

Abstract
Background
Advanced-stage neuroblastomas are often resistant to chemotherapy. Heat shock protein (Hsp) 90 is a molecular chaperone that maintains the stability of important signal transduction proteins. We have previously reported that geldanamycin (GA), Hsp90 inhibitor, decreases Raf-1 and Akt protein expression and induces apoptosis in neuroblastoma cells. We sought to determine the in vivo effects of Hsp90 inhibitor compounds on human neuroblastomas.
Materials and Methods
Human neuroblastoma (LAN-1 and SK-N-SH) xenografts (4 mm3 tumor implants) were placed in the flanks of athymic nude mice. Mice received either Hsp90 inhibitors (17-AAG or EC5) or vehicle (control). The tumor dimensions were measured twice weekly. Proteins were extracted for Western immunoblotting.
Results
Hsp90 inhibitor compounds significantly blocked both LAN-1 and SK-N-SH neuroblastoma growth in vivo. Drug-treated tumors showed decreases in Raf-1 and cleaved PARP expression.
Conclusions
Hsp90 inhibitors may prove to be important novel therapeutic agents for patients with advanced-stage neuroblastoma who fail to respond to current treatment regimens.
Keywords: Geldanamycin, Hsp90 inhibitor, neuroblastoma, apoptosis
INTRODUCTION
Neuroblastoma is the most common extracranial solid tumor of childhood and accounts for 15% of cancer-related deaths (1). Despite recent advances in multi-modality treatment protocols, the advanced-stage tumors remain aggressive and frequently resistant to chemotherapeutic regimens with an overall 5-year survival rate of only 30–40%. Altered cellular responses to apoptosis are thought to play an important role in drug resistance in high-risk neuroblastomas (2).Therefore, novel drug therapy targeting cellular signal transduction pathways regulating the apoptotic cascade may be crucial to the treatment of drug-resistant neuroblastomas.
Heat shock protein (Hsp) 90 is a ubiquitously-expressed molecular chaperone that is required for the conformational maturation and stability of a range of client proteins. Hsp90 clients include key mediators of signaling cascades that regulate cellular proliferation as well as block apoptotic signaling (3). Inhibition of Hsp90 results in simultaneous disruption of several key signaling pathways, thus providing a potential target for new chemotherapeutic drugs. Geldanamycin (GA), a benzoquinone ansamycin antibiotic, is an Hsp90 inhibitor that binds to the N-terminal ATP-binding pocket of Hsp90; it inhibits ATP binding and ATP-dependent Hsp90 chaperone activity (4, 5) and directs the proteasomal degradation of Hsp90 clients (6). Recent studies have shown that Hsp90 is in an “activated” state in tumor cells and therefore, exhibiting high-affinity binding to Hsp90 inhibitor drugs, which allows for specific accumulation in tumors and not in normal tissues, where Hsp90 is in a “latent” or inactive state (7). Taken together, this suggests that Hsp90 represents a selective anti-cancer drug target and that Hsp90 inhibition can lead to degradation of key oncogenic proteins.
We recently reported that GA causes increased apoptosis in neuroblastoma cells in vitro by inhibition of the Raf-1 and Akt pathways (8). However, the effects of Hsp90 inhibition on human neuroblastoma growth in vivo have not been described. A geldanamycin-derived Hsp90 inhibitor, 17-allylamino-demethoxygeldanamycin (17-AAG), which lacks the clinical toxicity of GA (9), has been shown to demonstrate potent antitumor activity in other preclinical models (10, 11), and is currently in Phase II clinical trials. Therefore, the purpose of this study was to determine the effects of 17-AAG and another novel ansamycin Hsp90 inhibitor, EC5 on the growth of human neuroblastoma xenografts in athymic nude mice.
MATERIALS AND METHOD
Reagents and antibodies
The ansamycin Hsp90 inhibitor drugs, 17-AAG and EC5, were synthesized by Conforma Therapeutics (San Diego, CA) as previously described (12). Anti-Raf-1, and Hsp90 were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-poly ADP-ribose polymerase (PARP) antibody was purchased from Cell Signaling (Beverly, MA). Anti-β-actin was obtained from Sigma (St. Louis, MO). All secondary antibodies against mouse, rabbit and goat IgG were purchased from Santa Cruz.
In vivo experiments
Human neuroblastoma cell line, SK-N-SH, was purchased from American Type Culture Collection (Manassas, VA), and LAN-1 was a gift from Dr. Robert C. Seeger (Univ. of Southern California, Los Angeles, CA). First, we established xenografts in athymic nude mice (Harlan Sprague Dawley, Indianapolis, IN) by injecting SK-N-SH cells (1×107 cells per injection) into the subcutaneous flanks. Once xenografts were established, we then transferred tumor implants (4 mm3) into the bilateral flanks of male athymic nude mice. One week later, the mice were randomized into two experimental groups (3–5 mice/group): group 1, (control) receiving vehicle solution alone; group 2, receiving three consecutive days per week intraperitoneal injections of 17-AAG (60 mg/kg/day). Dosage of 17-AAG for our study was chosen based on previous studies (3, 12).
For the second set of experiments, the effects of 17-AAG on xenografts established from another neuroblastoma cell line, LAN-1, were assessed. Lastly, we determined apoptotic effects of 17-AAG and EC5 (40mg/kg/day) on LAN-1 neuroblastomas. For all experiments, drug treatments were delivered by daily intraperitoneal injections three consecutive days per week. Tumor growth was assessed biweekly by measuring the two greatest perpendicular tumor dimensions with vernier calipers (Mitutoyo, Tokyo, Japan). Mice were weighed weekly. Tumor volumes were calculated as follows: tumor volume (mm3) = [tumor length (mm) × tumor width (mm)2]/2. At sacrifice, tumors were resected, weighed and snap frozen in liquid nitrogen for storage at −70ºC.
Western blot analysis
Tumor sections were lysed with buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.5 mM NP40, 50 mM NaF, 1 mM sodium orthovanadate, 1 mM DTT, 1 mM phenylmethylsulfonyl fluoride, and 25 μg/ml each of aprotinin, leupeptin, and pepstatin A on ice. Lysates were centrifuged at 15,000 × g for 30 min at 4°C. After protein concentrations were determined, equal amounts of proteins (100 μg) were resolved on NuPAGE Novex 4–12% Bis-Tris Gel (Invitrogen, Carlsbad, CA) and electrophoretically transferred to immunoblot polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA). Membranes were incubated overnight at 4°C in a blocking solution (Tris-buffered saline containing 5% nonfat dried milk and 0.05% Tween 20), followed by a 3 h incubation with primary antibodies, washed three times in Tris-buffered saline containing 1% nonfat dried milk and 0.05% Tween 20 and incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. After three additional washes, the immune complexes were visualized by the enhanced chemiluminescence (ECL) detection system (Amersham, Piscataway, NJ).Densitometric analyses were performed using Kodak 1D Image Analysis Software Version 3.6.
Statistical analysis
Tumor size was analyzed using analysis of variance for a two-factor experiment with repeated measures on time. The two factors were Hsp90 inhibitors and day. The first-order autoregressive covariance was used for a covariance structure. All effects were assessed at the 0.05 level of significance and all interactions of the effects were assessed at the 0.15 level of significance as the experiment-wise error rates. Fisher’s least significant difference procedure was used for multiple comparisons with 0.005 as the comparison-wise error rate. Average tumor weight of two tumors from each animal was analyzed using the Kruskal-Wallis test. Data analysis was conducted using PROC MIXED with LSMEANS option and Satterthwaite approximation for the denominator degrees of freedom in SAS®, Release8.2 [R1].
RESULT
17-AAG inhibits SK-N-SH neuroblastoma growth
To examine whether Hsp90 inhibitors block neuroblastoma tumor growth in vivo, we first determined the effects of 17-AAG. The mean tumor volume in either treatment group was not different before drug treatment when compared with control group (day 1; p value > 0.755). As shown in Figure 1A, 17-AAG significantly inhibited the growth of SK-N-SH neuroblastoma xenografts by treatment day 21 when compared to control mice receiving vehicle alone. This result corroborates our previous report demonstrating inhibition of human neuroblastoma cell growth by GA in vitro (8).
Figure 1.
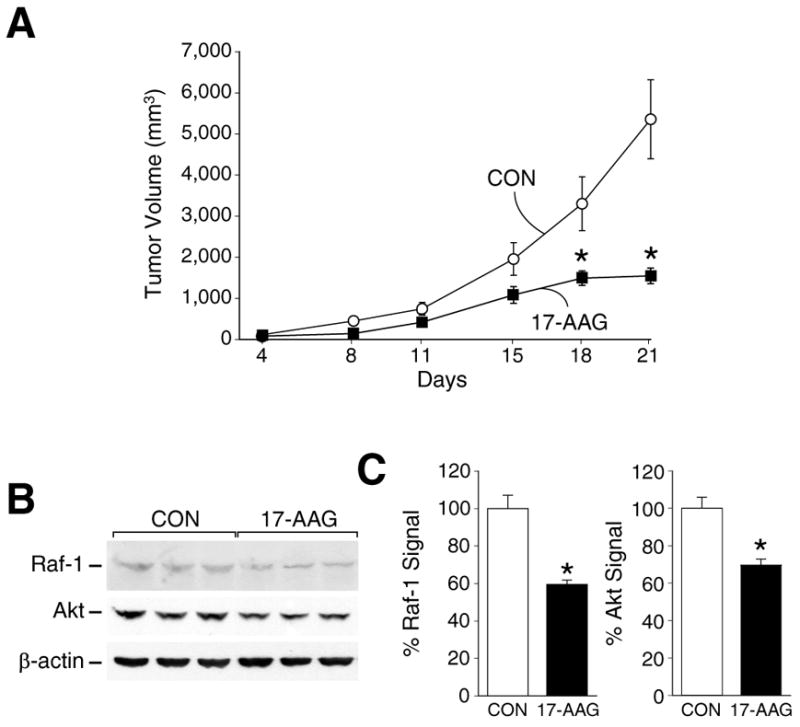
(A) Tumor volumes in nude mice bearing SK-N-SH human neuroblastoma during treatment with 17-AAG. (*=p<0.05 vs. control). (B) Western immunoblots demonstrate depletion in Raf-1 and Akt expression in 17-AAG treated group. β-actin shows relative equal protein loading. Three representative tumors from each group are shown. (C) Percentage of protein measurement of Raf-1 and Akt are presented. Values were normalized with the expression of β-actin. (*=p<0.05 vs. control).
Depletion of the Hsp90 client proteins Raf-1 and Akt, as a consequence of GA treatment, has been demonstrated in various cell lines such as MCF-7 breast cancer cells, CHP100 neuroepithelioma cells and NIH3T3 mouse fibroblast cells (13, 14). As shown in Figure 1B and 1C, 17-AAG treatment also reduced the levels of Raf-1 and Akt expression in SK-N-SH xenografts.
17-AAG inhibits LAN-1 neuroblastoma growth
To ensure that the in vivo inhibitory effect of Hsp90 inhibitors was not unique to SK-N-SH neuroblastomas, which express low levels of N-myc, we next examined the effects of 17-AAG on another human neuroblastoma xenograft established from LAN-1 cell line with high N-myc expression. Similar to the results noted with SK-N-SH xenografts in Fig. 1, LAN-1 tumor growth was also markedly inhibited by treatment with 17-AAG when compared to the control group (Fig. 2A). As shown in Fig. 2B and 2C, LAN-1 neuroblastoma xenografts also showed reduction in Raf-1 and Akt expression after 17-AAG treatment.
Figure 2.
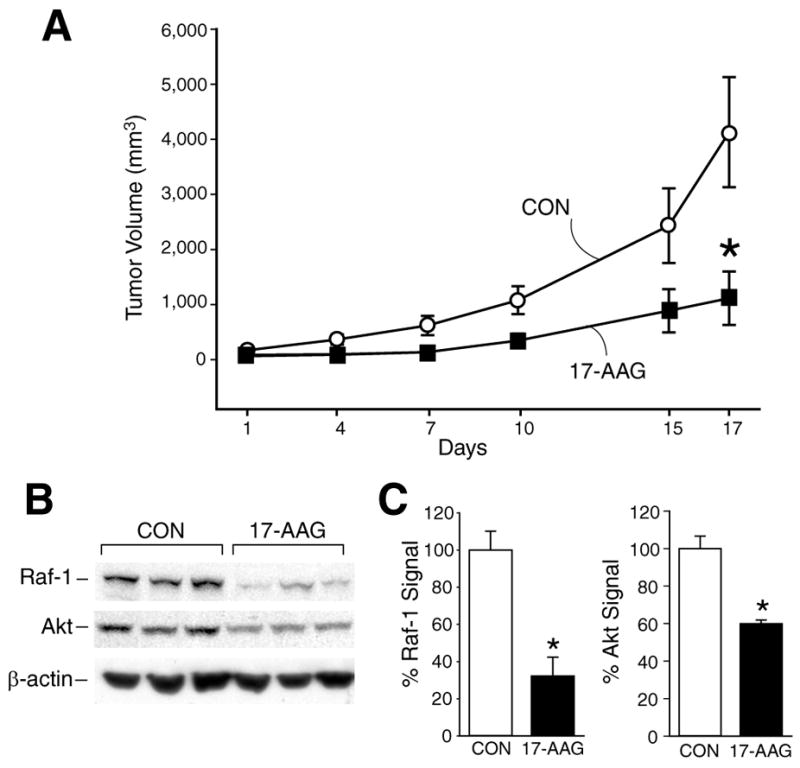
(A) Treatment with 17-AAG resulted in inhibition of LAN-1 neuroblastoma growth (*=p<0.05 vs. control). (B) Western immunoblots show reduction in Raf-1 and Akt protein levels in 17-AAG treated group. β-actin shows relative equal protein loading.(C) Percentages of Raf-1 and Akt protein measurements as in B (*=p<0.05 vs. control).
EC5 and 17-AAG induce apoptosis in neuroblastoma xenografts
Similar to the results noted with 17-AAG (Fig. 1 and Fig. 2), EC5 also significantly inhibited LAN-1 tumor growth when compared to control group (Fig. 3A). Consistent with their inhibitory effects on tumor volume, tumor weight in both 17-AAG and EC5 treated groups was also markedly less when compared to control group at the time of sacrifice (Fig. 3B). Interestingly, there was no significant difference between the two drugs used on neuroblastoma tumor growth inhibition. All mice tolerated intraperitoneal injections of either compound without obvious systemic toxicities; however, a slight weight loss (12%) was noted in mice receiving EC5 (data not shown).
Figure 3.
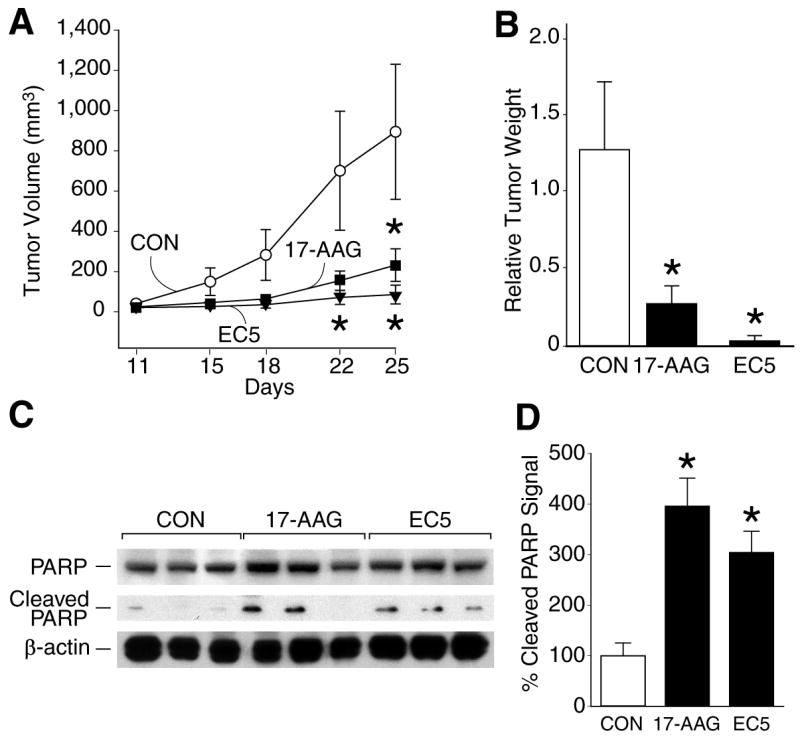
(A) Growth inhibition of LAN-1 neuroblastoma xenografts with 17-AAG and EC5 treatment (*=p<0.05 vs. control). (B) Effect of treatment with 17-AAG and EC5 on tumor weight of LAN-1 human neuroblastoma xenografts. (*=p<0.05 vs. control). (C) Western blots show induction in cleaved PARP expression with 17-AAG or EC5 treatment. β-actin shows relative equal protein loading. (D) Percentage of cleaved PARP protein measurements as in C (*=p<0.05 vs. control).
To determine whether the anti-tumor effect of 17-AAG or EC5 is associated with apoptosis, LAN-1 tumors were harvested at sacrifice and protein extracted to determine cleavage products of PARP, a marker of cells undergoing apoptosis. Treatment with both 17-AAG and EC5 increased protein expression of cleaved PARP, suggesting an induction of apoptosis in LAN-1 tumors (Fig. 3C and 3D). Taken together, these results further confirm that treatment with Hsp90 inhibitor drugs reduce growth rate of neuroblastomas, irrespective of N-myc amplification status.
DISCUSSION
Little progress has been made in improving the overall prognosis of patients with advanced-stage neuroblastomas (15). In this report, we examined the effects of the Hsp90 inhibitors 17-AAG, which is currently in phase II clinical trials (16), and EC5 on LAN-1 and SK-N-SH human neuroblastoma xenografts. We found that both Hsp90 inhibitors were effective in blocking neuroblastoma tumor growth in vivo. Furthermore, Hsp90 inhibitors induced apoptosis in neuroblastoma xenografts and decreased the expression of Raf-1 and Akt protein levels. Our results are consistent with the findings of other in vivo experiments, where 17-AAG resulted in the inhibition of prostate cancer xenografts (10), and EC5 compound decreased growth of head and neck squamous cell carcinoma xenografts (12).
Hsp90 is a critical component of the multi-chaperone complexes that regulate the activity of a wide range of signal transduction proteins within cells (17). Hsp90 clients include key components of the mitogenic signaling pathways (such as Raf-1/MAPK pathway) that drive cell cycle progression, as well as survival signaling pathways (such as the PI3-K/Akt pathways) that inhibit apoptosis (3, 8). In addition, Hsp90 clients include proteins that contribute to important functions in growth signaling, evasion of apoptosis, sustained angiogenesis, tissue invasion and metastasis (18). Thus, the ability of Hsp90 inhibitors to simultaneously destabilize many oncoproteins in multiple signaling pathways and block key features of malignancy is likely to be a major factor in their effectiveness against cancer cells.
GA and 17-AAG have been shown to destabilize Hsp90-associated client proteins by increasing their degradation via the ubiquitin-proteasome pathway (19, 20). Recently, 17-AAG has completed Phase I clinical trials as the first in-class Hsp90 inhibitor (18, 21). In an attempt to identify useful markers for monitoring the activity of Hsp90 inhibitors in pediatric cancer patients enrolled in clinical trials, we evaluated the effect of Hsp90 inhibitors on two client proteins that are important in childhood solid tumors, Akt and Raf-1. Because of its role in anti-apoptotic signaling, Akt was selected as a potential marker of the activity of Hsp90 inhibitors. Akt functions as a critical protein in the activation of various survival pathways (22). Depletion of Raf-1 expression with GA treatment has been described in various cell lines, including the murine neuroblastoma N2A (23). Consistent with these reports, we also found that treatment with 17-AAG and EC5 led to reduction of Raf-1 and Akt protein expression in human neuroblastoma xenografts in athymic nude mice. Moreover, increased cleavage products of PARP suggest that the tumor growth inhibitory actions of Hsp90 inhibitors may involve regulation of apoptotic pathways. Our future studies will involve further elucidating this potential mechanism of action for Hsp90 inhibitors on neuroblastomas.
17-AAG has some limitations as a therapeutic agent due to poor solubility (24); therefore, we also examined the effects of another analog, EC5, a novel ansamycin-based compound designed to stabilize the drug-target interaction for increased effectiveness (12) on neuroblastoma growth. In our study, both EC5 and 17-AAG compounds significantly inhibited neuroblastoma growth. Additionally, anti-tumor actions of these compounds were equally effective against high (LAN-1) as well as low (SK-N-SH) N-myc expressing neuroblastomas. This finding is particularly important for potential clinical applications since aggressive tumor behavior refractory to current treatment modalities is associated with a spectrum of neuroblastoma N-myc amplification status.
In conclusion, we have found that 17-AAG and additional ansamycin Hsp90 inhibitor, EC5, block human neuroblastoma growth in vivo. Our study suggests that targeting Hsp90, as an important signaling protein stabilizer, could prove to be a novel therapy in advanced-stage neuroblastomas.
Acknowledgments
The authors thank Karen Martin for manuscript preparation and Tatsuo Uchida for statistical analyses. This work was supported by the grants RO1 DK61470, RO1 CA104748, RO1 DK48498 and PO1 DK35608 from the National Institutes of Health.
References
- 1.Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald B. Terminology and morphologic criteria of neuroblastic tumors: recommendations by the International Neuroblastoma Pathology Committee. Cancer. 1999;86:349–363. [PubMed] [Google Scholar]
- 2.Teitz T, Lahti JM, Kidd VJ. Aggressive childhood neuroblastomas do not express caspase-8: an important component of programmed cell death. J Mol Med. 2001;79:428–436. doi: 10.1007/s001090100233. [DOI] [PubMed] [Google Scholar]
- 3.Kamal A, Boehm MF, Burrows FJ. Therapeutic and diagnostic implications of Hsp90 activation. Trends Mol Med. 2004;10:283–290. doi: 10.1016/j.molmed.2004.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neckers L, Ivy SP. Heat shock protein 90. Curr Opin Oncol. 2003;15:419–424. doi: 10.1097/00001622-200311000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Panaretou B, Prodromou C, Roe SM, O’Brien R, Ladbury JE, Piper PW, Pearl LH. ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. Embo J. 1998;17:4829–4836. doi: 10.1093/emboj/17.16.4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mimnaugh EG, Chavany C, Neckers L. Polyubiquitination and proteasomal degradation of the p185c-erbB-2 receptor protein-tyrosine kinase induced by geldanamycin. J Biol Chem. 1996;271:22796–22801. doi: 10.1074/jbc.271.37.22796. [DOI] [PubMed] [Google Scholar]
- 7.Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 8.Kim S, Kang J, Hu W, Evers BM, Chung DH. Geldanamycin decreases Raf-1 and Akt levels and induces apoptosis in neuroblastomas. Int J Cancer. 2003;103:352–359. doi: 10.1002/ijc.10820. [DOI] [PubMed] [Google Scholar]
- 9.Supko JG, Hickman RL, Grever MR, Malspeis L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother Pharmacol. 1995;36:305–315. doi: 10.1007/BF00689048. [DOI] [PubMed] [Google Scholar]
- 10.Solit DB, Zheng FF, Drobnjak M, Munster PN, Higgins B, Verbel D, Heller G, Tong W, Cordon-Cardo C, Agus DB, Scher HI, Rosen N. 17-Allylamino-17-demethoxygeldanamycin induces the degradation of androgen receptor and HER-2/neu and inhibits the growth of prostate cancer xenografts. Clin Cancer Res. 2002;8:986–993. [PubMed] [Google Scholar]
- 11.Solit DB, Basso AD, Olshen AB, Scher HI, Rosen N. Inhibition of heat shock protein 90 function down-regulates Akt kinase and sensitizes tumors to Taxol. Cancer Res. 2003;63:2139–2144. [PubMed] [Google Scholar]
- 12.Yin X, Zhang H, Burrows F, Zhang L, Shores CG. Potent activity of a novel dimeric heat shock protein 90 inhibitor against head and neck squamous cell carcinoma in vitro and in vivo. Clin Cancer Res. 2005;11:3889–3896. doi: 10.1158/1078-0432.CCR-04-2272. [DOI] [PubMed] [Google Scholar]
- 13.Schulte TW, An WG, Neckers LM. Geldanamycin-induced destabilization of Raf-1 involves the proteasome. Biochem Biophys Res Commun. 1997;239:655–659. doi: 10.1006/bbrc.1997.7527. [DOI] [PubMed] [Google Scholar]
- 14.Schulte TW, Blagosklonny MV, Ingui C, Neckers L. Disruption of the Raf-1-Hsp90 molecular complex results in destabilization of Raf-1 and loss of Raf-1-Ras association. J Biol Chem. 1995;270:24585–24588. doi: 10.1074/jbc.270.41.24585. [DOI] [PubMed] [Google Scholar]
- 15.Cotterill SJ, Parker L, More L, Craft AW. Neuroblastoma: changing incidence and survival in young people aged 0–24 years. A report from the North of England Young Persons’ Malignant Disease Registry. Med Pediatr Oncol. 2001;36:231–234. doi: 10.1002/1096-911X(20010101)36:1<231::AID-MPO1056>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 16.Ivy PS, Schoenfeldt M. Clinical trials referral resource. Current clinical trials of 17-AG and 17-DMAG. Oncology (Williston Park) 2004;18:610, 615, 619–620. [PubMed] [Google Scholar]
- 17.Chiosis G, Vilenchik M, Kim J, Solit D. Hsp90: the vulnerable chaperone. Drug DiscovToday. 2004;9:881–888. doi: 10.1016/S1359-6446(04)03245-3. [DOI] [PubMed] [Google Scholar]
- 18.Maloney A, Workman P. HSP90 as a new therapeutic target for cancer therapy: the story unfolds. Expert Opin Biol Ther. 2002;2:3–24. doi: 10.1517/14712598.2.1.3. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y, Ding J. Heat shock protein 90: novel target for cancer therapy. Ai Zheng. 2004;23:968–974. [PubMed] [Google Scholar]
- 20.Bagatell R, Whitesell L. Altered Hsp90 function in cancer: a unique therapeutic opportunity. Mol Cancer Ther. 2004;3:1021–1030. [PubMed] [Google Scholar]
- 21.Banerji U, Judson I, Workman P. The clinical applications of heat shock protein inhibitors in cancer - present and future. Curr Cancer Drug Targets. 2003;3:385–390. doi: 10.2174/1568009033481813. [DOI] [PubMed] [Google Scholar]
- 22.Agarwal A, Das K, Lerner N, Sathe S, Cicek M, Casey G, Sizemore N. The AKT/I kappa B kinase pathway promotes angiogenic/metastatic gene expression in colorectal cancer by activating nuclear factor-kappa B and beta-catenin. Oncogene. 2005;24:1021–1031. doi: 10.1038/sj.onc.1208296. [DOI] [PubMed] [Google Scholar]
- 23.Lopez-Maderuelo MD, Fernandez-Renart M, Moratilla C, Renart J. Opposite effects of the Hsp90 inhibitor Geldanamycin: induction of apoptosis in PC12, and differentiation in N2A cells. FEBS Letters. 2001;490:23. doi: 10.1016/s0014-5793(01)02130-5. [DOI] [PubMed] [Google Scholar]
- 24.Workman P. Auditing the pharmacological accounts for Hsp90 molecular chaperone inhibitors: unfolding the relationship between pharmacokinetics and pharmacodynamics. Mol Cancer Ther. 2003;2:131–138. [PubMed] [Google Scholar]