMMA particles (Polysciences, Inc., Warrington, PA) 1–10 µm in diameter (6.0 µm mean diameter, 95%<10µm) were used for all experiments as previously reported. Particles were rinsed in ethanol four times, sterilized in ethanol overnight and then rinsed 4 times with PBS. Particles were resuspended in serum-free MEM and stored at −20°C. All particle preparations tested negative for endotoxin contamination with a Limulus Amebocyte Lysate assay (BioWhittaker, Inc.). For cell culture experiments the optimal particle concentration (0.5mg/ml) represents 2.6×107 particles per 5×105 plated cells.
Animals
C57Bl/6 3–4 week old male mice were purchased from Jackson labs and housed at the Washington University School of Medicine barrier facility. Approval was obtained from Institutional Animal Care and Use Committee prior to performing this study.
Cell isolation and purification
Bone marrow macrophages (osteoclast precursors) were isolated from whole bone marrow of 4–6 wk old mice, purified over Ficoll-Hypaque gradient and cultured with 10ng/ml M-CSF and 10% heat-inactivated fetal bovine serum.
Osteoclast generation
Osteoclasts were generated by culturing purified precursor cells in the presence of M-CSF and soluble RANKL (10 ng/ml each) for 4 days. Bona fide osteoclasts develop on days 3–4 of culture and cells are then fixed and TRAP-stained or subjected to further treatments such as exposure to NFAT inhibitors and stimulation with PMMA. These TRAP-positive (purple color) multinucleated cells (> 3 nuclei/cell) are bona fide osteoclast-like cells capable of resorbing bone wafers. Cells are counted per surface area under light microscope.
Immunoblotting
Total cell lysates were boiled in the presence of 2xSDS-sample buffer (0.5 M Tris-HCl [pH 6.8], 10% (w/v) SDS, 10% glycerol, 0.05% (w/v) bromophenol blue, distilled water) for 5 min and subjected to electrophoresis on 8–12% SDS-PAGE. Proteins were transferred to nitrocellulose membranes using a semi-dry blotter (Bio-Rad, Richmond, CA) and incubated in blocking solution (10% skim milk prepared in PBS containing 0.05% Tween-20), to reduce non-specific binding. Membranes were washed with PBS/Tween buffer and exposed to primary antibodies (1 hr at room temperature), washed again four times and incubated with the respective secondary HRP-conjugated antibodies (1 hr at room temperature). Membranes were washed extensively (5 × 15 min), and an ECL detection assay (Pierce, Rockford, IL) was performed following manufacturer’s directions.
Electrophoretic mobility shift assay (EMSA) for NFAT DNA binding activity
Nuclear fractions were prepared as previously described [26]. In brief, cells were washed twice with ice-cold phosphate-buffered saline. Cells were then lifted from the dish by treating with 5 mM EDTA and 5 mM EGTA in PBS, resuspended in hypotonic lysis buffer A (10 mM HEPES (pH 7.8) 10 mM KCl, 1.5 mM MgCl, 0.5 mM dithiothreitol 0.5 mM AEBSF, 5 µg/ml Leupeptin) and incubated on ice for 15 min. NP-40 was added to a final concentration of 0.64% and samples were vortexed. Nuclei were pelleted and the cytosolic fraction was carefully removed. The nuclei were then resuspended in nuclear extraction buffer B (20 mM HEPES (pH 7.8), 420 mM NaCl, 1.2 mM MgCl, 0.2 mM EDTA 25% glycerol, 0.5 mM dithiothreitol, 0.5 mM 4-(2-aminoethyl) benzenesulfonyl fluoride (AEBSF), 5 mg/ml Pepstatin A, 5 µg/ml Leupeptin), vortexed for 30s and rotated for 30 min in 4°C. The samples were then centrifuged and the nuclear proteins in the supernatant were transferred to fresh tubes and protein content was measured using standard BCA kit (Pierce, Rockford, IL). Nuclear extracts (10 µg) were incubated with an end-labeled double stranded oligonucleotide probe commercially available from Promega (Madison, WI) containing the sequence 5′-CAACGCCCTGACCACCGATAG-3′. The reaction was performed in a total of 20 µl of binding buffer (20 mM HEPES (pH 7.8), 100 mM NaCl, 0.5 mM dithiothreitol, 1 µg poly dI-dC, and 10% glycerol) for 15–20 min at room temperature. After incubation with the labeled probe for 30 min, samples were fractionated on a 4% polyacrylamide gel and visualized by exposing dried gel to film.
Statistics
All results are representative of at least three independent experiments with similar findings. Treatment conditions are compared to control conditions with a Student t-test in appropriate experiments.
Results
PMMA particles stimulate accumulation, nuclear translocation and activation of NFAT2
PMMA and other inducers/mediators of osteolysis utilize complex signaling cascades. We have established that PMMA particles activate NF-κB, JNK and c-jun/AP-1 in osteoclast precursors. In the current experiments we examined activation of the NFAT2 pathway in osteoclast precursors by PMMA particles. Our data show that protein (1A) expression levels of NFAT are progressively enhanced following treatment of osteoclast precursors with PMMA particles for one day. More importantly, PMMA particles induce nuclear translocation of NFAT2. The data indicate that NFAT2 levels were marginally increased in the nuclei after 1 hour treatment with PMMA and robustly after 24 hour treatment in response to 0.5mg/ml and higher doses of PMMA particles (figure 1A). We further examined activation of the transcription factor. Electrophoretic mobility shift assay indicates that similar to RANKL, PMMA particles induce DNA-binding activity of NFAT in nuclei of osteoclast progenitors (figure 1B). Activation with PMMA particles was maximal at 24 hours post exposure (figure 1B).
Figure 1. Cytosolic induction and nuclear accumulation and activation of NFAT in response to PMMA particles.

A) Cytosolic and nuclear protein extracts were isolated from bone marrow macrophages after treatment with the control media or PMMA (0.1mg/ml) for the time points indicated. Equal amounts of extract protein were electrophoresed, transferred to a nitrocellulose membrane and analyzed by immunoblotting with anti-NFAT2, beta-actin, and histone-1 antibodies. B) Osteoclast precursor cells were cultured and treated with control and PMMA (0.1mg/ml). Cells were lysed 1 hour and 24 hours after PMMA treatment and were subjected to electrophoretic mobility shift assay (EMSA) using radiolabeled NFAT consensus oligonucleotide.
Cyclosporine-A (CsA), FK506, and VIVIT block PMMA particle-stimulated osteoclastogenesis
NFAT2 activation is regulated by the phosphatase calcineurin which dephosphorylates multiple phosphoserines on NFAT2 leading to its nuclear translocation and activation. The immunosuppressive drugs cyclosporine-A (CsA) and FK506 inhibit calcineurin activity. Thus, we asked whether selective inhibition of calcineurin impacts PMMA-induced osteoclastogenesis. To this end, osteoclasts were generated in vitro with RANKL (10ng /ml for 3 days) and then treated for an additional 24 hours with PMMA in the absence or presence of increasing doses of CsA or FK506. In some experiments, media was refreshed following 24 hour incubation with inhibitors to examine possible cytotoxicity. The data indicate that whereas PMMA mounts a strong osteoclastogenic response, as measured by number of TRAP-positive osteoclasts (4–5 fold increase, p<0.001), inclusion of calcineurin inhibitor significantly and dose-dependently reduced this response. Specifically, CsA at 0.5 µM and higher concentrations (1, 2 µM) reduced PMMA-induced osteoclastogenesis (figure 2A–B). CsA blocked TNF-α-induced osteoclastogenesis at the concentration of 0.1µM and higher (0.5, 1, 2 µM) (figure 2B). FK506 blocked osteoclast formation significantly at the concentration of 100, 500nM under both TNF-α and PMMA particle-induced osteoclastogenesis (figure 3A–B). To exclude non-specific or cytotoxic effects of the chemical compound, one half of the control and inhibitor-treated cultures were replenished with fresh media and stimulated with PMMA particles for an additional 48 hours. The data confirm that osteoclastogenesis resumes following removal of the inhibitor (figure 3C–D). Osteoclast recovery was measured at approximately 80% of the control (PMMA-treated) levels (figure 3D). Thus, we provide evidence that inhibition of PMMA-induced osteoclastogenesis by calcineurin inhibitor is specific and reversible thereby precluding non-specific or cytotoxic effects.
Figure 2. CsA dose-dependently inhibits PMMA-induced osteoclastogenesis.

Osteoclast precursor cells were isolated and maintained for three days in media supplemented with M-CSF (10ng/ml) and RANKL (10ng/ml). A) Cultures were then treated with control media or PMMA (0.1mg/ml) in the presence or absence of CsA (500nM) for 24 hours. B) CsA treatment with the concentrations indicated (0.01µM, 0.1µM, 0.5µM, 1µM and 2µM) started 30 minutes prior to TNF (10 ng/ml) or PMMA exposure (both TNF and PMMA were added on day 3 post RANKL treatment). All conditions were run in quadruplicate. Significance compared to control is * p<0.001 and compared to TNF or PMMA alone is **p<0.05. CsA blocked osteoclast formation significantly at the concentration of 0.1, 1, 2 μM on TNF and 0.1, 0.5, 1, 2 μM on PMMA.
Figure 3. FK506 dose-dependently inhibits PMMA-induced osteoclastogenesis in a reversible manner.

Osteoclast precursor cells were isolated and maintained for three days in media supplemented with M-CSF (10ng/ml) and RANKL (10ng/ml). A) Cultures were then treated with control media or PMMA (0.1mg/ml) with or without FK506 (NFAT inhibitor) for 24 hours. Arrows point to osteoclasts. B) Cells were treated with concentrations indicated (1nM, 10nM, 100nM, and 500nM) starting 30 minutes prior to TNF or PMMA exposure. All conditions were run in quadruplicate. C) Similar conditions to those shown in part B were used. FK506 was washed out after 24 hours and cell cultures (all conditions) were supplemented with fresh media with RANKL and M-CSF for 2 additional days. Arrows point to osteoclasts. All conditions were run in quadruplicate. Significance compared to control * p<0.001 and compared to TNF or PMMA alone **p<0.05. D) Quantification of osteoclast counts from panel C. Asterisk represents p<0.01 (FK506 compared with PMMA and FK506-withdrawn compared with FK506 treated conditions). FK506 blocked osteoclast formation significantly at the concentration of 100, 500nM both TNF and PMMA.
Finally, we show that VIVIT, a NFAT inhibitor, blocks PMMA particle-stimulated osteoclastogenesis. Under similar experimental conditions described above, VIVIT (500nM) was added on day 0 and 2 or day 2 or day3. Duplicate cultures were treated with PMMA particles on day 3 for an additional day. VIVIT blocked osteoclast formation in a time dependent manner (figure 4). Efficiency of blockade of osteoclastogenesis was high when VIVIT was added at early stages of osteoclastogenesis (up to 2 days) and decreased thereafter, indicating that NFAT2 is essential for the early determination stages of osteoclastogenesis. Similar to FK506, osteoclastogenesis resumed following withdrawal of VIVIT (not shown), suggesting that the effect of this peptide is reversible.
Figure 4. VIVIT inhibits basal and PMMA-induced osteoclastogenesis at early stages.
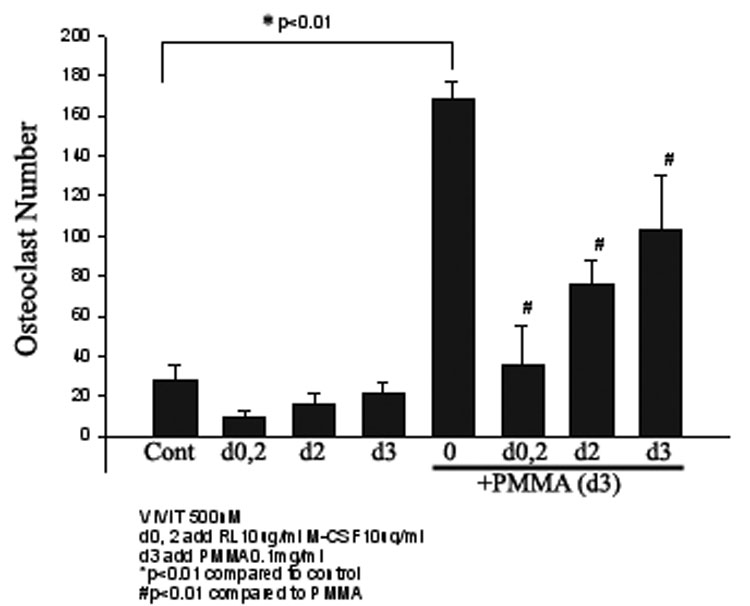
Osteoclast precursor cells were isolated and maintained for three days in media supplemented with M-CSF (10ng/ml) and RANKL (10ng/ml). The NFAT inhibitor VIVIT (500nM) was added at the indicated time points of the culture. Cultures were then treated with control media or PMMA (0.1mg/ml) on day 3 as shown. All conditions were run in quadruplicates. Significance compared to control ** p<0.01. # reflects p<0.05 compared with PMMA treated conditions. VIVIT blocked osteoclast formation by a time-dependent manner (VIVIT added on d0, 2 significantly blocked osteoclastogenesis).
CsA and FK506 inhibit NFAT DNA binding activity
To further establish that the inhibition of PMMA-induced osteoclastogenesis by these inhibitors is NFAT-specific event, we examined NFAT-DNA binding activity following PMMA stimulation of osteoclast precursor cells. The data depicted in figure 5 demonstrates that CsA and FK506 inhibit NFAT binding activity to DNA when used at the lowest concentration (100nM) shown to have an impact on osteoclastogenesis (see figure 2, figure 3). These observations provide direct evidence that CsA and FK506 inhibit PMMA-induced NFAT and subsequent osteoclastogenesis.
Figure 5. CsA and FK506 inhibit PMMA-induced NFAT activity.
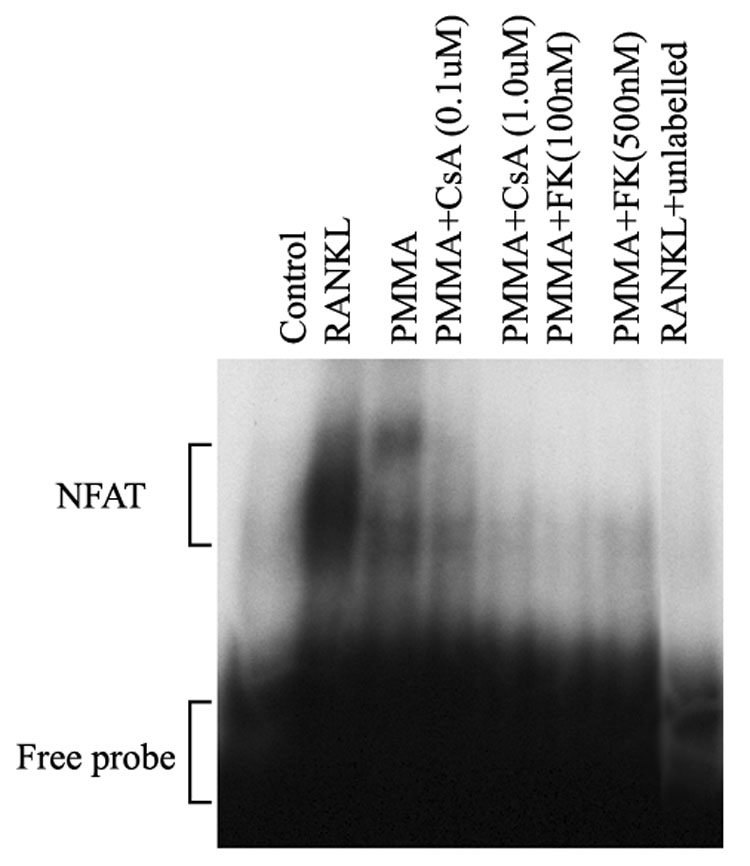
Osteoclast precursors were treated with RANKL (24hr), PMMA (24hr), or PMMA in the presence of CsA and FK506 as indicated. NFAT inhibitors were added to cells 30 minutes prior to stimulation with PMMA particles. Nuclear extracts were then prepared and subjected for EMSA using radiolabeled NFAT consensus oligonucleotide. Specificity of the shifted band was confirmed using a competition study with excess unlabeled probe (compare second lane with far right lane).
PMMA particles induce NFAT2 associates with MAP kinase components
We have shown previously that PMMA particles activate several MAP kinase pathways [27], and that the AP1/JNK MAP kinase pathway is an essential mediator of PMMA-induced inflammatory osteoclastogenesis and in vivo osteolysis. Thus, we tested whether PMMA particles induce binding of NFAT2 to components of the AP1/JNK pathway. Co-immunoprecipitation studies show that c-Jun and c-Fos both bind to NFAT2 following exposure of osteoclast precursors to PMMA particles (figure 6).
Figure 6. PMMA particles induce binding of NFAT to c-Jun and c-Fos proteins.
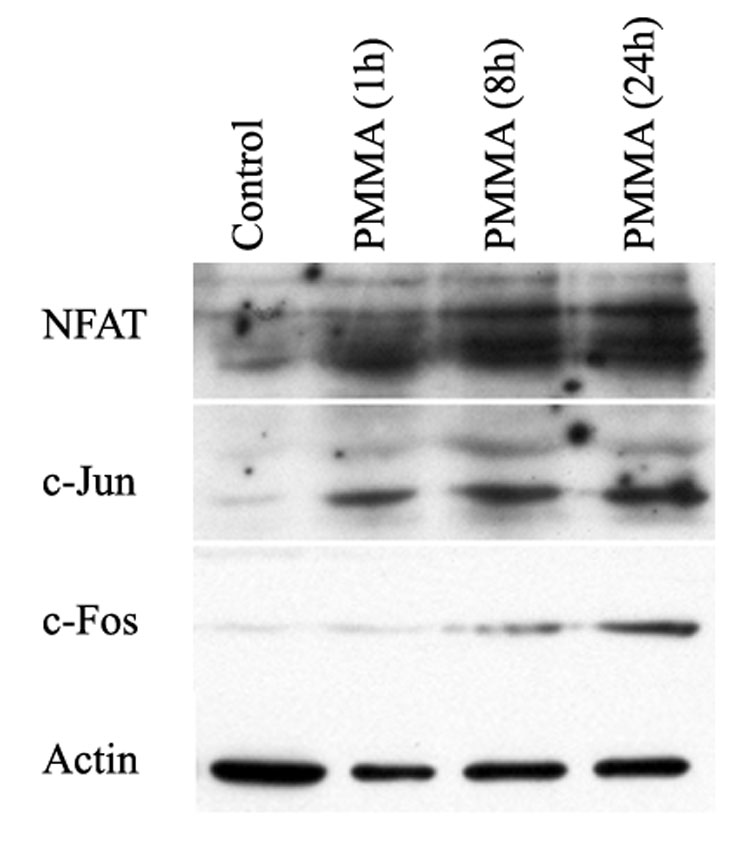
Osteoclast precursors were treated with PMMA particles (0.1 mg/ml) for the time points indicated. Cells were then lysed and reciprocal immunoprecipitations were carried out with anti-NFAT2. Co-immunoprecipitations were detected with immunoblots using appropriate antibodies (as shown). All samples were pre-incubated with non-immune IgG as a negative control (not-shown). Beta-actin expression was detected in cell lysates obtained from various treatment conditions prior to immunoprecipitation and serves as a control.
Discussion
Several studies have described the effect of orthopedic particles on osteoclast differentiation and activation [2, 28–31]. These studies provided an initial understanding of possible mechanisms that may facilitate the action of orthopedic particles in osteolysis. However, the mechanisms by which particles regulate osteoclastogenesis and osteolysis have not been fully clarified. It has been reported that PMMA particles exert a pathological effect in part via intermediate pro-inflammatory molecules [2]. In this regard, we have reported that TNF family members, especially RANKL and TNF are essential mediators of particle osteolysis [18, 32]. Furthermore, we have reported that the transcription factor NF-κB appears to be central to particle induction of osteolysis [19, 20]. Specifically, we have established that PMMA particles activate NF-κB and that introduction of NF-κB inhibitors arrest PMMA-induced osteoclastogenesis and in vivo osteolysis. We further reported that the Map kinase JNK mediates PMMA induction of osteoclasts. Blockade of c-jun/AP-1 signaling pathway by SP600125 abolished activation of JNK and c-jun/AP-1, and inhibited differentiation of macrophages into osteoclasts [25, 27].
Similar to NF-κB and JNK/c-jun pathway, the NFAT pathway is also considered as an important participant in osteoclastogenesis. Initially identified in T cells, NFAT has become an important determinant of osteoclast commitment and plays an essential role in regulating osteoclastogenesis and bone homeostasis [16, 17, 24, 33]. Recent studies have demonstrated that RANKL induces NFAT2 expression in bone marrow macrophages and showed that NFAT2 is an essential factor in osteoclastogenesis in vitro. Further, introduction of NFAT2 promoted the differentiation of bone marrow macrophages into TRAP-expressing osteoclast-like cells [16, 33]. These findings established a key role for NFAT, like RANKL, as an essential mediator of osteoclastogenesis.
In this study, we demonstrate that PMMA particles induce expression and activation of NFAT2 (Fig 1A–B). Further, we find that PMMA particles stimulate nuclear translocation of NFAT2. The data indicates that NFAT2 levels were marginally increased in the nuclei and after 1 hour with PMMA and significantly after 24 hours (Fig. 1A). Nuclear translocation was further confirmed by electrophoretic mobility shift assay (Fig. 1B). In the absence of clear understanding of how PMMA particles transmit cellular signals, the mechanisms underlying PMMA-induction of NFAT2 remain unclear. However, it is likely that cellular mediators intercede the effect of PMMA particles. We then tested specificity of PMMA’s response on induction of NFAT2 and osteoclastogenesis using appropriate inhibitors. NFAT activation is controlled by calcineurin [24]. Calcineurin deposphorylates NFAT and induces its translocation to nuclei. When present, the calcineurin/NFAT inhibitors, cyclosporine-A (CsA) and FK506, blocked PMMA-particle stimulated osteoclastogenesis in a dose dependent manner (Fig. 2, fig 3). CsA blocked TNF-induced osteoclast formation significantly at the concentration of 0.1, 1, 2 µM. However, five fold concentration of CsA was required to block PMMA-induced osteoclast formation (at least 0.5 µM and higher) (Fig. 2). This observation suggests the possibility of different signaling pathway between TNF and PMMA at least mediated by calcineurin/NFAT. The relatively higher dose (5 fold) of CsA required for inhibiting PMMA-induced NFAT further suggests that PMMA-induced signals are more complex and may involve several additive or synergistic pathways leading to a more potent activation and amplification of the NFAT pathway.
We further provide direct evidence by using the NFAT specific inhibitor VIVIT. VIVIT peptide has been developed as an optimized peptide that inhibits the NFAT family. VIVIT does not affect phosphatase activity of calcineurin and is a more specific inhibitor for NFAT than cyclosporine-A or FK506 [34]. Interestingly, VIVIT blocked osteoclast formation in a time-dependent manner. The peptide significantly blocked osteoclastogenesis when added at early stages of the culture (days 0, 2) (Fig. 4), suggesting an important role at the osteoclast determination phase.
The specificity of the inhibitory effect of NFAT inhibitors on PMMA-induced osteoclastogenesis was further verified using two approaches. First, we demonstrate that following withdrawal of the inhibitors from cultures, osteoclastogenesis resumes and reaches significant levels (80% compared with control), excluding toxic or non-specific effects. Second, administration of NFAT inhibitors abolishes PMMA-induced NFAT DNA binding activity (Fig. 5). Of the three NFAT inhibitors tested, the effect of FK506 and VIVIT were significantly reversed, whereas no definitive conclusion was reached regarding CsA. The reason for these differences is unclear, nonetheless our finding suggest that using specific and selective NFAT2 inhibitors such as FK506 and VIVT may benefit PMMA-induced osteolysis.
Recently, molecular signaling studies have demonstrated that c-jun/AP-1 may have a critical role in osteoclastogenesis by directly interacting with NFAT2. Over expression of dominant negative c-jun indicates that c-jun impacts the transcription regulatory role of NFAT2 [35]. Similar to interplay between JNK/c-jun/AP-1 and NF-κB [36, 37], regulation of NFAT2 by AP-1 components (JNK/c-Jun/c-Fos) may not be isolated or incidental. It has been reported that AP-1 and NFAT2 interaction is crucial for RANKL regulation of osteoclastogenesis [35]. Further, it has been suggested that NFAT forms ternary complex with c-jun and c-fos in response to RANKL and activates the transcriptional machinery [38–40]. In this regard, it is plausible to suggest that these transcription factors may cooperate in mediating orthopedic particle induced osteoclastogenesis and osteolysis. Indeed, we provide initial and novel evidence demonstrating PMMA-induced binding of NFAT, c-Jun and c-Fos. This finding underscores the possible mechanism by which PMMA particles may contribute to auto-amplification of the NFAT pathway leading to heightened inflammatory and osteolytic responses. Further effort is underway to establish the significance of this interaction in osteolysis. In support of this cooperative scheme typical to complex signaling pathways, we have unpublished evidence that selective inhibitors of certain MAP kinases partially block particle-induced NF-κB activation. Future studies will elucidate the molecular interaction between the various signaling pathways in particle-induced osteoclastogenesis and in vivo osteolysis.
In summary, our observations demonstrate a major function of NFAT2 during orthopedic particle - induced osteolysis. The fact that inhibition of calcineurin/NFAT and direct blockade of NFAT abolishes PMMA-induced osteoclastogenesis in vitro, presents the NFAT2 activation pathway as a potential target for intervening in particle-induced osteolysis. More importantly, our findings suggest that PMMA particles co-stimulate major signaling pathways including NFAT2, NF-κB and MAP kinases, the integration of which appears to be essential for stimulation of inflammatory osteolysis.
Supplementary Material
Acknowledgments
This study was supported in part by NIH grants AR 47443, AR 49192, grants from the Shriners Hospital for Children (YAA), and Zimmer grant (for JCC).
References
- 1.Abu-Amer Y. Mechanisms of inflammatory mediators in bone loss diseases. In: N.Rosier R, H.Evans C, editors. Molecular biology in orthopedics: AAOS. 2003. pp. 229–239. [Google Scholar]
- 2.Abu-Amer Y, Darwech I, Clohisy JC. Aseptic loosening of total joint replacements: mechanisms underlying osteolysis and potential therapies. Arthritis research & therapy. 2007;9:S6. doi: 10.1186/ar2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akatsu T, Tamura T, Takahashi N, Udagawa N, Tanaka S, Sasaki T, et al. Preparation and characterization of a mouse osteoclast-like multinucleated cell population. Journal of Bone & Mineral Research. 1992;7:1297–1306. doi: 10.1002/jbmr.5650071109. [DOI] [PubMed] [Google Scholar]
- 4.Merkel KD, Erdmann JM, McHugh KP, Abu-Amer Y, Ross FP, Teitelbaum SL. Tumor necrosis Factor-α mediates orthopedic implant osteolysis. American Journal of Pathology. 1999;154:203–210. doi: 10.1016/s0002-9440(10)65266-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ritchlin CT, Schwarz EM, O'Keefe RJ, Looney RJ. RANK, RANKL and OPG in inflammatory arthritis and periprosthetic osteolysis. Journal of Musculoskeletal Neuronal Interactions. 2004;4:276–284. [PubMed] [Google Scholar]
- 6.Horowitz SM, Purdon MA. Mechanisms of cellular recruitment in aseptic loosening of prosthetic joint implants. CalcifTissue Int. 1995;57:301–305. doi: 10.1007/BF00298886. [DOI] [PubMed] [Google Scholar]
- 7.Purdue PE, Koulouvaris P, Potter HG, Nestor BJ, Sculco TP. The cellular and molecular biology of periprosthetic osteolysis. Clinical orthopaedics and related research. 2007;454:251–261. doi: 10.1097/01.blo.0000238813.95035.1b. [DOI] [PubMed] [Google Scholar]
- 8.Hallab NJ, Anderson S, Stafford T, Glant T, Jacobs JJ. Lymphocyte responses in patients with total hip arthroplasty. Journal of orthopaedic research : official publication of the Orthopaedic Research Society. 2005;23:384–391. doi: 10.1016/j.orthres.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 9.Arora A, Song Y, Chun L, Huie P, Trindade M, Smith RL, et al. The role of the TH1 and TH2 immune responses in loosening and osteolysis of cemented total hip replacements. Journal of biomedical materials research Part A. 2003;64:693–697. doi: 10.1002/jbm.a.10200. [DOI] [PubMed] [Google Scholar]
- 10.Ritchlin CT, Haas-Smith SA, Li P, Hicks DG, Schwarz EM. Mechanisms of TNF-alpha-and RANKL-mediated osteoclastogenesis and bone resorption in psoriatic arthritis. Journal of Clinical Investigation. 2003;111:821–831. doi: 10.1172/JCI16069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haynes DR, Crotti TN. Regulation of bone lysis in inflammatory diseases. Inflammopharmacology. 2003;11:323–331. doi: 10.1163/156856003322699500. [DOI] [PubMed] [Google Scholar]
- 12.Jones DH, Kong YY, Penninger JM. Role of RANKL and RANK in bone loss and arthritis. Annals of the Rheumatic Diseases. 2002;61:ii32–ii39. doi: 10.1136/ard.61.suppl_2.ii32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khosla S. Minireview: the OPG/RANKL/RANK system. Endocrinology. 2001;142:5050–5055. doi: 10.1210/endo.142.12.8536. [DOI] [PubMed] [Google Scholar]
- 14.Wei S, Kitaura H, Zhou P, Ross FP, Teitelbaum SL. IL-1 mediates TNF-induced osteoclastogenesis. J Clin Invest. 2005;115:282–290. doi: 10.1172/JCI23394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abu-Amer Y. Advances in osteoclast differentiation and function. Current drug targets Immune, endocrine and metabolic disorders. 2005;5:347–355. doi: 10.2174/1568008054863808. [DOI] [PubMed] [Google Scholar]
- 16.Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889–901. doi: 10.1016/s1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- 17.Takayanagi H. Mechanistic insight into osteoclast differentiation in osteoimmunology. Journal of Molecular Medicine. 2005;83:170–179. doi: 10.1007/s00109-004-0612-6. [DOI] [PubMed] [Google Scholar]
- 18.Clohisy J, Teitelbaum S, Chen S, Erdmann J, Abu-Amer Y. TNF mediates PMMA particle-induced NF-kB activation in osteoclast precursor cells. JOrthopRes. 2002;20:174–181. doi: 10.1016/S0736-0266(01)00088-2. [DOI] [PubMed] [Google Scholar]
- 19.Clohisy J, Roy B, Biondo C, Frazier E, Willis D, Teitelbaum S, et al. Direct Inhibition of NF-kB Blocks Bone Erosion Associated with Inflammatory Arthritis. The Journal of Immunology. 2003;171:5547–5553. doi: 10.4049/jimmunol.171.10.5547. [DOI] [PubMed] [Google Scholar]
- 20.Clohisy J, Hirayama T, Frazier E, Han S, Abu-Amer Y. NF-kB signaing blockade abolishes implant particle-induced osteoclastogenesis. JOrthopRes. 2004;22:13–20. doi: 10.1016/S0736-0266(03)00156-6. [DOI] [PubMed] [Google Scholar]
- 21.Abu-Amer Y, Faccio R. Therapeutic approaches in bone pathogeneses: targeting the IKK/NF-kB axis. Future Medicine. 2006;1:133–146. [Google Scholar]
- 22.Dai S, Hirayama T, Abbas S, Abu-Amer Y. The IkB Kinase (IKK) Inhibitor, NEMO-binding Domain Peptide, Blocks Osteoclastogenesis and Bone Erosion in Inflammatory Arthritis. Journal of Biological Chemistry. 2004;279:37219–37222. doi: 10.1074/jbc.C400258200. [DOI] [PubMed] [Google Scholar]
- 23.Clohisy JC, Yamanaka Y, Faccio R, Abu-Amer Y. Inhibition of IKK activation, through sequestering NEMO, blocks PMMA-induced osteoclastogenesis and calvarial inflammatory osteolysis. Journal of orthopaedic research : official publication of the Orthopaedic Research Society. 2006;24:1358–1365. doi: 10.1002/jor.20184. [DOI] [PubMed] [Google Scholar]
- 24.Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes and Development. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 25.Yamanaka Y, Abu-Amer Y, Faccio R, Clohisy JC. Map kinase c-JUN N-terminal kinase mediates PMMA induction of osteoclasts. Journal of orthopaedic research : official publication of the Orthopaedic Research Society. 2006;24:1349–1357. doi: 10.1002/jor.20199. [DOI] [PubMed] [Google Scholar]
- 26.Abu-Amer Y. IL-4 abrogates osteoclastogenesis through STAT6-dependent inhibition of NF-kB. J Clin Invest. 2001;107:1375–1385. doi: 10.1172/JCI10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abbas S, Clohisy JC, Abu-Amer Y. Mitogen-activated protein (MAP) kinases mediate PMMA-induction of osteoclasts. J Orthop Res. 2003;21:1041–1048. doi: 10.1016/S0736-0266(03)00081-0. [DOI] [PubMed] [Google Scholar]
- 28.Sabokbar A, Pandey R, Athanasou NA. The effect of particle size and electrical charge on macrophage-osteoclast differentiation and bone resorption. Journal of materials science Materials in medicine. 2003;14:731–738. doi: 10.1023/a:1025088418878. [DOI] [PubMed] [Google Scholar]
- 29.Neale SD, Athanasou NA. Cytokine receptor profile of arthroplasty macrophages, foreign body giant cells and mature osteoclasts. Acta orthopaedica Scandinavica. 1999;70:452–458. doi: 10.3109/17453679909000980. [DOI] [PubMed] [Google Scholar]
- 30.Gallo J, Kamâinek P, Tichâa V, Rihâakovâa P, Ditmar R. Biomedical papers of the Medical Faculty of the University Palackây. Vol. 146. Czechoslovakia: Olomouc; 2002. Particle disease. A comprehensive theory of periprosthetic osteolysis: a review; pp. 21–28. [DOI] [PubMed] [Google Scholar]
- 31.Haynes DR, Crotti TN, Zreiqat H. Regulation of osteoclast activity in peri-implant tissues. Biomaterials. 2004;25:4877–48855. doi: 10.1016/j.biomaterials.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 32.Clohisy J, Frazier E, Hirayama T, Abu-Amer Y. RANKL is an essential cytokine mediator of PMMA particle-induced osteoclastogenesis. JOrthopRes. 2003;21:202–212. doi: 10.1016/S0736-0266(02)00133-X. [DOI] [PubMed] [Google Scholar]
- 33.Asagiri M, Sato K, Usami T, Ochi S, Nishina H, Yoshida H, et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. The Journal of Experimental Medicine J Exp Med. 2005;202:1261–1269. doi: 10.1084/jem.20051150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aramburu J, Yaffe MB, Lâopez-Rodrâiguez C, Cantley LC, Hogan PG, Rao A. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science (New York, NY) 1999;285:2129–2133. doi: 10.1126/science.285.5436.2129. [DOI] [PubMed] [Google Scholar]
- 35.Ikeda F, Nishimura R, Matsubara T, Tanaka S, Inoue Ji, Reddy SV, et al. Critical roles of c-Jun signaling in regulation of NFAT family and RANKL-regulated osteoclast differentiation. Journal of Clinical Investigation. 2004;114:475–484. doi: 10.1172/JCI19657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang G, Minemoto Y, Dibling B, Purcell NH, Li Z, Karin M, et al. Inhibition of JNK activation through NF-kappaB target genes. Nature. 2001;414:313–317. doi: 10.1038/35104568. [see comment]. [DOI] [PubMed] [Google Scholar]
- 37.Meyer C, Wang X, Chang C, Templeton D, Tan T. Interaction between c-Rel and the mitogen activated protein kinase kinase kinase 1 signaling cascade in mediating kB enhancer activation. JBiolChem. 1996;271:8971–8976. doi: 10.1074/jbc.271.15.8971. [DOI] [PubMed] [Google Scholar]
- 38.Macian F. NFAT proteins: key regulators of T-cell development and function. Nature reviews Immunology. 2005;5:472–484. doi: 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- 39.Maciâan F, Lâopez-Rodrâiguez C, Rao A. Partners in transcription: NFAT and AP-1. Oncogene. 2001;20:2476–2489. doi: 10.1038/sj.onc.1204386. [DOI] [PubMed] [Google Scholar]
- 40.Maciâan F, Garcâia-Rodrâiguez C, Rao A. Gene expression elicited by NFAT in the presence or absence of cooperative recruitment of Fos and Jun. The EMBO journal. 2000;19:4783–4795. doi: 10.1093/emboj/19.17.4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.