

SUMMARY
Non-availability of adult worms from living hosts remains a key problem in population genetic studies of schistosomes. Indirect sampling involving passage through laboratory animals presents significant ethical and practical drawbacks, and may result in sampling biases such as bottlenecking processes and/or host-induced selection pressures. The novel techniques reported here for sampling, storage and multi-locus microsatellite analysis of larval Schistosoma mansoni, allowing genotyping of up to 7 microsatellite loci from a single larva, circumvent these problems. The utility of these assays and the potential problems of laboratory passage, were evaluated using 7 S. mansoni population isolates collected from school-children in the Hoima district of Uganda, by comparing the associated field-collected miracidia with adult worms and miracidia obtained from a single generation in laboratory mice. Analyses of laboratory-passaged material erroneously indicated the presence of geographical structuring in the population, emphasizing the dangers of indirect sampling for population genetic studies. Bottlenecking and/or other sampling effects were demonstrated by reduced variability of adult worms compared to their parent field-collected larval samples. Patterns of heterozygote deficiency were apparent in the field-collected samples, which were not evident in laboratory-derived samples, potentially indicative of heterozygote advantage in establishment within laboratory hosts. Genetic distance between life-cycle stages in the majority of isolates revealed that adult worms and laboratory-passaged miracidia clustered together whilst segregating from field miracidia, thereby further highlighting the utility of this assay.
Keywords: schistosome, sampling, laboratory passage, 3 Rs, bottlenecking, miracidia, cercariae, population genetics, multiplex
INTRODUCTION
Schistosomiasis is a macroparasitic disease of profound medical and veterinary importance, with some 600 million people exposed and 200 million infected at any time throughout the tropical world (Chitsulo et al. 2000). The causative agent, Schistosoma spp. (Platyhelminth; Trematoda), has an indirect life-cycle involving obligatory alternation of generations between a mammalian definitive host and a molluscan intermediate host. Transmission between hosts occurs via free-swimming larval stages, miracidia (infective to the mollusc) and cercariae (infective to the mammal). Molecular epidemiological studies of schistosomes provide exciting opportunities to investigate many important topics, such as the contribution of parasite genetics to variation in disease burden and pathology (Brouwer et al. 2003), the genetic consequences of various control activities for the parasite population (Curtis et al. 2002), patterns of recruitment and transmission in endemic areas (Sire et al. 1999) and the potential for the circulation of strains differentially adapted to human and reservoir hosts, particularly important in the zoonotic Schistosoma japonicum (Shrivastava et al. 2005). Curtis and Minchella (2000) stressed the need for “estimates of schistosome genetic diversity ... over a broad area and in different infection foci with varying transmission dynamics”. To help achieve this, microsatellite markers have been developed for 2 of the major schistosomes of humans, S. mansoni and S. japonicum (Durand et al. 2000; Blair et al. 2001; Curtis et al. 2001; Rodrigues et al. 2002; Shrivastava et al. 2003).
A key problem for schistosome molecular epidemiological studies, however, is that adult worms are not available for direct sampling due to their location in the mesenteric system surrounding the intestine (S. mansoni and S. japonicum) or bladder (S. haematobium). Researchers have taken various approaches to circumvent these difficulties, the most common being to use indirect sampling, analysing adult worms following a single, or occasionally multiple, generation(s) of laboratory passage (Curtis et al. 2002; Rodrigues et al. 2002; Stohler et al. 2004). In such cases, isolation of the parasite depends on hatching eggs and infecting laboratory snails with miracidia and subsequently exposing laboratory mammals to cercariae from the infected snails. Such methods not only present evident disadvantages in terms of animal usage, but are also extremely time consuming, requiring several months for passage. Moreover, laboratory passage has the potential to introduce sampling error, bottlenecks and selection biases, such that the resulting isolates may not be representative of genetic variation in the original population. Indeed, previous studies have demonstrated that long-established laboratory schistosome strains may only represent 10-15% of the diversity present in that of recent field isolations, with the absence in particular of many rare alleles (Rodrigues et al. 2002; Stohler et al. 2004), although the severity of this problem in a single life-cycle passage is less established (LoVerde et al. 1985; Shrivastava et al. 2005). An alternative approach has been to characterize the schistosome populations of natural non-human hosts such as wild rats in Guadeloupe (Sire et al. 2001; Prugnolle et al. 2002), which, whilst such lethal sampling allows for direct characterization of adult worms, it carries the caveat that schistosome epidemiology may be quite different in human populations due, in part, to potential strain adaptations (LoVerde et al. 1985) and patterns of host movement (Curtis et al. 2002; Davies et al. 1999). Finally, Brouwer and co-workers (Brouwer et al. 2001, 2003) made elegant use of the amplification inherent within the schistosome life-cycle to generate sufficient parasite samples for genetic analysis through, in their case, collection of S. haematobium miracidia from schoolchildren in Zimbabwe and subsequent single miracidial infections of snails. Cercarial samples arising from asexual reproduction in the snail host, representing clonal copies of the infecting miracidium, were then used for their Randomly Amplified Polymorphic DNA (RAPD) analyses. However, such methodology still necessitates infection of laboratory or field-collected snails, thereby introducing potentially serious bottlenecking and selective pressures, such as variations in snail-schistosome compatibility (Webster and Davies, 2001). Indeed, this may be particularly relevant to this system since the snail-schistosome interaction is characterized by a high level of strain-specificity, where the likelihood of establishment in a given laboratory or field snail strain is directly influenced by both parasite (Cohen and Eveland, 1988; Davies et al. 2001) and snail host genetics (Manning et al. 1995; Morand et al. 1996; Webster et al. 2004).
An ethically and biologically superior alternative to laboratory passage must thereby consist of the development and application of novel methodologies for the field collection, storage and microsatellite analysis of larval schistosome samples directly collected from natural infections. However, these are not without practical difficulties, due primarily to the small size (S. mansoni and S. haematobium - 180 μm×80 μm, S. japonicum - 80×63 μm) and limited life-span (hours) of schistosome larvae (Rollinson and Johnston, 1996). We have previously reported a methodology for microsatellite amplification of single S. japonicum larvae (Shrivastava et al. 2005), which represented a substantial epidemiological and practical improvement over previous sampling techniques requiring laboratory passage. Nevertheless, that methodology requires samples to be frozen on collection. Moreover, it is limited to single locus analyses per individual parasite larva, which not only requires prior information about parasites in the region regarding the presence of linkage (dis)equilibrium between the loci used (Rosenberg et al. 2002), but also requires large numbers of larval samples per individual host (the number required being the product of the sample size per locus and the number of loci). Furthermore, it is not possible to assess the multilocus genotype of an individual larval stage.
The aim of the current study was therefore to develop a substantially refined alternative methodology which allows for larval sampling, room temperature-based storage, and PCR amplification of, in this case, up to 7 microsatellite loci from individual S. mansoni larvae collected directly from naturally infected humans. Furthermore, we aimed to demonstrate the utility of this novel assay, and the potential influence of the stage of sampling and isolate production on genetic data through comparative population genetic analyses of field-collected S. mansoni miracidia obtained from Ugandan schoolchildren and the resulting adult worms and miracidia obtained when these isolates were passaged in the laboratory.
MATERIALS AND METHODS
Development and optimization of assays for storage and PCR amplification of field-collected miracidia
For DNA storage, following hatching of eggs, individual miracidia were picked up using a glass pipette under the binocular microscope and washed twice by successive transfer to a new Petri dish containing autoclaved deionized (in the laboratory) or bottled spring water (in the field), in order to minimize the presence of contaminants. Single miracidia were transferred to Whatman FTA® indicator cards in a volume of 5 μl of deionized autoclaved water and allowed to dry for 1 h. Addition of the sample activates chemicals in the cards that lyse cells, inactivate proteins and immobilize the genomic nucleic acids. For DNA extraction, a 2·0 mm sample disk was removed from the Whatman FTA® cards using a Harris Micro Punch and incubated for 5 min in FTA® purification reagent (Whatman plc, Maidstone, Kent). The FTA® purification reagent was removed and replaced twice for a total of 3 washes. This was followed by 2×5 min incubations in TE buffer. Samples were dried for 10 min at 56 °C before use in PCR reactions.
A novel multiplex PCR assay using 7 previously published S. mansoni primers (Durand et al. 2000; Blair et al. 2001; Curtis et al. 2001; see Table 1) was developed. Considerable time was taken during assay development in investigating and optimizing the effects of reaction conditions and the combination of individual and different groups of primers on the results obtained using laboratory stock solutions of DNA originally derived from adult worms. The final amplification and thermal cycling protocol selected was as follows. Forward primers were fluorescently labelled using 6-FAM, TET and NED dyes (Applied Biosystems, Cheshire, UK), using different colours for alleles with overlapping size ranges. PCR reactions were performed on Gene Amp® PCR System 9700 (Applied Biosystems, Cheshire, UK). Amplifications were performed in 25 μl reactions containing template ≤1 μg DNA, 0·02 μm of each primer, 3 mm MgCl2, ultra-pure quality dNTP Mix and HotStarTaq DNA Polymerase (Qiagen® Multiplex PCR kit, West Sussex, UK). Thermal cycling was performed with a step-down PCR beginning with an initial hot-start activation of 15 min at 95 °C, followed by 40 cycles of 30 sec at 94 °C, 90 sec at annealing temperature (2 cycles at each temperature from 58 to 48 °C followed by 20 cycles at 48 °C), 60 sec at 72 °C, with a final extension at 60 °C for 30 min. Products were diluted in N, N’-dimethyl formamide with Genescan®-500 [ROX 500] size standard and analysed using an ABI 377 automated sequencer and Genescan v. 3.7 (PE Applied Biosystems). Allele sizes were calculated using ABI PRISM Genescan v 2.7 and Genotyper v 2.7 software (Applied Biosystems).
Table 1.
Descriptions of microsatellite loci that can be amplified on larval stages of Schistosoma mansoni, highlighting (in bold) the loci used in this study
| Locus | Reference | GenBank Acc no. | Repeat motif | Size range (bp) |
|---|---|---|---|---|
| SMDA28 | Curtis et al. (2001) | AF325695 | GATA | 92-128 |
| SMD43 | Curtis et al. (2001) | AF325697 | GATA | 162-166 |
| SMD011 | Curtis et al. (2001) | AF325698 | GATA | 314-363 |
| SMD25 | Durand et al. (2000) | AF202965 | CA | 272-312 |
| SMD28 | Durand et al. (2000) | AF202966 | CAA | 230-245 |
| SMD89 | Durand et al. (2000) | AF202968 | TC | 138-169 |
| SMU31768 | Durand et al. (2000) | U31768 | GAT | 179-247 |
| SCGA3 | Blair et al. (2001) | AF629514 | CT | 167-207 |
| SATA12 | Blair et al. (2001) | AI395718 | TA | 303-343 |
| CA11-1 | Blair et al. (2001) | AI068336 | GA, GT | 191-231 |
| SMS9-1 | Blair et al. (2001) | AF330106 | GT | 178-208 |
| SMS6-1 | Blair et al. (2001) | AF330104 | GT | 148-188 |
| SMS7-1 | Blair et al. (2001) | AF330105 | CA | 164-204 |
A random selection of 20 adult schistosome worms from Ugandan isolates (see below) were also used to conduct single locus amplifications of each of the 7 primers used in the multiplex assays, under identical reaction and thermocycling conditions in order to verify the robustness of the multiplex reaction. This was not possible for miracidial samples, which could only be used in a single reaction.
Assessment of utility of developed assays
Seven ‘population’ isolates were collected from schoolchildren in the Hoima district of Uganda in June 2004; 4 consisted of pooled isolates from 20 children at 4 randomly selected primary schools and the remaining 3 were isolates from individual children. Miracidia were also collected from a fourth infected child, but isolate establishment in the laboratory host failed in this case (Table 2: see Fig. 1 for location of schools). Infected children were identified by positive Kato-Katz smears and all examined children were treated with praziquantel at 40 mg/kg. Miracidia were hatched from faecal samples in bottled spring water. Samples were divided and 48 to 150 miracidia used for the immediate exposure of laboratory bred Biomphalaria glabrata (strain NHM2), at a dose of 6 miracidia per snail. The remaining samples were stored on Whatman FTA® Indicator cards (Whatman plc, Maidstone, UK). The number of snails exposed varied according to the number of miracidia hatched (Table 2). Snails were transported to the UK, where they were maintained in the laboratory in bottled spring water (Sainsburys Supermarkets plc, London, UK) and fed ad libitum on freshly washed lettuce. The laboratory was maintained at 27 °C and subject to a light regime of 12 h light and 12 h darkness, with a 30-min gradual transition at ‘dawn’ and ‘dusk’. Snails were maintained for 35-40 days, the pre-patent period during which larval development to the cercarial stage takes place. Following this time, cercariae from each snail were induced to shed by placing snails in the dark for 24 h and subsequently exposing them, at 10 am to an overhead light source (100 W) for 2 h in vials containing 16 ml of water. Cercariae from the snails representing one isolate were pooled and used to infect 5-10 laboratory mice (strain TO) per isolate at a dose of 220 cercariae per animal by paddling for 0·5 h in 30 ml of infected water. At 7 weeks post-infection, before signs of pathology had occurred, mice were asphyxiated using a rising concentration of carbon dioxide. Adult schistosomes (which varied in number from 24 to several hundred established worms per 4-mouse isolate) were recovered by a modified hepatic perfusion technique (Smithers and Terry, 1965). Thirty randomly selected adult worms were stored in 100% ethanol until required for genetic analysis, except for isolate Run-1 where only 24 adult worms were recovered and stored. Miracidia from eggs in each of the livers were stimulated to hatch by maceration through a sieve and exposure to a bright light (100 W) source for 30 min in 100 ml of deionized water. Liver samples of each isolate separately, were pooled and 30-50 miracidia per isolate stored for PCR analysis on Whatman FTA® Indicator cards (Whatman plc, Maidstone, UK), with the exception again of isolate Run-1 where a total of only 25 miracidia were successfully hatched.
Table 2.
Details of isolates collected from Hoima district of Uganda
| Isolate name | Location | Children | No. of miracidia stored on Whatman paper | No. of snails infected | Miracidial dose/snail | No. of snails surviving to patency | No. of snails shedding | Observed patency (%) |
|---|---|---|---|---|---|---|---|---|
| Kas-1 | Kasenyi Primary School (PS) | Child 712 | 52 | 8 | 6 | 8 | 4 | 50 |
| Run-P | Runga community | 20 pooled | 45 | 25 | 6 | 21 | 11 | 52 |
| Run-1 | Runga PS | Child 04-661 | 48 | 25 | 6 | 22 | 6 | 27 |
| Kas-2 | Kasenyi PS | Child 815 | none | 8 | 3 (4 snails) 2 (4 snails) |
7 | 0 | 0 |
| Kas-P | Kasenyi PS | 20 pooled | 48 | 25 | 6 | 5* | 2 | 40 |
| Kas-3 | Kasenyi PS | Child 802 | 60 | 8 | 6 | 4 | 2 | 50 |
| Kib-P | Kibiro PS | 20 pooled | 30 | 25 | 6 | 18 | 12 | 67 |
| Ton-P | Tonya PS | 20 pooled | 45 | 25 | 6 | 22 | 6 | 27 |
Very high snail mortality during exposure as accidentally left in sun.
Fig. 1.
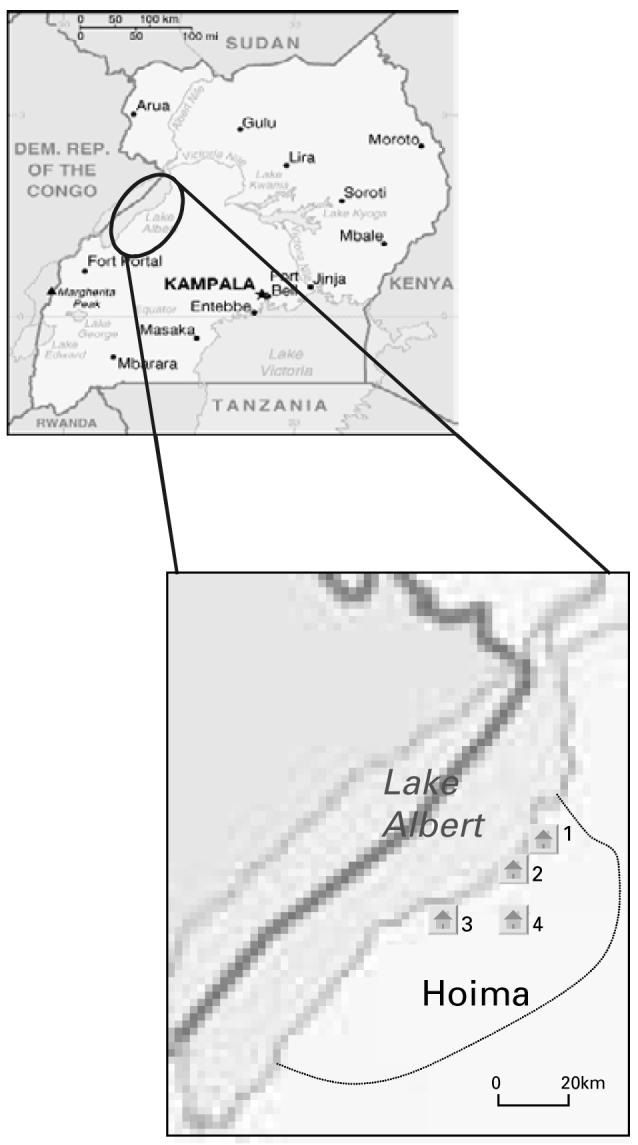
Map of Uganda showing Hoima district and the sample sites for this study (1- Runga community, 2-Kibiro primary school, 3-Tonya primary school, 4-Kasenyi primary school).
DNA was extracted from the 30 (or 24 for Run-1) adult worms using phenol chloroform and ethanol precipitation (Davies et al. 1999) and from 30 field and 30 laboratory miracidial samples (25 for Run-1) as described above. All samples were subject to multiplex PCR for 7 microsatellite loci as described above. A sample size of 30 was selected as this represented the smallest field miracidial sample (isolate Kib-P).
Data analyses
Allelic diversity
Genetic data analysis (GDA) version 1.1 (Lewis and Zaykin, 2001) was used to calculate the total number of alleles per locus and the number of private alleles per locus within individual isolates (i.e. alleles exclusive to a sample type). Differences in the mean number of polymorphic loci and number of private alleles per population were investigated using multivariate analysis of variance (Minitab, Minitab Inc, State College, PA) using the 7 microsatellite loci as dependent variables and sample type (field-collected miracidia, adult worms or laboratory derived miracidia), isolate identity and their interactions as independent variables. The analysis was repeated using the number of unique multi-locus genotypes as a covariate, to control for any potential differences in effective population sizes since adult worms could be clonal. The significance of differences was assessed using Pillai-Bartlett’s trace test statistic.
Heterozygosity
Hardy-Weinberg equilibrium analyses, testing the hypothesis that observed diploid genotypes are the product of random union of gametes (i.e. that the population is randomly mating), were carried out using Arlequin 2.000 (Schneider et al. 2000). To detect significant departure from this equilibrium, a modified version of the Markov-chain random walk algorithm described by Guo and Thompson (1992) was used, a test analogous to Fisher’s exact test using a contingency table of observed allele frequencies and the number of alleles through 100 000 permutations.
Representation of parent populations by laboratory-passaged samples
As a measure of genetic distance between populations, for each of the isolates in turn, a matrix of Cavalli-Sforza and Edwards’ chord distances (Cavalli-Sfoza and Edwards, 1967) was estimated between the sample types using POPULATIONS 1.2.28 (Langella, 1999) and visualized using Unweighted Pair Group Method with Arithmetic Mean (UPGMA). The reliability of phenograms was assessed by bootstrapping over loci with 10 000 replications.
Evidence of population structure between the isolates
Using each of the 3 sample types (field-collected miracidia, adult worms or laboratory-derived miracidia) in turn, Fst statistics measuring evidence of genetic differentiation between the isolates were calculated in Arlequin 2.000 (Schneider et al. 2000). P-values were calculated by 100 000 random permutations. UPGMA clustering was used to visually represent the data using Molecular Evolutionary Genetic Analysis (MEGA) version 3 (Kumar et al. 2004). For each of the 3 datasets individually, the correlation between the matrix of (Fst-1/Fst) genetic distance and geographical distance (as measured by the shortest straight line distance on a map) was investigated using Mantel tests. These analyses were conducted in XLSTAT-Pro 7.5 (Adinsoft Inc, New York, USA).
RESULTS
Development of assays for storage and PCR amplification of field-collected larval schistosomes
The successful development and application of novel techniques which provide a filter-paper based, room temperature storage system for schistosome larval DNA and a multiplex assay for multi-locus genotyping of single larvae with microsatellite markers, which were able to perform amplification of up to 7 loci from a single miracidium, are described (Table 1). For the 20 adult worms subject to both single locus and multiplex amplification there was 94% agreement between the reactions. Of the 140 loci, there were 3 that failed to amplify in the multiplex reaction (2 involving the same adult template DNA), 2 which failed to amplify in the single locus reaction and 4 genotyping errors (2·85%). This represented 100% concordance for primers s9-1 and smd28, 95% for primers ca11-1 and smd25, 90% for primers smd89 and smu31768 and 85% for primer smda28. In each of the 4 genotyping errors the sequencer traces were very similar but had a large stutter band which varied in height between the multiplex and single locus reaction resulting in the genotyping error.
Assessment of utility of developed assays: population genetic analysis of samples from Uganda
From 8 attempted population isolations, only 7 isolates were successfully recovered (Table 2). Due to the low number of miracidia present in isolate Kas-2, only snail infections were conducted with this isolate, and these infections failed to establish. Thus potential genetic data from this isolate were lost. Table 2 also illustrates that the frequency of patent infections differed significantly between isolates (χ2=76·8, P<0·001), as did the successful establishment in vertebrate hosts as evidenced by the limited number of samples obtained from isolate Run-1.
Allelic diversity
There was a significant difference in the total number of alleles per locus (A) between sample types (F=3·07, P=0·04), being most diverse in field miracidia (Table 3 and Fig. 2A). The number of private alleles (i.e. alleles exclusive to 1 sample type within the isolate - Ap) was significantly higher in field miracidia than in the passaged adult worms and laboratory miracidia (F=4·71, P=0·003; see Table 3 and Fig. 2B). This was also true when controlling for the unique multi-locus genotype sample size (F=3·61, P=0·02). However, there was no evidence of any difference in allelic diversity between the 7 isolates (A: F=0·87, P=0·68; Ap: 0·86, P=0·70).
Table 3.
Total number of alleles (A) and number of alleles exclusive to a subpopulation within isolates (Ap) observed for 7 microsatellite loci amplified from field-collected miracida (FM) and adult worms (AW) and resultant miracidia (LM) derived from a single generation of laboratory passage
| ca11-1 |
smd25 |
smd89 |
smu31768 |
s9-1 |
smda28 |
smd28 |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | G | A | Ap | A | Ap | A | Ap | A | Ap | A | Ap | A | Ap | A | Ap | ||
| Kas-1 | AW | 24 | 20 | 2 | 1 | 4 | 0 | 5 | 0 | 10 | 3 | 3 | 1 | 11 | 4 | 3 | 2 |
| LM | 25 | 25 | 7 | 6 | 10 | 5 | 6 | 0 | 9 | 2 | 9 | 4 | 10 | 3 | 9 | 5 | |
| FM | 29 | 25 | 4 | 3 | 13 | 7 | 7 | 1 | 9 | 3 | 5 | 1 | 9 | 2 | 8 | 4 | |
| Run-P | AW | 29 | 27 | 2 | 1 | 8 | 1 | 3 | 0 | 16 | 9 | 4 | 1 | 9 | 2 | 2 | 0 |
| LM | 23 | 23 | 2 | 1 | 13 | 6 | 4 | 0 | 12 | 4 | 7 | 4 | 9 | 2 | 12 | 10 | |
| FM | 24 | 24 | 1 | 0 | 8 | 2 | 6 | 1 | 4 | 0 | 3 | 0 | 14 | 9 | 1 | 0 | |
| Run-1 | AW | 22 | 17 | 1 | 0 | 5 | 1 | 6 | 2 | 16 | 12 | 5 | 3 | 8 | 4 | 1 | 0 |
| LM | 17 | 13 | 1 | 0 | 4 | 0 | 6 | 2 | 5 | 2 | 3 | 2 | 3 | 1 | 4 | 1 | |
| FM | 30 | 28 | 2 | 1 | 14 | 9 | 6 | 2 | 9 | 6 | 6 | 4 | 6 | 4 | 8 | 5 | |
| Kas-P | AW | 27 | 9 | 2 | 0 | 5 | 0 | 3 | 1 | 7 | 1 | 6 | 4 | 7 | 1 | 2 | 1 |
| LM | 27 | 21 | 2 | 1 | 7 | 2 | 4 | 3 | 13 | 5 | 4 | 2 | 14 | 7 | 12 | 11 | |
| FM | 29 | 22 | 2 | 0 | 12 | 6 | 5 | 3 | 8 | 3 | 2 | 0 | 14 | 9 | 2 | 1 | |
| Kas-3 | AW | 27 | 13 | 2 | 1 | 6 | 0 | 4 | 0 | 12 | 10 | 3 | 1 | 10 | 3 | 2 | 0 |
| LM | 21 | 17 | 2 | 1 | 5 | 0 | 4 | 0 | 6 | 3 | 3 | 1 | 8 | 2 | 3 | 1 | |
| FM | 22 | 20 | 2 | 1 | 10 | 6 | 6 | 2 | 12 | 8 | 5 | 4 | 6 | 4 | 5 | 4 | |
| Kib-P | AW | 29 | 25 | 1 | 0 | 7 | 0 | 4 | 2 | 9 | 6 | 4 | 2 | 11 | 4 | 1 | 0 |
| LM | 24 | 24 | 4 | 3 | 10 | 3 | 11 | 0 | 8 | 2 | 5 | 2 | 13 | 5 | 6 | 5 | |
| FM | 30 | 23 | 2 | 1 | 13 | 7 | 6 | 6 | 10 | 5 | 7 | 6 | 2 | 1 | 5 | 4 | |
| Ton-P | AW | 29 | 21 | 1 | 0 | 6 | 1 | 1 | 0 | 5 | 2 | 4 | 1 | 14 | 7 | 1 | 0 |
| LM | 25 | 21 | 3 | 2 | 7 | 2 | 5 | 1 | 4 | 0 | 4 | 1 | 13 | 4 | 1 | 0 | |
| FM | 27 | 27 | 4 | 3 | 14 | 8 | 6 | 2 | 10 | 5 | 4 | 4 | 5 | 2 | 17 | 16 | |
The number of individuals successfully typed (N) and the number of unique multi-locus genotypes (G) recorded are shown.
Fig. 2.

Comparison of mean (±standard error of the mean) (A) total number of alleles and (B) private alleles (exclusive to a particular sample type within isolates) for field miracidia (miracidia collected directly in the field), and adult worms and miracidia (laboratory miracidia) following a single generation of laboratory passage.
Heterozygosity
Patterns of heterozygosity are shown in Table 4 and Fig. 3. Comparison of the expected and the observed heterozygosity within isolates demonstrated a significant deficiency of heterozygotes in field miracidia; this pattern was also observed to a lesser extent in the laboratory-derived miracidia. However, little difference between observed and expected heterozygosity was apparent in adult worms. Moreover, in field miracidia there was variation in this pattern according to the isolate type, and the heterozygote deficiency was stronger in isolates from single children than in pooled isolates of 20 children. There was no difference in patterns of heterozygosity between the isolates in adult worms or laboratory-derived miracidia.
Table 4.
Expected (He), and observed heterozygosity (Ho) for 7 microsatellite loci amplified from field-collected miracidia (FM) and adult worms (AW) and resultant miracidia (LM) derived from a single generation of laboratory passage
| ca11-1 |
smd25 |
smd89 |
smu31768 |
s9-1 |
smd28 |
smda28 |
mean |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Isolates | He | Ho | He | Ho | He | Ho | He | Ho | He | Ho | He | Ho | He | Ho | He | Ho | |
| Kas-1 | AW | 0·04 | 0·04 | 0·72 | 1·00 | 0·62 | 0·63 | 0·78 | 0·90 | 0·52 | 0·38* | 0·07 | 0·07 | 0·87 | 0·72* | 0·52 | 0·53 |
| LM | 0·41 | 0·13* | 0·86 | 0·81* | 0·74 | 0·52* | 0·92 | 0·91 | 0·94 | 0·60* | 0·59 | 0·28* | 0·85 | 0·67* | 0·76 | 0·56 | |
| FM | 0·20 | 0·06* | 0·97 | 0·54* | 0·22 | 0·14 | 0·92 | 0·46* | 1·00 | 0·25* | 0·56 | 0·18* | 0·81 | 0·28* | 0·67 | 0·27 | |
| Run-P | AW | 0·13 | 0·10 | 0·79 | 0·50* | 0·54 | 0·57 | 0·93 | 1·00 | 0·55 | 0·27 | 0·13 | 0·10 | 0·84 | 0·75* | 0·56 | 0·47 |
| LM | 0·10 | 0·05 | 0·86 | 0·48* | 0·54 | 0·41 | 0·91 | 0·86* | 0·90 | 0·63* | 0·67 | 0·48 | 0·88 | 0·43* | 0·69 | 0·47 | |
| FM | mono | 0·87 | 0·42* | 0·28 | 0·23 | 0·79 | 1·00 | 0·80 | 0·33 | mono | 0·89 | 0·38* | 0·72 | 0·47 | |||
| Run-1 | AW | mono | 0·69 | 0·75 | 0·70 | 0·88 | 0·92 | 0·94 | 0·62 | 0·33 | mono | 0·71 | 0·50 | 0·73 | 0·68 | ||
| LM | mono | 0·67 | 0·81 | 0·78 | 0·62 | 0·87 | 0·40 | 1·00 | 0·50 | 0·58 | 0·20 | 0·79 | 0·33 | 0·78 | 0·48 | ||
| FM | 0·12 | 0·06 | 0·89 | 0·34* | 0·24 | 0·21 | 0·97 | 0·50* | 1·00 | 0·50 | 0·60 | 0·21* | 0·73 | 0·11* | 0·65 | 0·28 | |
| Kas-P | AW | 0·10 | 0·07 | 0·68 | 0·73 | 0·32 | 0·33 | 0·76 | 0·50 | 0·58 | 0·53 | 0·07 | 0·03 | 0·83 | 0·47* | 0·48 | 0·38 |
| LM | 0·22 | 0·59 | 0·91 | 0·44* | 0·45 | 0·32 | 0·93 | 0·87 | 0·69 | 0·14* | 0·84 | 0·57* | 0·87 | 0·31* | 0·70 | 0·46 | |
| FM | 0·06 | 0·03 | 0·89 | 0·40* | 0·31 | 0·30 | 0·83 | 0·60* | 0·73 | 0·33 | 0·07 | 0·04 | 0·99 | 0·55* | 0·56 | 0·32 | |
| Kas-3 | AW | 0·10 | 0·07 | 0·81 | 0·79* | 0·40 | 0·37 | 0·90 | 0·76* | 0·70 | 0·63 | 0·10 | 0·07* | 0·89 | 0·50* | 0·56 | 0·45 |
| LM | 0·66 | 0·00 | 0·87 | 0·40 | 0·43 | 0·42 | 0·82 | 0·60 | 0·67 | 0·20 | 0·32 | 0·33 | 0·84 | 0·64 | 0·66 | 0·37 | |
| FM | 0·35 | 0·06 | 0·91 | 0·77 | 0·42 | 0·17* | 0·89 | 0·73* | 1·00 | 0·25* | 0·59 | 0·13* | 0·88 | 0·29* | 0·72 | 0·34 | |
| Kib-P | AW | mono | 0·70 | 0·83 | 0·25 | 0·24 | 0·78 | 0·92 | 0·56 | 0·22* | mono | 0·92 | 0·68* | 0·64 | 0·58 | ||
| LM | 0·47 | 0·21* | 0·81 | 0·50* | 0·47 | 0·41 | 0·83 | 0·46* | 0·78 | 0·86 | 0·36 | 0·15* | 0·89 | 0·77 | 0·66 | 0·48 | |
| FM | 0·12 | 0·06 | 0·87 | 0·48* | 0·43 | 0·46 | 0·85 | 0·63* | 1·00 | 0·40* | 0·63 | 0·25 | 0·31 | 0·00 | 0·60 | 0·33 | |
| Ton-P | AW | mono | 0·64 | 0·97 | mono | 0·62 | 1·00 | 0·35 | 0·22 | mono | 0·86 | 0·71* | 0·62 | 0·72 | |||
| LM | 0·41 | 0·08* | 0·78 | 0·46 | 0·31 | 0·29 | 0·60 | 0·90 | 0·80 | 0·67 | mono | 0·88 | 0·70 | 0·63 | 0·52 | ||
| FM | 0·37 | 0·12* | 0·92 | 0·65* | 0·30 | 0·28* | 0·85 | 0·67* | 1·00 | 0·33 | 0·81 | 0·56 | 0·68 | 0·38 | 0·71 | 0·43 | |
Significant differences between observed and expected values are highlighted (P<0·01). Tests were not done where populations were monomorphic for a particular locus (mono).
Fig. 3.

Comparison of mean (±standard error of the mean) observed (Ho) and expected heterozygosity (He) (±standard error of mean) across 7 microsatellite loci for (A) field miracidia, (B) adult worms and (C) laboratory-derived miracidia.
Representation of parent populations by laboratory-passaged samples
There was evidence of low yet significant genetic differentiation between the parent field-collected miracidia and their derived laboratory-passaged samples. When each isolate was considered separately, for all isolates, field miracidia segregated from clusters of adult worms and laboratory-passaged miracidia, and the amount of variation differed between the isolates (Fig. 4A-G).
Fig. 4.

UPGMA depicting Cavalli-Sforza Edwards chord measured separately for 7 Schistosoma mansoni isolates between field-collected miracidia (FM), and adult worms (AW) and miracidia (LM) obtained following a single generation of laboratory passage.
Evidence of population structure between the isolates
In order to determine if the use of laboratory-passaged material affected conclusions regarding the presence of population differentiation between isolates, Fst statistics were calculated between isolates using any single life-cycle stage at a time. These matrices revealed differences in the presence or absence of population structure and in the clustering patterns between isolates (Fig. 5A-C). As shown in Fig. 5A, there was little evidence of differentiation between the original field miracidial populations (Fst values ranged from zero to 0·13 with mean of 0·014; 4 of 21 pairwise comparisons were significant at ά=0.05) and no evidence of an association with geographical distance. This was supported by a non-significant Mantel test (r=−0·053, P=0·60). In contrast, using adult worms, Fst values ranged from 0 to 0·23 with a mean value of 0·08. Nineteen of 21 pairwise comparisons were significant at ά=0.05 (data not shown). An UPGMA phenogram (Fig. 5B) suggested separation between populations and there was evidence of samples from the same geographical area clustering together. There was a positive correlation between the matrix of the measure Fst/(1-Fst) and geographical distance (r=0·56, P=0·004). The phenogram using laboratory-derived miracidia was similar to that of the adult worms, although in general distances between isolates were reduced (mean=0·02, range=0 to 0·07).
Fig. 5.
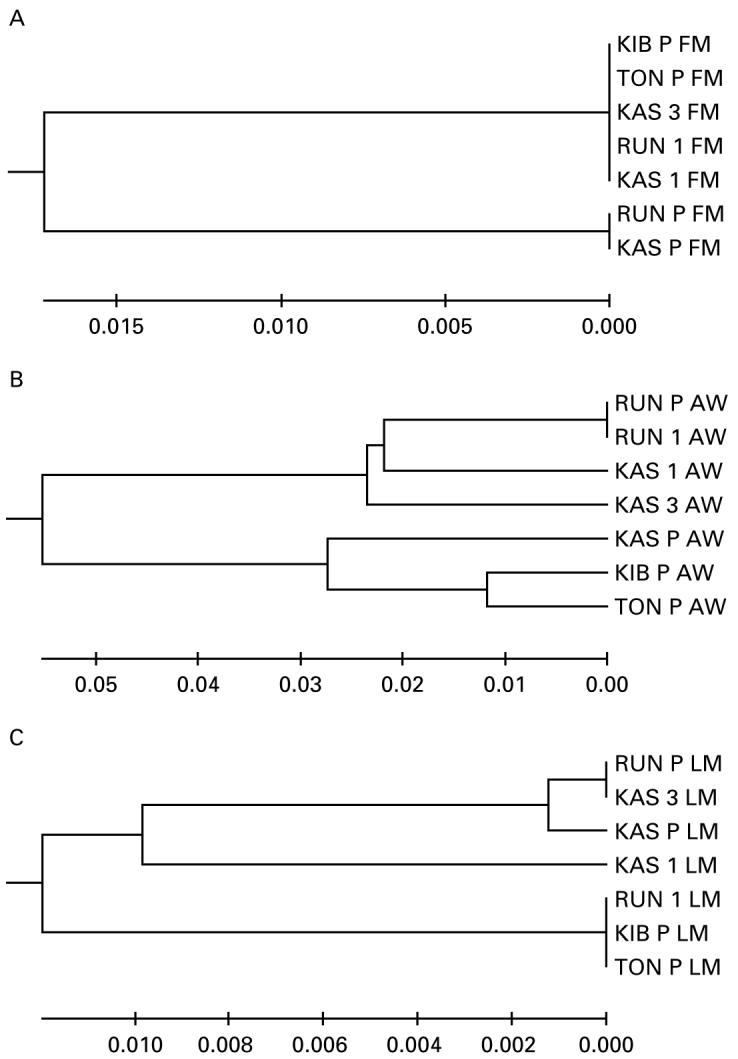
Comparison of UPGMA phenograms depicting Fst distance between 7 Schistosoma mansoni isolates from 4 geographical locations using (A) field-collected miracidia (FM), (B) adult worms (AW) and (C) laboratory-passaged miracidia (LM). Details of isolates are shown in Table 2 and P values are reported in the text.
DISCUSSION
The techniques described here present novel methodologies for both the simple, long-term, room temperature storage of field-collected larval schistosome samples on Whatman FTA® cards and for multiplex PCR analysis leading to multilocus genotyping of single larval samples. This complements and expands upon our previously described techniques which enabled genotyping of individual S. japonicum larval samples stored by freezing (Shrivastava et al. 2005), and presents a further improvement in allowing multilocus analysis, in this case for up to 7 microsatellite loci, from a single larva. It is suggested that these novel techniques will be particularly valuable for field situations, where access to cold storage is often difficult, and for analyses of schistosome samples in cases of low infection intensity or post-treatment situations. The methodology does, nevertheless, have a few limitations - in particular since only a single PCR reaction is possible from a single parasite, one cannot verify the percentage genotyping error by repeat genotyping a particular individual, and data are lost if PCR amplification fails. A further caveat is that the set of loci used here were not sufficiently polymorphic for full discrimination, as can be seen in Table 3 where the number of miracidia typed in most cases exceeded the number of unique multi-locus genotypes observed. However, comparison of patterns of population structure assessed using laboratory-derived miracidia were similar to those generated using adult worms, indicating that miracidia are a good representation of their parent worm population in this respect and rare alleles, in particular, were evidently lost during transfer to laboratory passage. Overall, such refined methodology thereby represents a significant advance in the technical capacity available for molecular epidemiological studies of Schistosoma spp. and should prompt similar research in other trematode species.
Inherent within our study we also directly assessed the advantages of these larval assays through investigation of the potential for sampling error, bottlenecking and other biases ordinarily experienced during a single generation of laboratory passage to obtain adult worms, the standard prior technique used in population genetic studies of schistosomes. Indeed, we demonstrate here, for the first time, evidence that laboratory passage can seriously bias interpretation of patterns of population structure between endemic regions. Fst statistics, Mantel correlations and UPGMA phenograms generated by using adult worms indicated population structuring and clustering based on the geographical origin of the isolates, but this pattern was not evident in phenograms generated by the parent field-collected miracidia. Hence, if inferences were to be drawn regarding the presence of population structure in the schistosome population using only (adult) derived samples, serious errors in interpretation may result. Well-documented characteristics of the schistosome life-cycle almost inevitably predict that isolations are likely to be subject to sampling error and/or be non-random. For example, infection rates of snails from single miracidia range from 10% to 80% depending on the snail-schistosome strain combination, and have been demonstrated to be directly influenced by parasite genetics (Davies et al. 2001) and host and parasite geographical strain (Morand et al. 1996). In our study, isolate Kas-2 was lost because of its non-establishment in laboratory bred snails and there existed a significant difference between isolates in the frequency of patent infections, such that the degree of bottlenecking experienced might be expected to vary. Infection of definitive mammalian hosts also exhibits high levels of redundancy and is directly influenced by parasite strain (Nelson and Saoud, 1968; Thiongo et al. 1997). Moreover, differential host-induced selection has already been demonstrated according to the species of definitive host used (LoVerde et al. 1985; Shrivastava et al. 2005). Differences between geographical areas, for example, in the presence and species of reservoir hosts might also be expected to have a strong effect on the randomness of laboratory isolations. We suggest that geographical differences in compatibility with the laboratory snails used for isolation collection could be one mechanism for the erroneous conclusion of geographical subdivision of populations using passaged samples in this study. Even without the presence of host-induced selection, bottlenecks and founder effects during isolate establishment are likely to bias sample sizes and hence interpretation of derived data; a situation made all the more complex by potential relatedness of adult worms whose pre-cursors are generated by asexual reproduction in the snail host. Successful infection of, and survival in, laboratory snails was indeed observed to be a highly significant factor in our study, reducing effective sample sizes from 50-150 naturally collected parasites to as few as 12 potential genotypes following passage in some isolates.
There was also evidence of reduced allelic variability in the passaged samples, even when correcting for potential sample size variation between sample types due to possible clonal relatedness between adult worms. This could be due to founder effects, bottlenecking and/or selection during laboratory passage. Such losses can arise due to failure of miracidia to establish in snails, pre-patent mortality of snails infected with different genotypes, competitive interactions between larvae within snails, failure of cercariae to infect rodents or failure of cercariae to establish and mature to adult worms, or most likely a combination of these factors. Unique multi-locus genotype variation in the passaged samples was relatively high, although many genotypes differed at a single or small number of loci. The number of multi-locus genotypes in passaged adult worms reflects the number of miracidia establishing and successfully developing in snails and then in the definitive hosts, and suggests that initial snail infection was a major bottleneck point. However, mutation can occur during asexual reproduction in the snail host although this is likely to occur in only a single locus or small number of loci for a particular individual. Any such mutation will tend to increase the number of multilocus genotypes observed in adult worms, but will result in MLGs differing only by a single/small number of loci as indeed we observed in this study. Genetic diversity was highest in the original parent field-collected miracidia, particularly in terms of the presence within isolates of private alleles in this group. However, smaller numbers of private alleles were also present in the derived populations, suggesting that miracidial sample sizes must be kept high to represent most of the parasite genotypes present and is complimentary to our previous results (Shrivastava et al. 2005). Higher levels of total diversity were apparent in the laboratory-derived miracidia than in the adult worms, which could be indicative of high levels of new mutation. This might also reflect a reduced specificity in the microsatellite assay of larval stages, although this is less likely since if this were the case, the number of private alleles would also be expected to be higher in laboratory-derived miracidia than adult worms, where in fact no significant difference was observed. Sampling error may also affect such patterns, and thus caution in the interpretation of this diversity data is necessary.
Comparison of observed heterozygosity to the values expected from the allele frequencies assuming Hardy-Weinberg equilibrium revealed a significant deficiency of heterozygotes in the field-collected miracidia, particularly in those isolates generated and collected from single individuals, perhaps due to pairing between related worms and/or structuring in the population at the level of individual hosts. However, this heterozygote deficiency was apparently lost in the adult worms. Since adult worms are generated from miracidia solely by asexual reproduction, maturation and development, one could speculate that this may indicate some form of heterozygote advantage during either miracidial establishment or progression through the schistosome life-cycle. It is plausible that more genetically diverse individuals are better able to infect hosts as has been suggested in models of host-parasite coevolution (Thompson, 1994) and indeed a correlation between heterozygosity and clone size has been observed in S. mansoni (Prugnolle et al. 2004) in rats. This merits further investigation, since changes in heterozygosity are a potential outcome of mass chemotherapy programmes. A non-biological explanation for the decreased observed heterozygosity is that multiplex PCR from low yields of miracidial DNA results in null amplification. The pattern arising from allelic diversity and heterozygosity estimates were also reflected in the direct evidence from UPGMA phenograms that passaged material was variably genetically differentiated from the naturally collected field miracidia in all 7 isolates studied, and again argues that material collected from even a single generation of laboratory passage may be poorly representative of natural infections.
To conclude, novel techniques for sampling, long-term room temperature storage and multi-locus genotypic analysis of larval stages of schistosomes have been developed. This methodology allows the replacement of laboratory animals used in sample collection, and is thus in keeping with the ‘3Rs’ (the reduction, refinement and replacement of animal use), the cornerstone of animal experimentation ethics (Wolfensohn and Lloyd, 1999). Moreover, such techniques are logistically easier and more efficient than previous methods, allowing collection and analysis of samples within a matter of days. Since there is a substantial amount of genetic variation present in schistosome miracidia, these assays on larval stages will prove extremely helpful in understanding the epidemiology of this system. Finally, we have used these techniques to investigate the potential for sampling error and biases introduced by transfer of parasite isolates to laboratory passage and demonstrate that there is a high probability of problems in this approach. Our results overall thereby reveal the epidemiological, ethical and practical advantages of examining larval samples, directly collectable from natural infections, in order to conduct population genetic studies of schistosomes.
Acknowledgments
We are very grateful to the children and teachers of the Kibiro, Tonya and Kasenyi Primary Schools and the children and adults of the Runga community for taking part in this survey, to the technical team of the Vector Control Division, Kampala and collaboration with Professor Alan Fenwick and the ongoing SCI programme for enabling field collections, to Dr Russell Stothard for assistance with field work and for the development of the field miracidia hatching protocol, to Nicole Bilek and Julia Llewellyn-Hughes for assistance with Genescan reactions, to Mike Anderson and Jayne King for assistance with life-cycle maintenance, and to two anonymous referees for comments on the text. This research was funded by grants from the Wellcome Trust (C.M.G., D.R., J.P.W. grant no. GR063774), The Felix Foundation (J.S.), The Medical Research Council (P.H.L.L., J.P.W.), the Royal Society (J.P.W.), and the Bill and Melinda Gates Schistosome Control Initiative (N.K., P.H.L.L., J.P.W.). Ethical approval for this study (application 03.36) was granted by National Health System Local Research Ethics Committee (NHS-LREC) of St Mary’s Hospital, London as well as approval from the Uganda Ministry of Health, Kampala.
REFERENCES
- Blair L, Webster JP, Barker GC. Isolation and characterisation of polymorphic microsatellite markers in Schistosoma mansoni from Africa. Molecular Ecology Notes. 2001;1:93–95. [Google Scholar]
- Brouwer KC, Ndhlovu P, Munatsi A, Shiff CJ. Genetic diversity of a population of Schistosoma haematobium derived from schoolchildren in east central Zimbabwe. Journal of Parasitology. 2001;87:762–769. doi: 10.1645/0022-3395(2001)087[0762:GDOAPO]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Brouwer KC, Ndhlovu PD, Wagatsuma Y, Munatsi A, Shiff CJ. Urinary tract pathology attributed to Schistosoma haematobium: does parasite genetics play a role? American Journal of Tropical Medicine and Hygiene. 2003;68:456–462. [PubMed] [Google Scholar]
- Cavalli-Sforza LL, Edwards AWF. Phylogenetic analysis: models and estimation procedures. American Journal of Human Genetics. 1967;19:233–257. [PMC free article] [PubMed] [Google Scholar]
- Chitsulo L, Engels D, Montressor A, Savioli L. The global status of schistosomiasis and its control. Acta Tropica. 2000;77:41–51. doi: 10.1016/s0001-706x(00)00122-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen LM, Eveland LK. Schistosoma mansoni: characterisation of clones maintained by the microsurgical transplantation of sporocysts. Journal of Parasitology. 1988;74:963–969. [PubMed] [Google Scholar]
- Curtis J, Minchella DJ. Schistosome population genetic structure: when clumping worms is not just splitting hairs. Parasitology Today. 2000;16:68–71. doi: 10.1016/s0169-4758(99)01553-7. [DOI] [PubMed] [Google Scholar]
- Curtis J, Sorensen RE, Minchella DJ. Schistosome genetic diversity: the implications of population structure as detected with microsatellite markers. Parasitology. 2002;125:S51–S59. doi: 10.1017/s0031182002002020. [DOI] [PubMed] [Google Scholar]
- Curtis J, Sorensen RE, Page LK, Minchella DJ. Microsatellite loci in the human blood fluke Schistosoma mansoni and their utility for other schistosome species. Molecular Ecology Notes. 2001;1:143–145. [Google Scholar]
- Davies CM, Webster JP, Kruger O, Munatsi A, Ndamba J, Woolhouse MEJ. Host-parasite population genetics: a cross-sectional comparison of Bulinus globosus and Schistosoma haematobium. Parasitology. 1999;119:295–302. doi: 10.1017/s0031182099004722. [DOI] [PubMed] [Google Scholar]
- Davies CM, Webster JP, Woolhouse ME. Trade-offs in the evolution of virulence in an indirectly transmitted macroparasite. Proceedings of the Royal Society of London, B. 2001;268:251–257. doi: 10.1098/rspb.2000.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand P, Sire C, Theron A. Isolation of microsatellite markers in the digenetic trematode Schistosoma mansoni from Guadeloupe island. Molecular Ecology. 2000;9:997–998. doi: 10.1046/j.1365-294x.2000.00939-4.x. [DOI] [PubMed] [Google Scholar]
- Guo S, Thompson E. Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics. 1992;48:361–372. [PubMed] [Google Scholar]
- Kumar S, Tamura K, Nei M. MEGA3: integrated software for Molecular Evolutionary Genetic Analysis and sequence alignment. Briefings in Bioinformatics. 2004;5:150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- Langella O. Populations 1.2.28 (12/5/2002), CNRS UPR9034. 1999. [Google Scholar]
- Lewis PO, Zaykin D. Genetic Data Analysis: computer program for the analysis of allelic data. Version 1.0 (d16c) 2001. Free program distributed by the authors over the internet from http://lewis.eeb.uconn.edu/lewishome/software.html.
- Loverde PT, Dewald J, Minchella DJ, Bosshardt SC, Damian RT. Evidence for host-induced selection in Schistosoma mansoni. Journal of Parasitology. 1985;71:297–301. [PubMed] [Google Scholar]
- Manning SD, Woolhouse MEJ, Ndamba J. Geographic compatibility of the freshwater snail Bulinus globusus and schistosomes from the Zimbabwe Highveld. International Journal for Parasitology. 1995;25:37–42. doi: 10.1016/0020-7519(94)00097-8. [DOI] [PubMed] [Google Scholar]
- Morand S, Manning SD, Woolhouse MEJ. Parasite-host coevolution and geographic patterns of parasite infectivity and host susceptibility. Proceedings of the Royal Society of London, B. 1996;263:119–128. doi: 10.1098/rspb.1996.0019. [DOI] [PubMed] [Google Scholar]
- Nelson GS, Saoud MFA. A comparison of the pathogenicity of two geographical strains of Schistosoma mansoni in rhesus monkeys. Journal of Helminthology. 1968;17:339–362. doi: 10.1017/s0022149x00017946. [DOI] [PubMed] [Google Scholar]
- Prugnolle F, De Meeus T, Durand P, Sire C, Theron A. Sex-specific genetic structure in Schistosoma mansoni: evolutionary and epidemiological implications. Molecular Ecology. 2002;11:1231–1238. doi: 10.1046/j.1365-294x.2002.01518.x. [DOI] [PubMed] [Google Scholar]
- Prugnolle F, Choisy M, Theron A, Durand P, De Meeus T. Sex-specific correlation between heterozygosity and clone size in the trematode Schistosoma mansoni. Molecular Ecology. 2004;13:2859–2864. doi: 10.1111/j.1365-294X.2004.02273.x. [DOI] [PubMed] [Google Scholar]
- Rodrigues NB, Filho PC, Souza CP, Passos LKJ, Dias-Neto E, Romanha AJ. Population structure of Schistosoma mansoni assessed by DNA microsatelites. International Journal for Parasitology. 2002;32:843–851. doi: 10.1016/s0020-7519(02)00031-0. [DOI] [PubMed] [Google Scholar]
- Rollinson D, Johnston DA. Schistosomiasis: a persistent parasitic disease. Interdisciplinary Science Reviews. 1996;21:140–154. [Google Scholar]
- Rosenberg NA, Pritchard JK, Weber JL, Cann HM, Kidd KK, Zhivotovsky LA, Feldman MW. Genetic structure of human populations. Science. 2002;298:2381–2383. doi: 10.1126/science.1078311. [DOI] [PubMed] [Google Scholar]
- Schneider S, Roesli D, Excoffier L. Arlequin ver 2.000: A Software for Population Genetic Data Analysis. Genetics and Biometry Laboratory, University of Geneva; Switzerland: 2000. [Google Scholar]
- Shrivastava J, Barker GC, Johansen MV, Xiaonong Z, Aligui GD, Mcgarvey ST, Webster JP. Isolation and characterisation of polymorphic DNA microsatellite markers from Schistosoma japonicum. Molecular Ecology Notes. 2003;3:406–408. [Google Scholar]
- Shrivastava J, Gower CM, Balolong EJ, Wang TP, Qian BZ, Webster JP. Population genetics of multihost parasites - the case for natural sampling of Schistosoma japonicum larval stages. Parasitology. 2005;31:617–626. doi: 10.1017/S0031182005008413. [DOI] [PubMed] [Google Scholar]
- Sire C, Durand P, Pointier JP, Theron A. Genetic diversity and recruitment pattern of Schistosoma mansoni in a Biomphalaria glabrata snail population: a field study using random-amplified polymorphic DNA markers. Journal of Parasitology. 1999;85:436–441. [PubMed] [Google Scholar]
- Sire C, Durand P, Pointier JP, Theron A. Genetic diversity of Schistosoma mansoni within and among individual hosts (Rattus rattus): infrapopulation differentiation at microspatial scale. International Journal for Parasitology. 2001;31:1609–1616. doi: 10.1016/s0020-7519(01)00294-6. [DOI] [PubMed] [Google Scholar]
- Smithers SR, Terry RJ. The infection of laboratory hosts with cercariae of Schistosoma mansoni and the recovery of adult worms. Parasitology. 1965;55:695–700. doi: 10.1017/s0031182000086248. [DOI] [PubMed] [Google Scholar]
- Stohler RA, Curtis J, Minchella DJ. A comparison of microsatellite polymorphism and heterozygosity among field and laboratory populations of Schistosoma mansoni. International Journal for Parasitology. 2004;34:595–610. doi: 10.1016/j.ijpara.2003.11.026. [DOI] [PubMed] [Google Scholar]
- Thiongo FW, Madsen H, Ouma JH, Andreassen J, Christensen NO. Host-parasite relationships in infections with two Kenyan isolates of Schistosoma mansoni in NMRI mice. Journal of Parasitology. 1997;83:330–332. [PubMed] [Google Scholar]
- Thompson JN. The Coevolutionary Process. Chicago University Press; Chicago: 1994. [Google Scholar]
- Webster JP, Davies CM. Coevolution and compatibility in the snail-schistosome system. Parasitology. 2001;123:S41–S56. doi: 10.1017/s0031182001008071. [DOI] [PubMed] [Google Scholar]
- Webster JP, Gower CM, Blair L. Do hosts and parasites coevolve?: empirical support from the Schistosoma system. American Naturalist. 2004;164:S33–S51. doi: 10.1086/424607. [DOI] [PubMed] [Google Scholar]
- Wolfensohn S, Lloyd M. Handbook of Laboratory Animal Management and Welfare. Blackwell Science; Oxford: 1999. [Google Scholar]