

Abstract
Differentiating between injurious and reparative factors facilitates appropriate therapeutic intervention. We evaluated the role of tumor necrosis factor α (TNFα) in parenchymal brain pathology resolution following virus‐induced encephalitis from a picornavirus, Theiler's murine encephalomyelitis virus (TMEV). We infected the following animals with TMEV for 7 to 270 days: B6/129 TNF−/− mice (without TNFα expression), B6/129 TNFR1−/− mice (without TNFα receptor 1 expression), and B6/129 TNFR2−/− mice (without TNFα receptor 2 expression). Normal TNFα‐expressing controls were TMEV‐infected B6, 129/J, B6/129F1 and B6/129F2 mice. Whereas all strains developed inflammation and neuronal injury in the hippocampus and striatum 7 to 21 days postinfection (dpi), the control mice resolved the pathology by 45 to 90 dpi. However, parenchymal hippocampal and striatal injury persisted in B6/129 TNF−/− mice following infection. Treating virus‐infected mice with active recombinant mouse TNFα resulted in less hippocampal and striatal pathology, whereas TNFα‐neutralizing treatment worsened pathology. T1 “black holes” appeared on MRI during early infection in the hippocampus and striatum in all mice but persisted only in TNF−/− mice. TNFR1 mediated hippocampal pathology resolution whereas TNFR2 mediated striatal healing. These findings indicate the role of TNFα in resolution of sublethal hippocampal and striatal injury.
Keywords: cytokine, neuroinflammation, neuropathology, neuroprotection, Theiler's murine encephalomyelitis virus, virus persistence
INTRODUCTION
Differentiating between injurious and reparative factors is critical for appropriate therapeutic intervention. Immune response factors are implicated in injury; nevertheless, immune response components aid central nervous system (CNS) healing 4, 22, 33. Natural human autoantibodies enhance remyelination in toxic and viral models (39) of multiple sclerosis (MS), and cytokines and interleukins exhibit CNS‐reparative potential (7). Several models demonstrate the role of interferon‐γ in neuronal protection. Other cytokines may protect neurons following virus injury.
The experiments in this manuscript focus on the cytokine tumor necrosis factor α (TNFα), upregulated in several CNS disorders (9). Macrophages (34) and astrocytes produce TNFα during injury. In tissue culture, TNFα elicits toxic or protective effects dependent upon the target cell. TNFα also activates two receptor subtypes, p55 (TNFα receptor type 1/TNFR1) and p75 (TNFα receptor type 2/TNFR2), which mediate function. In some systems, TNFα‐related ligand interactions sequentially activate pro‐survival and pro‐apoptotic pathways in the same cell (21). These neuroprotective/neurotoxic effects may be age and concentration dependent (38).
There have been some animal model studies to examine TNFα in injury vs. healing in vivo 2, 3, 5, 14, 32. In EAE, TNF appears to affect the initiation of deficits (14), although it had no effect on demyelination. Using transgenic animals in which glia expressed TNF (2), the authors have demonstrated apoptosis of oligodendrocytes. We investigated whether this pro‐inflammatory molecule also had neuroprotective properties in vivo. We utilized B6/129 TNF−/−mice (without TNFα expression), B6/129 TNFR1−/− mice (without TNFR1 expression), and B6/129 TNFR2−/− mice (without TNFR2 expression) to study the role of TNFα in CNS pathology. Controls were B6, 129/J, B6/129F1 and B6/129F2 mice from which the genetic models were derived. By examining these four control strains, we felt we could exclude other genetic influences related to the B6/129 background of the TNF “knockout mice.” We investigated virus clearance, persistence and CNS injury following intracerebral TMEV injection (8). During days 7 to 21 of infection, virus replicates primarily in the hippocampus, striatum, cortex and anterior horn cells of the spinal cord and then clears rapidly 7, 11. These parenchymal injuries to the hippocampus and striatum occur in all strains and are usually spontaneously resolved by 45 to 90 days after infection. These changes in the brain occur irrespective of mouse major histocompatibility complex (MHC) haplotype. We define healing in this paper as the resolution of the inflammatory necrotic lesions observed in the model soon after infection in the hippocampus and striatum.
After intracranial TMEV infection, we examined viral replication and hippocampal morphology. Absence of TNFα led to persistent hippocampal and striatal injury dependent upon different TNF receptors. Moreover, recombinant mouse TNFα treatment alleviated neuronal injury, while TNFα‐neutralizing antibody treatment exacerbated injury. We conclude, as have others before us 3, 5, 32, that TNFα is critical in CNS restoration or healing. In addition, we demonstrate the differential effect of TNFR1 and TNFR2 in injury resolution in the hippocampus and striatum.
MATERIALS AND METHODS
Virus
The Daniel's strain of TMEV was used for all experiments (17).
Mice
Mice were bought directly from the Jackson Laboratories, Bar Harbor, ME, USA; no further inbreeding of mice was done at the Mayo Clinic. We utilized B6/129 TNF−/−, B6/129 TNFR1−/−and B6/129 TNFR2−/− mice infected with TMEV from 7 to 270 days. As controls, we studied B6, 129/J, B6/129F1 and B6/129F2 mice following TMEV infection to exclude for other genetic influences on the phenotype. B6/129F1 or B6/129F2 mice are the control mice recommended by the Jackson Laboratories for companions to the TNF “knockout” mice. All mice were of the identical H‐2b haplotype. For some experiments, we used SJL/J mice to determine whether the effect of TNF on hippocampal and striatal injury was also found in a separate independent strain of mouse. All experiments were approved by the Mayo IACUC and conformed to the National Institute of Health guidelines for the care of animals. Mice were followed daily until they were either moribund or dead. All mice survived the acute infection and were sacrificed on day 7, 21, 35, 45, 90, 120, 180, 270 or 330 after infection (various endpoints of the study) for pathology and virus antigen expression.
Infection and harvesting of the CNS for morphology
At 4–6 weeks of age, mice were intracerebrally infected with 2 × 105 plaque‐forming units (PFUs) of TMEV in a total volume of 10 µL. At various times after infection, mice were perfused via intracardiac puncture with 50 mL of Trump's fixative. Spinal cords and brains were removed and postfixed for 24–48 h in Trump's fixative in preparation for morphologic analysis.
Treatment with recombinant TNFα or monoclonal antibody to TNFα
Recombinant murine TNFα (10 µg/dose) was kindly provided by Genentech (San Francisco, CA). Mice were injected by the intraperitoneal route in 200 µL starting 12 h before TMEV infection and then three times a week until the day of sacrifice. This dosing regimen was identical to that used previously (23) to show protective effects of TNFα in demyelination. Dr. R. L. Coffman from DNAX (Palo Alto, CA, USA) kindly provided the monoclonal antibody to mouse TNFα (clone XT22‐11, Lot # SP028‐042) 1, 26. This antibody was used at 2.0 mg/dose 1 day prior to infection and again 15 days after infection. Isotype control antibody to beta‐galactosidase (clone GL113, DNAX) was used as a negative control (2.0 mg/dose). For these experiments, SJL mice were used because the strain had been used previously to determine the effect of TNF on demyelination. We also wanted to determine whether the effects of TNFα on CNS pathology were present in another mouse strain.
Spinal cord morphology
Spinal cords were removed from spinal columns and cut into 1 mm coronal blocks with every third block postfixed in osmium and embedded in glycol methacrylate. Two‐micron‐thick sections were prepared and stained with a modified erichrome/cresyl violet stain (24). Morphological analysis was performed on 12 to 15 sections per mouse as previously described (31). Briefly, each quadrant from every coronal section from each mouse was graded for the presence or absence of gray matter disease, meningeal inflammation and demyelination. The score was expressed as the percentage of spinal cord quadrants examined with the pathologic abnormality. A maximum score of 100 indicated a particular pathologic abnormality in every quadrant of all spinal cord sections of a given mouse. All grading was performed without knowledge of the experimental group. Additional spinal cord blocks were embedded in paraffin for immunocytochemistry.
Brain pathology
Brain pathology was assessed on coded sections at various timepoints following infection using our previously described technique (24). Following perfusion with Trump's fixative, two coronal cuts were made in intact brain at time of removal from the skull (one section through the optic chiasm and a second section through the infundibulum). As a guide, we used the Atlas of the Mouse Brain and Spinal Cord corresponding to sections #220 and #350 7, 36. This resulted in three blocks that were then embedded in paraffin to allow for systematic analysis of the pathology of the cortex, corpus callosum, hippocampus, brainstem, striatum and cerebellum. The resulting slides were stained with hematoxylin and eosin. Pathology scores were assigned on coded sections without knowledge of the experimental group to the following areas of the brain: cortex, corpus callosum, hippocampus, brainstem, striatum and cerebellum. Each area of brain was graded on a 4‐point scale: 0 = no pathology; 1 = no tissue destruction and only minimal inflammation; 2 = early tissue destruction (loss of architecture) and moderate inflammation; 3 = definite tissue destruction (neuronal loss, demyelination, parenchymal damage, cell death, neurophagia, neuronal vacuolation); 4 = necrosis (complete loss of all tissue elements with associated cellular debris). Meningeal inflammation was assessed and graded as follows: 0 = no inflammation; 1 = one cell layer of inflammation; 2 = two cell layers of inflammation; 3 = three cell layers of inflammation; 4 = four or more cell layers of inflammation. The area with maximum tissue damage within a section was used for assessment of each brain region. For statistical analyses, the scores of the brain area for a mouse strain at a specific timepoint were compared using nonparametric statistical methods. Because the major differences between strains were found in the hippocampus and striatum, the pathologic scores for these areas were separately analyzed statistically for comparisons among the groups.
Immunostaining for virus antigen, cell markers and TNF receptors
Immunocytochemistry was performed on paraffin‐embedded sections as previously described. Slides were deparaffinized in xylene and rehydrated through an ethanol series (absolute, 95%, 70%, 50%). Virus antigen staining was carried out using polyclonal antisera to TMEV‐DA (28) that reacts strongly with the capsid proteins of TMEV. Following incubation with a secondary biotinylated antibody (Vector Laboratories, Burlingame, CA, USA), immunoreactivity was detected using the avidin–biotin immunoperoxidase technique (Vector Laboratories). The reaction was developed using Hanker–Yates reagent with hydrogen peroxide as the substrate (Polysciences, Warrington, PA, USA). Slides were lightly counterstained with Mayer's hematoxylin. The data were expressed as the number of virus antigen‐positive cells per 40× high power field (h.p.f.) in areas of maximal pathology in the cerebellum, brain stem, cortex, hippocampus, striatum and corpus callosum at day 7 and day 45 following virus infection. We used a similar methodology to stain for astrocytes [glial fibrillary acidic protein (GFAP)], microglia (F4/80) and immunoglobulin (Ig) deposition (anti‐mouse IgG and IgM) and for TNFR1 and TNFR2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
RNA isolation
The brain and spinal cords were removed from TMEV‐infected animals. Total RNA was extracted from the brain and spinal cord (4). Briefly, the tissues were frozen and stored in liquid nitrogen. Tissues samples were homogenized in the RNA STAT‐60™ (1 mL/100 mg tissue) (Tel‐Test, Inc., Friendswood, TX, USA) with a homogenizer, and total RNA was isolated according to the manufacturer's recommendations. The RNA concentrations were determined by spectrophotometry. The RNA samples were equilibrated to a concentration of 0.25 µg/µL and stored at −80°C.
RT‐PCR and real‐time analysis for virus RNA
The VP2 viral capsid region of TMEV was amplified by RT‐PCR using gene‐specific primers (31). The primer pair sequences for VP2 were as follows: forward (5′‐TGGTCGACTCTGTG GTTACG‐3′) and reverse (5′‐GCCGGTCTTGCAAAGATAGT‐3′). Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was used as a control for intersample variability. The sequences used for assaying the presence of GAPDH were forward (5′‐ACCACCA TGGAGAAGGC‐3′) and reverse (5′‐GGCATGGACTGTGGT CATGA‐3′). Sizes of PCR products amplified with primers were 238 base pairs for VP2 and 236 base pairs for GAPDH. Gene copy standards were generated with each set of samples. Standards were generated by serial 10‐fold dilutions of plasmid cDNA. Standards were amplified in parallel with unknown samples by real‐time quantitative RT‐PCR using the LightCycler® (Roche, Indianapolis, IN, USA). Standard curves were generated using LightCycler 4 software. Negative controls (omitting input cDNA) were also used in each PCR run to confirm the specificity of the PCR products. PCR product curves were linear across serial 10‐fold dilutions, and the melting curve analysis indicated synthesis of a single homogeneous product of the expected melting temperature. The reactions were done in 20‐µL capillaries using 7.0 mM Mg2+, 10 pM concentrations of each forward and reverse primer, 4.0 µL of LightCycler‐RT‐PCR Reaction Mix SYBR Green I (LightCycler®‐RNA Amplification Kit SYBR Green I), 2 µL of resolution solution, 0.4 µL of LightCycler‐RT‐PCR Enzyme Mix, sterile H2O, and 0.5 µg total RNA. Reaction conditions for RT‐PCR for VP2 and GAPDH were as follows: reverse transcription at 55°C for 10 minutes, followed by denaturation at 95°C for 2 minutes, followed by 40 cycles of amplification. Amplification conditions were as follows: denaturation at 95°C at 20°C/s without plateau phase, annealing at 57°C for 7 s, and extension at 72°C for 15 s. The accumulation of products was monitored by SYBR Green fluorescence at the completion of each cycle. There was a direct relationship between the cycle number at which accumulation of PCR products became exponential and the log concentration of RNA molecules initially present in the RT‐PCR reaction. The reaction conditions for melting curve analysis were as follows: denaturation to 95°C at 20°C/s without plateau phase, annealing at 60°C for 5 s and denaturation to 95°C at 0.1°C/s, with continuous monitoring of SYBR Green fluorescence. RNA samples (n = 76) from infected mice were analyzed for GAPDH mRNA to determine the levels of mRNA per sample and the technical reproducibility. The GAPDH mRNA level per sample was log10 7.19 ± 0.02 (mean ± SEM). Therefore, the marked variations in viral RNA levels in individual specimens could not be attributed to differences in amplifiable material. The amount of viral RNA was expressed as log10 VP2 copy number per 0.5 µg total RNA.
MRI methods
Five B6/129 F1 (TNF+/+) and five TNF−/− mice were studied with MRI at timepoints 0, 7 and 45 days after intracranial infection with TMEV. MRI was performed at 7 T on a Bruker Avance™ 300 MHz vertical bore NMR spectrometer (Bruker Biospin, Billerice, MA, USA), equipped with “mini‐imaging” accessories. The animal body temperature was maintained at 37°C by a thermocouple‐based system with continuous flow of conditioned air. During the imaging, each mouse was anesthetized with inhalational isoflurane anesthesia (2.5% in oxygen) delivered and maintained via a nose cone. We used a T1‐weighted 3D spin‐echo sequence to image the mouse brain and proximal cervical spinal cord (TR 200 ms, TE 9.7 ms, NA 1, FOV 4 cm × 2 cm × 2 cm, matrix: 256 × 128 × 128; acquisition time: 57 minutes). By using 3D datasets, we were able to generate equivalently oriented slices. Image analysis and slice selection were done in the Bruker Para Vision™ software package, supplied with the spectrometer.
Statistics
Data were analyzed using either the Student's t‐test for normally distributed data or the Mann–Whitney rank sum test for data not normally distributed. ANOVA was used for comparisons of more than one group. Dunn's method was used for all pair‐wise multiple comparisons. Proportional data were evaluated using the chi‐square test. Level for significance was set as P < 0.05 for all tests.
RESULTS
TNFα deficiency does not alter resistance to chronic demyelinating disease in mice of the H‐2b haplotype
Mice of the H‐2b haplotype (prototypic strain C57BL/6) are resistant to chronic demyelination following TMEV infection. These mice replicate virus in the brain during the first 2 weeks of infection, which results in inflammatory lesions in the hippocampus, striatum and cortex at 7 to 21 days after infection. Neurons of the hippocampus and striatum also die apoptotically during this time (7). However, by 45 days after infection, the brain clears the virus, and there is remarkable resolution of injury of the brain pathology with almost complete morphologic restoration. In mice of the H‐2bhaplotype, the virus does not persist in the spinal cord white matter, and therefore, no demyelination occurs from 45 to 270 days after infection.
We asked if depletion of TNFα abrogated resistance and converted the mice to a demyelinating phenotype. This was important because other immune system alterations within the context of the H‐2b haplotype have resulted in demyelination. For example, interferon‐γ‐deficient mice of the H‐2b haplotype develop prominent spinal cord white matter demyelination, and immunosuppression with total body irradiation results in demyelination in H‐2b mice (30). Likewise, depletion of the MHC class I immune‐mediated response via genetic deletion of β2‐microglobulin also converts these mice to a susceptible phenotype. We studied B6/129 TNF−/−, B6/129 TNFR1−/−and B6/129 TNFR2−/− mice infected with TMEV for 7 to 270 days. As controls, we studied B6, 129/J, B6/129J F1 and B6/129F2 mice following TMEV infection to exclude for other genetic influences on the phenotype. We did not observe demyelination in any strain (1, 2). As a positive control, we infected SJL/J mice, which are known to be susceptible to demyelination. All SJL/J mice showed demyelination from 21 to 330 days after infection (1, 2).
Figure 1.
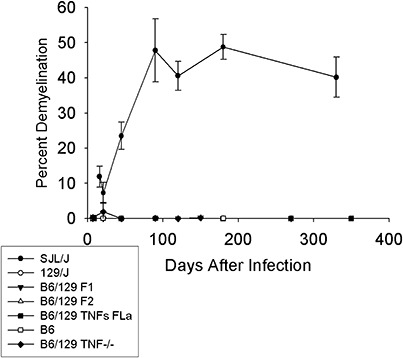
Deletion of tumor necrosis factorα (TNFα) does not predispose mice to develop chronic demyelination in the spinal cord following Theiler's murine encephalomyelitis virus infection. Percent spinal cord quadrants with demyelination in SJL/J (n = 71), 129/J (n = 58), B6/129F1 (n = 64), B6/129F2 (n = 66), B6 (n = 78) and B6/129 TNF−/− (n = 66) mice; “n” is the number of mice studied by pathology per strain. Mice were studied for chronic demyelination for up to 350 days after infection.
Figure 2.

Pathology in the spinal cord following Theiler's murine encephalomyelitis virus (TMEV) infection. Spinal cord sections were embedded in methacrylate and stained for myelin using a modified erichrome stain. A. Demyelination in an SJL/J mouse chronically infected with virus (positive control). B. Absence of demyelination in a B6/129F1 mouse chronically infected with TMEV (negative control). C. Absence of demyelination in a B6/129 TNF−/− mouse chronically infected with TMEV (experimental group). These mice were infected for 270 days. Bar = 100 µm. Likewise, no demyelination was observed in TMEV‐infected B6, 129/SJ and B6/129F2 mice.
We also examined the extent of gray matter spinal cord pathology in each strain at various timepoints. For these analyses, “n” represents the number of mice examined. At the 7‐day timepoint, the percent of quadrants with gray matter pathology was 15.5% ± 5.3% in B6/129 TNF−/− mice (n = 10), which contrasted with 10.6% ± 3.4% in the B6/129F1 controls (n = 21) and 3.1% ± 2.5% in the B6/129F2 controls (n = 8). There was no statistical significance by rank order sum test in comparing gray matter spinal cord pathology in B6/129 TNF−/− mice with B6/129F1 mice (P = 0.123). However, there was a statistical difference by Student's t‐test in the increased gray matter pathology of B6/129 TNF−/− mice as compared with B6/129F2 mice (P < 0.001). At the 7‐day timepoint, the gray matter pathology score in the spinal cord of B6 mice was 9.3% ± 1.9% (n = 25); for 129/J mice, the score was 2.1% ± 1.2% (n = 9). The values for SJL mice at the 21‐day timepoint were 0.0 ± 0.0 for gray matter, 7.5 ± 2.3 for meningeal inflammation and 7.2 ± 3.0 for demyelination. Nonetheless, all strains cleared spinal cord gray matter pathology after 21 days irrespective of TNFα expression.
B6, 129/J, B6/129F1, B6/129 and B6/129 TNF−/− strains of mice have similar brain injury at 7 and 21 days after TMEV infection
We asked whether the presence or absence of TNFα influenced the extent of early virus‐induced encephalitis. We compared the extent of brain pathology in the cortex, corpus callosum, hippocampus, brainstem, striatum and cerebellum of mice following TMEV infection. We used a 4‐point scale as described in Materials and Methods to evaluate the effect of TNFα on early encephalitis. There was no difference in the extent or distribution of CNS injury when comparing B6, 129/J, B6/129F1, B6/129F2 and B6/129 TNF−/− mice (Figure 3). Mice of each strain were studied at each of the timepoints for sacrifice. To determine the effect of TNFα on overall brain pathology, we compiled the brain pathology scores from all the brain regions into datasets segregated by strain and by time after infection (7 or 21 days). Data were expressed as mean ± SEM for each dataset; “n” for these analyses represents the total number of brain regions analyzed per genetic strain at a specific time after infection. At 7 days postinfection (dpi) brain scores for various strains were not statistically different (B6, 2.00 ± 0.15; n = 119), (129/J, 2.05 ± 0.22; n = 63), (B6/129F1, 2.00 ± 1.8; n = 70), (B6/129F2, 2.27 ± 0.24; n = 56), (B6/129 TNF−/−, 2.39 ± 0.19; n = 70). At 21 dpi, brain scores also did not differ statistically (B6, 1.32 ± 0.14; n = 140), (129/J, 0.78 ± 0.18; n = 63), (B6/129F1, 0.69 ± 1.5; n = 70), (B6/129F2, 0.91 ± 0.16; n = 77), (B6/129 TNF−/−, 0.97 ± 0.19; n = 70). Similar results were obtained for B6/129 TNFR1−/− mice at 7 days (2.29 ± 0.24; n = 56) and 21 days (0.40 ± 0.18; n = 35). We then asked whether any statistical differences in the pathologic scores in the hippocampus and striatum occurred independently, because these areas subsequently (see later discussions) showed differences in degree of healing between the TNF+/+ mice as compared with the TNF−/− mice. For these analyses, we studied the pathologic scores from the hippocampus or the striatum of individual mice at 7 and 21 dpi (early phase of disease). By ANOVA on ranks, there was no statistical difference in the pathologic scores between the various genetic strains in the hippocampus (P = 0.853) or in the striatum (P = 0.584). These results demonstrate that the presence or absence of TNFα does not alter the normal course of early encephalitis observed following TMEV infection.
Figure 3.
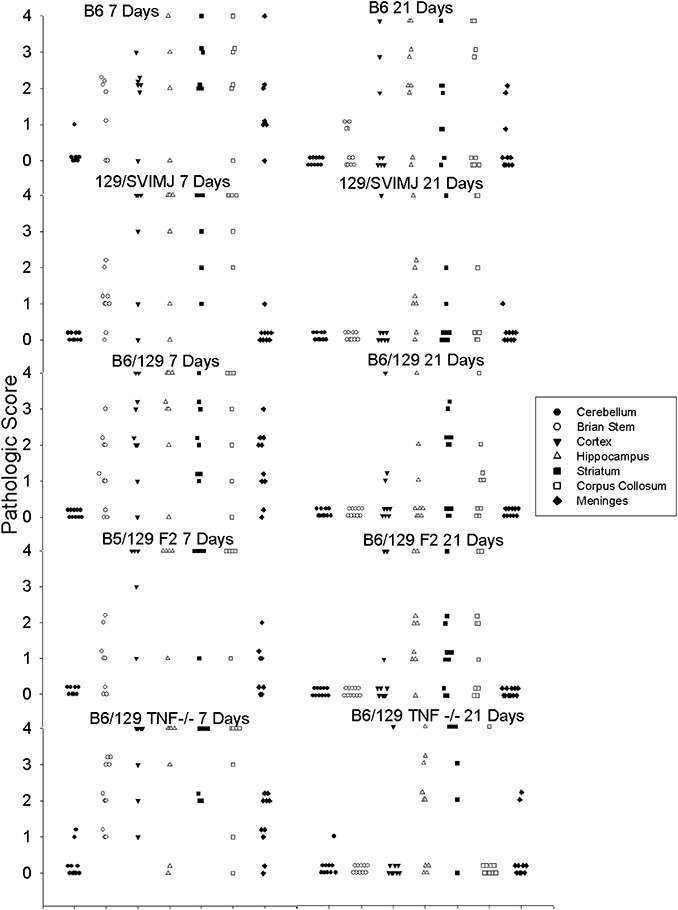
Pathology scores in the brain following early Theiler's murine encephalomyelitis virus (TMEV) infection. Morphologic analysis was performed on coded samples of brain areas (cerebellum, brain stem, cortex, hippocampus, striatum, corpus callosum and meninges) at 7 and 21 days following TMEV infection. Data are shown for the following mouse strains: B6, 129, B6/129F1, B6/129F2, B6/129 TNF−/−. Pathology scores from 0 to 4 are described in Materials and Methods. Each point represents one mouse.
TNFα is required for rapid resolution of the lesions in hippocampus and striatum following acute infection
A characteristic feature of mice infected with TMEV is resolution of encephalitis and reconstitution of almost normal brain morphology 45 days after infection. This resolution of injury is rapid and dramatic. At 7 or 21 days, many animals showed pathology scores demonstrating severe neuronal loss with inflammation (pathology score of 3) or frank complete destruction of the brain parenchyma (pathology score of 4) in the hippocampus or striatum (Figure 3). In contrast, following day 45, most mice with normal expression of TNFα showed resolution of this pathology, with many animals showing only minimal inflammation (pathology score of 1) or no pathology (pathology score of 0). The brain pathology scores in mice with normal expression of TNFα (B6/129F1) continued to decrease at 45, 90 to 120 and 150 to 270 days after infection (Figure 4). By comparison, B6/129 TNF−/− mice (45, 90 to 120 and 150 to 270 days after infection) showed some resolution of lesions but with a slower response than that observed in mice of similar genetic background but with normal expression of TNFα (Figure 4). For these experiments, 10 mice of each strain were studied at each time of sacrifice. This slowed reparative response caused persistent injury to parenchyma of the hippocampus and the striatum (Figure 4). For statistical analyses, we first studied if TNFα played a general role in influencing resolution of brain injury. For these analyses, we compiled the pathology scores from all the various regions of the brain into datasets segregated by strain and time following virus infection. We compared data using the rank order Mann–Whitney test, as the data were ordinal but not randomly distributed; “n” for these analyses was the total number of brain areas analyzed per strain and per time of infection. The brain pathology scores (mean ± SEM) for B6/129F1 mice at day 7 (2.00 ± 0.18, n = 70) or day 21 (0.69 ± 0.15, n = 70) did not differ statistically compared with B6/129 TNF−/− mice at day 7 (2.39 ± 0.19, n = 70) or day 21 (0.97 ± 0.19, n = 70). However, there was a statistical difference (P = 0.012) in decreased brain pathology scores at day 45 in B6/129F1 mice (0.62 ± 0.17, n = 70) as compared with B6/129 TNF−/− mice (1.15 ± 0.19, n = 67). In support of this conclusion, on day 45 there was a statistical difference (P < 0.001) between brain scores of B6 mice (0.32 ± 0.09, n = 126) and B6/129 TNF−/− mice (1.15 ± 0.19, n = 67). A similar statistical trend (P = 0.078) was observed at the 90‐ to 120‐day timepoint, with decrease of the brain pathology scores in B6/129F1 mice (0.14 ± 0.18, n = 35) as compared with B6/129 TNF−/− mice (0.81 ± 0.13, n = 147). We examined the proportion of total number of brain regions with a score of >0 (thus demonstrating some degree of pathological abnormality) in B6/129F1 mice (22/168, 13.1%) as compared with B6/129 TNF−/− mice (84/351, 23.9%). This difference was also statistically significant (P = 0.024) by chi‐square with Yates's correction for continuity.
Figure 4.
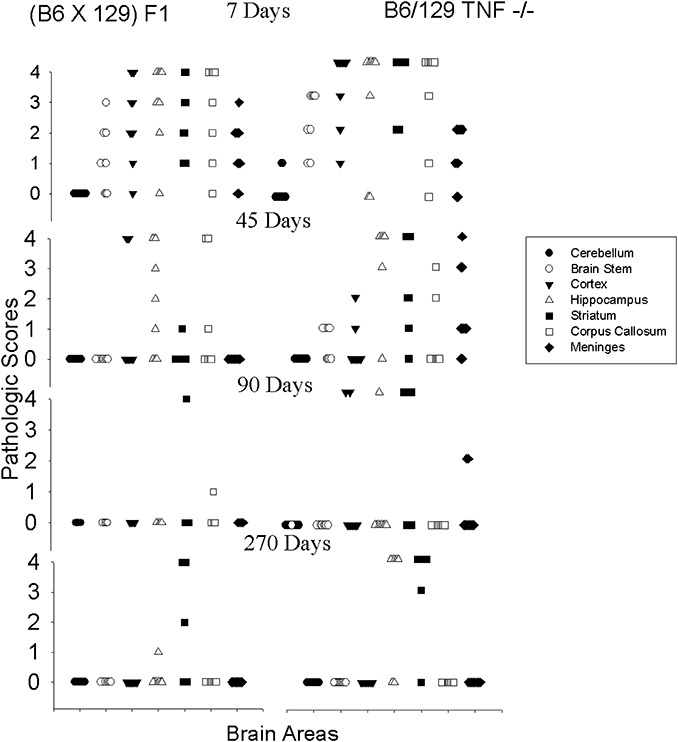
Pathology scores in the brain following chronic Theiler's murine encephalomyelitis (TMEV) infection. Morphologic analysis was performed on coded samples of brain areas (cerebellum, brain stem, cortex, hippocampus, striatum, corpus callosum and meninges) at 7 to 270 days following TMEV infection. A comparison of brain pathology scores is shown for B6/129F1 and B6/129 TNF−/− mice. Pathology scores from 0 to 4 are described in Materials and Methods. Each point represents one mouse. We chose to show B6/129F1 in the Figure as representative of the control mice (B6, 129/J, B6/129F2), which all behaved similarly in contrast to B6/129 TNF−/− mice.
Paraffin‐embedded sections (Figure 5) showed similar extent of hippocampal pathology in the control mice (B6, 129/J, B6/129F1 or B6/129F2) as compared with the B6/129 TNF−/− mice at 7 dpi. In control mice at 45 dpi, the hippocampal pathology had essentially resolved, whereas in the B6/129TNF−/− mice, the injury to the pyramidal layer of the hippocampus persisted.
Figure 5.

Tumor necrosis factorα (TNFα) is required to heal the hippocampus following chronic infection with Theiler's murine encephalomyelitis (TMEV). Paraffin‐embedded sections were stained with hematoxylin–eosin stain. A. Severe hippocampal pathology with necrosis of the pyramidal layer in a B6/129F1 mouse (7 days). B. Absence of hippocampal pathology in a B6/129F1 mouse by 45 dpi. C. Severe hippocampal pathology in a B6/129 TNF−/− mouse (7 days). D. Severe hippocampal pathology in the pyramidal layer in a B6/129 TNF−/− mouse at 45 dpi. Arrowheads point to the dentate gyrus and arrows point to the pyramidal cell layer of the hippocampal formation. The dentate gyrus is preserved following TMEV infection. In contrast, the pyramidal layer is markedly injured in both B6/129F1and B6/129 TNF−/− mice at 7 dpi. The pyramidal layer is restored to preinfection state in B6/129F1 mice (B) but not in B6/129 TNF−/− mice (D) by 45 dpi. B6/129F1 mice were chosen to illustrate the pathology in TMEV‐infected “control” mice. Similar pathology was observed in the other “control mice” (B6, 129/SJ, B6/129F2).
We then asked specifically if TNFα deletion delayed the resolution of injury of the hippocampus and striatum, the two brain regions with the worst brain pathology following TMEV infection. For statistical comparisons, we compiled by genetic strain all pathology scores from the early disease (<45 days after infection) as compared with the late disease (>45 days after infection). All strains were studied for up to 270 days after infection. Ten animals per genetic strain were studied at the different timepoints after infection. The hippocampus and striatum datasets were examined separately for this analysis. As expected, there was a statistically significant reduction (P < 0.001, ranks) in the extent of pathologic injury in the hippocampus when examining TNF+/+ (B6, B6/129F1 and B6/129F2) mice during the early disease (<45 days, 2.300 ± 0.238; n = 50) as compared with the late disease (>45 days, 0.322 ± 0.101; n = 59). A similar statistically significant result (P < 0.001, ranks) was found in the scores in the striatum during the early disease (<45 days, 1.859 ± 0.172; n = 85) as compared with the late disease (>45 days, 0.614 ± 0.136; n = 83). In contrast, TNF−/− mice showed no statistical difference (P = 0.445, ranks) in the pathologic scores in the hippocampus early (<45 days, 2.075 ± 0.259; n = 40) vs. late (>45 days, 2.250 ± 0.270; n = 48). Similarly, in TNF−/− mice, statistically nonsignificant results (P = 0.532, ranks) were found when comparing the pathologic scores in the striatum early in the disease (<45 days 3.029 ± 0.215; n = 35) with the pathologic scores late in disease (>45 days, 2.607 ± 0.233; n = 61). We concluded that TNFα was necessary to resolve hippocampal and striatal pathology following virus infection.
The resolution in hippocampal injury depends on TNFR2, whereas the resolution in striatal injury depends on TNFR1
Having established that deletion of TNFα altered the rate and degree of injury resolution in the hippocampus and striatum, we asked which of the known TNFα receptors caused this effect. We infected B6/129 TNFR1−/− mice and B6/129 TNFR2−/− mice with TMEV and studied the extent of brain pathology at 7 dpi (Figure 6A–D) and 45 dpi (Figure 6E–H). Analyses of the hippocampus and striatum of mice with deletions of TNFR1 and TNFR2 showed a similar extent of severe pathology at the 7‐day timepoint (Figure 6A–D). We chose the 45‐dpi timepoint for primary analysis of resolution of pathology because at this time, there was a statistical difference in degree of brain pathology when comparing TNF+/+ mice with TNF−/− mice. When the scores from all of the brain areas were combined for analyses, there was no statistical difference in degree or distribution of brain pathology in B6/129F1 mice (n = 70) as compared with B6/129 TNFR1−/− mice (n = 70, P = 0.977) or B6/129 TNFR2−/− mice (n = 49, P = 0.338).
Figure 6.

Resolution of inflammation and preservation of neurons in the hippocampus following virus infection is dependent on tumor necrosis factor receptor 2 (TNFR2) whereas healing of the striatum is dependent on TNFR1. Paraffin‐embedded sections were stained with hematoxylin–eosin stain. A,C,E,G. From mice expressing the TNFR1 receptor. B,D,F,H. From mice expressing the TNFR2 receptor. A–D. From mice infected for 7 days. E–H. From mice infected for 45 days. There is no difference in the extent of pathology at 7 days following infection irrespective of the expression of TNFR1 or TNFR2. In contrast, in the reparative phase (45 days following infection), there is repair in the striatum (E) but not in the hippocampus (G) in mice expressing only TNFR1. In mice expressing TNFR2, however, there is repair in the hippocampus (H) but not in the striatum (F). Arrows point to the dentate gyrus of the hippocampus, which is normally preserved after infection with TMEV. White stars show areas of necrosis (Bar = 500 µm).
Having previously established that the protective effect of TNF was primarily on the hippocampus and striatum, we analyzed these areas separately. In the hippocampus, the pathologic score in TNFR1−/− mice was 0.300 ± 0.213 (n = 10) whereas the score for TNFR2−/− mice was 1.267 ± 0.431 (n = 15). By ANOVA on ranks, there was a statistical difference (P = 0.005) between the scores in TNF−/−, TNFR1−/− and TNFR2−/− mice. Multiple pair‐wise comparisons using Dunn's method demonstrated statistically significant differences when comparing the pathologic scores during the late disease between TNF−/− mice (2.250 ± 0.270, n = 40) and TNFR1−/− mice (0.300 ± 0.213, n = 10). Therefore, the TNFR1−/− mice behaved similarly to the TNF+/+ mice in reference to the hippocampus neuronal preservation. This supported the hypothesis that the beneficial effect of TNFα. in the hippocampus functioned through the still‐active TNFR2 receptor present in the TNFR1−/− mice (Figure 6H).
However, study of the injury of the striatum in these mice yielded an unexpected result. The resolution of injury in the striatum occurred via TNFR1 (Figure 6E). In the striatum, the pathologic score in TNFR1−/− mice was 2.200 ± 0.533 (n = 10) whereas the score for TNFR2−/− mice was 1.188 ± 0.359 (n = 16). By ANOVA on ranks, there was a statistical difference (P = 0.009) between the scores in TNF−/−, TNFR1−/− and TNFR2−/− mice. Multiple pair‐wise comparison using Dunn's method demonstrated a statistically significant difference in the pathologic scores during the late disease between TNF−/− mice (3.029 ± 0.215, n = 35) and TNFR2−/− mice (1.188 ± 0.359, n = 16). This implied that the beneficial effect of TNFα in the striatum functioned through TNFR1 (Figure 6E). Thus, the results support the hypothesis that the CNS‐restorative effect of TNFα functions via a mechanism dependent on the p55 receptor (TNFR1) and the p75 receptor (TNFR2) for striatal and hippocampal resolution of pathology, respectively (Figures 6E–H).
Having established the different effects of TNFR1 and TNFR2 on the protection of the CNS from disease in the hippocampus and striatum, we stained for TNFR1 and TNFR2 in the CNS by immunocytochemistry. TNFR1 and TNFR2 appear to be equally expressed in the hippocampus and striatum. Therefore, the differential expression of the proteins in the hippocampus or striatum did not account for the differing effects of TNFR1 and TNFR2 in the protection of the CNS.
The effect of TNFα on speed and completeness of resolution of hippocampal and striatal injury is independent of virus antigen or virus RNA expression
Having established that TNF−/− mice had slower, less complete resolution of hippocampal and striatal injury than TNF+/+ mice, we asked if this was a result of failure to control virus antigen expression from the brain at these timepoints. We quantitated the number of virus antigen‐positive cells per 40× h.p.f. in the section from the cerebellum, brain stem, cortex, hippocampus, striatum and corpus callosum of B6, B6/129F1 and B6/129 TNF−/− mice at 7 and 45 days after infection (Table 1, Figure 5). We chose these timepoints because brain pathology was similar between these strains on day 7, whereas brain pathology was increased in B6/129 TNF−/−mice as compared with the control strains at 45 dpi. We quantitated virus antigen‐positive cells in areas of maximal pathology (5 to 11 specimens per experimental group). On day 7, there were, on average, 8 to 20 antigen‐positive cells per h.p.f. in the cortex. Statistics by ANOVA with multiple pair‐wise comparisons (Dunn's method) showed more virus antigen‐positive cells in the cortex of B6/129 TNF−/− mice than in either B6 or B6/129F1 mice. On day 7, there were, on average, 7 to 43 virus antigen‐positive cells per h.p.f. in the hippocampus. However, there were no statistical differences between the groups in virus antigen‐positive cells in the hippocampus. Similar numbers of virus antigen‐positive cells were observed in the striatum between the three strains. As expected, few antigen‐positive cells were observed in the cerebellum, brain stem and corpus callosum. We conclude that TNFα deletion only affects the expression of virus antigen in the cortex early after infection but does not affect the expression of virus antigen in the hippocampus or the striatum.
Table 1.
Virus antigen in TNF−/− mice. Abbreviation: TNF = tumor necrosis factor.
| Brain area | TNF−/− | B6 | B6/129F1 | |||
|---|---|---|---|---|---|---|
| Day 7 (n = 8) | Day 45 (n = 5) | Day 7 (n = 9) | Day 45 (n = 5) | Day 7 (n = 11) | Day 45 (n = 5) | |
| Cerebellum | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| Brain stem | 1.2 ± 1.3 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.6 ± 0.6 | 3.6 ± 2.9 | 0.0 ± 0.0 |
| Cortex | 20.0 ± 2.9 | 0.0 ± 0.0 | 8.2 ± 1.9 | 0.0 ± 0.0 | 13.0 ± 5.4 | 0.0 ± 0.0 |
| Hippocampus | 28.4 ± 7.1 | 0.0 ± 0.0 | 6.8 ± 5.3 | 1.6 ± 1.6 | 42.8 ± 6.9 | 1.8 ± 1.2 |
| Striatum | 7.0 ± 6.2 | 0.0 ± 0.0 | 6.8 ± 5.2 | 0.0 ± 0.0 | 5.2 ± 2.7 | 0.0 ± 0.0 |
| Corpus callosum | 0.0 ± 0.0 | 0.0 ± 0.0 | 4.8 ± 4.8 | 0.6 ± 0.4 | 0.0 ± 0.0 | 0.0 ± 0.0 |
Values are expressed as number of antigen‐positive cells per 40× high power field in area of maximal pathology.
Statistics by ANOVA of three strains at day 7: cortex, P < 0.001, Dunn's Method comparing TNF−/− vs. B6 and TNF−/− vs. B6/129F1; hippocampus Non‐Significant (NS) (P = 0.142); striatum NS (P = 0.759).
We determined whether the delayed resolution of pathology observed in B6/129 TNF−/− mice resulted from virus antigen persistence in those areas with persistent injury. Analysis of five B6/129 TNF−/− mice with severe CNS pathology at 45 days showed no antigen‐positive cells in any brain area examined (Table 1). This was the case even though four out of five mice had pathology scores of “4” indicative of necrosis (Figure 6) in the striatum and/or hippocampus. B6 and B6/129F1 mice with resolved pathology in the striatum and hippocampus also showed minimal or no virus antigen expression in the cortex, hippocampus and brain stem (Table 1, Figure 7). We conclude that delayed recovery of CNS pathology in TNFα‐deficient mice did not result from persistent virus antigen expression in cells in the CNS.
Figure 7.

Detection of virus antigen‐positive cells in the brain following Theiler's murine encephalomyelitis (TMEV) infection does not correlate with restoration of the CNS mediated by TNFα. Paraffin‐embedded sections were stained by immunoperoxidase with a rabbit antibody directed against capsid antigens of TMEV. A. Virus antigen‐positive cells in the pyramidal layer of the hippocampus of a B6/129F1 mouse (7 days). B. Absence of virus antigen‐positive cells in the hippocampus of a B6/129F1 mouse (45 days). C. Scattered virus antigen‐positive cells in pyramidal layer of the hippocampus of a B6/129 TNF−/− mouse (7 days). D. Absence of virus antigen‐positive cells in the hippocampus of a B6129 TNF−/− mouse (45 days). Arrowheads point to the dentate gyrus and arrows point to the pyramidal layer of the hippocampal formation. The dentate gyrus does not express virus antigen following TMEV infection. In contrast, the pyramidal layer shows multiple virus antigen‐positive cells in both B6/129F1and B6/129 TNF−/− mice after 7 days of infection. Neither B6/129F1 mice nor B6/129 TNF−/− mice show virus antigen in the pyramidal layer after 45 dpi. Even though there are more virus antigen‐positive cells in the pyramidal layer of the TNF+/+ mouse than in the TNF−/− mouse, there is almost complete resolution of pathology in the TNF+/+ mouse (B) but not in the TNF−/− mouse (D).
We also isolated RNA from the brain and spinal cord of TNF−/−mice (n = 13) and B6/129 F2 mice (n = 14) at 7, 21 and 45 dpi (Figure 8). Both TNF−/− and B6/129 F2 mice had high levels of viral RNA expression as measured by RT‐PCR for VP2 transcripts in the CNS at 7 dpi. However, by 21 dpi, both the TNF−/− and B6/129 F2 mice had controlled virus RNA in the CNS, and viral RNA levels were further reduced in both groups at 45 dpi. No differences by quantitative RT‐PCR for virus RNA were observed when comparing TNF−/− mice that showed persistent neurologic injury and B6/129 F2 mice that had cleared most of the CNS injury. This further supports the conclusion that virus clearance does not determine hippocampal and striatal resolution of inflammation and neuronal injury following TMEV infection.
Figure 8.
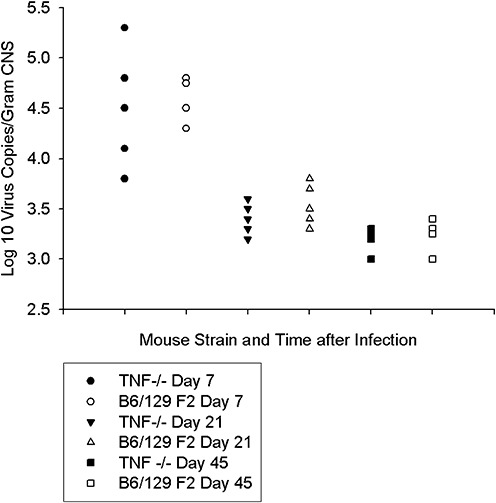
Viral RNA levels in B6/129F2 vs. B6/129 TNF−/− mice. The number of viral copies of RNA was determined by RT‐ PCR. Both B6/129 F2 mice and B6/129 TNF−/− mice had similar levels of virus replication early (7 days) after infection. By 21 and 45 dpi, the levels of viral RNA were decreased 100‐fold and were at the level of detection for this assay (approximately 1000 and 10 000 copies of viral RNA). There were no differences when comparing virus replication in tumor necrosis factor α (TNF)‐deficient versus TNF‐competent mice. For this experiment, the spinal cord and the brain from each animal were combined for extraction of total RNA. Each symbol represents an individual mouse.
Active treatment with recombinant mouse TNFα reduces the extent of injury in the brain following TMEV infection, whereas treatment with a monoclonal antibody to TNFα exacerbates brain disease
Having established that TNFα‐deficient mice had slowed healing of the CNS following TMEV infection, we asked whether active treatment with recombinant TNFα would accelerate restoration following injury. For these experiments, we used SJL/J (H‐2s) mice because previous experiments showed beneficial effects of recombinant TNFα on demyelination using this mouse strain following virus infection (23). We also wanted to determine if the effects of TNF were observed independently in another mouse strain. We first established that in the SJL/J strain, there was encephalitis after virus infection that was then rapidly resolved. We analyzed brain pathology scores of seven mice at 7 dpi and 14 mice at 35 dpi. Similar but to a lesser extent than what was observed in B6 (H‐2b) mice, SJL/J mice had CNS injury at 7 dpi and rapid resolution by 35 dpi. This resulted in brain scores of 1.55 ± 0.18 (n = 49, 7 days) and 0.45 ± 0.09 (n = 70, 35 days), with predominant pathology observed in the striatum during the early phase of the disease. The injury level in SJL mice was less than that observed in B6/129 mice but sufficient to test the effect of TNF on resolution of injury. We then tested whether treatment with recombinant mouse TNFα positively affected the resolution of brain pathology scores. All analyses were done on coded sections without knowledge of treatment groups. For these experiments, five animals were treated with murine recombinant TNFα (10 µg/dose), and eight animals were treated with PBS as the vehicle in which the recombinant TNFα was diluted. Recombinant TNF treatment was started 12 h prior to infection and then given three times a week until sacrifice at 14 dpi. The 14‐day timepoint was chosen because it was after the peak of virus‐induced brain injury (7 days) but before the almost complete spontaneous resolution of injury of the hippocampus, striatum and cortex (35 days). There was a statistically significant decrease in brain pathology score for recombinant mouse TNFα‐treated mice (0.32 ± 0.09) as compared with vehicle‐treated mice (1.23 ± 0.18). Of interest, the brain scores for recombinant TNFα‐treated mice at 14 dpi were similar to the brain scores of nontreated mice at 35 dpi. This confirmed that TNFα accelerated the rate of normal spontaneous resolution of injury following a virus infection. The resolution of injury in the mouse TNFα‐treated mice occurred predominantly in the hippocampus and the striatum (Figure 9). We addressed whether the beneficial effect of TNFα treatment was a result of enhanced virus clearance. This was important because these experiments used SJL/J mice that were susceptible to virus persistence. In a separate experiment (four animals per experimental group), mice were treated with either mouse TNFα or PBS and sacrificed at 7, 14 and 35 dpi. Brain and spinal cord were harvested and a virus plaque assay performed. There were no differences in the PFUs expressed as log10per gram of CNS. The number of plaques (log10) isolated was 4.9 ± 0.3 in PBS‐treated mice vs. 5.1 ± 0.3 in TNFα‐treated mice at 7 dpi, 3.6 ± 0.6 in PBS‐treated mice vs. 3.2 ± 0.2 in TNFα‐treated mice at 14 dpi and 3.5 ± 0.4 in PBS‐treated mice vs. 3.2 ± 0.1 in TNFα‐treated mice at 35 dpi. Therefore, we conclude that the beneficial effect of TNFα is independent of virus replication.
Figure 9.

Disease in the striatum of Theiler's murine encephalomyelitis (TMEV)‐infected mice treated with anti‐tumor necrosis factorα (TNFα) antibody or with recombinant TNFα. Arrows indicate third ventricle. A. Inflammation and early parenchymal disease in the striatum of an SJL mouse treated with isotype‐control antibody directed to beta‐galactosidase. This striatum received a pathology score of “1.”B. Moderately severe parenchymal disease in a mouse treated with anti‐TNF antibody. This striatum received a pathology score of “3.”C. Normal striatum in a mouse treated with recombinant mouse TNF. This striatum received a pathology score of “0.”
In a complementary set of separate experiments, we asked whether a neutralizing mouse monoclonal antibody to mouse TNFα would exacerbate brain pathology in SJL/J mice. Mice were treated 1 day prior to infection and 15 days following infection with either 2.0 mg of a neutralizing mouse monoclonal antibody to mouse TNFα or 2.0 mg of an isytope control mouse monoclonal antibody to beta‐galactosidase (1). This anti‐TNFα antibody had been used previously in vivo at similar dosages to neutralize TNFα in a murine model of inflammatory bowel disease. Mice were euthanized 35 days after TMEV infection and analyses performed of brain pathology as described earlier. We analyzed 20 mice treated with anti‐TNFα monoclonal antibody compared with 38 mice treated with the control anti‐beta‐galactosidase monoclonal antibody. All analyses were done without knowledge of treatment groups. As expected, pathologic scores in the brain of SJL mice were lower than those of B6 mice. Despite the overall lower scores, the data showed a statistical difference (P = 0.004, rank order sum test) with more brain pathology observed in the anti‐TNFα‐treated mice (0.83 ± 0.09, n = 139) than in the control antibody‐treated mice (0.44 ± 0.06, n = 218). Of note, these mice had a higher degree of pathology in the striatum rather than the hippocampus (Figure 9). Striatal values in the treated SJL group were 1.826 ± 0.312 (n = 23) as compared with a hippocampal score of 0.478 ± 0.176 (n = 23, P = 0.03, ranks). We examined the proportion of total number of brain regions with a score of >0 (thus demonstrating pathologic abnormality) in the anti‐TNFα‐treated mice (59/113, 52.1%) as compared with the control antibody‐treated mice (48/218, 22.9%). This difference was also statistically significant (P = 0.0004) by chi‐square analysis. These data support the protective role of TNFα in brain neuronal disease, particularly in the striatum. Viral‐load studies were not done with these experiments, but our previous experiments argue against a role for viral clearance as the mediator of the TNF effect.
MRI demonstrates the persistence of brain injury in TNF−/− mice following TMEV infection
Having established that TNF−/− and TNF+/+ mice showed a similar extent of hippocampal and striatal injury following early TMEV infection, we then determined whether TNF−/− mice followed longitudinally had persistent CNS injury while individual B6/129 F1 mice showed resolution of injury. We previously showed that hippocampal and striatal injury can be visualized as T1 “black holes” using MRI (25). For this experiment, five TNF−/−mice and five B6/129 F1 mice were imaged before virus infection and then at 7 and 45 dpi. We kept track of the individual mice and made sure that the mice were registered in the MRI machine identically at the 0‐, 7‐ and 45‐day timepoints (Figure 10). We considered this the best way to show the morphologic data as a cross‐sectional representation of the injury in individual mice. As expected, we observed no MRI lesions in mice before infection (Figure 10). All TNF−/− mice and B6/129 F1 mice showed variable sizes and locations of T1 black holes in the area of the hippocampus and the striatum (Figure 10). None of the B6/129 F1 mice had lesions at the 45‐day timepoint. In contrast, three out of five TNF−/− mice showed persistent T1 black holes in the hippocampus and/or striatum at the 45‐day timepoint (Figure 10). The pathology of all three mice with persistent T1 black holes showed severe, persistent injury of the hippocampus and striatum. In contrast, mice with resolving MRI showed only minor—if any—injury in the hippocampus or striatum. These data indicate that, in an individual mouse, the presence or absence of TNFα influences the degree of CNS injury vs. resolution.
Figure 10.

MRI shows “T1 black holes” in the hippocampus and striatum. A. Noninfected B6/129F1 mouse shows absence of imaging abnormalities in the brain. B. Age‐matched noninfected B6/129 TNF−/− mouse also shows absence of imaging abnormalities. C. Same mouse as in A shows a lesion in the left hippocampus following infection with Theiler's murine encephalomyelitis (TMEV) for 7 days. D. Same mouse as in B shows a larger lesion in the left hippocampus following infection with TMEV for 7 days. E. Same mouse as in A and C shows resolution of the imaging abnormality 45 days after virus infection. F. Same mice as in B and D show persistence of signal abnormality 45 days after virus infection. Figures on the right and the bottom of each image show the identical registration in the X, Y and Z planes.
DISCUSSION
In this paper, we demonstrate that genetic deletion of TNFα markedly delays healing of the hippocampus and the striatum following virus‐induced encephalitis. This observation is specific to TNFα and is likely not influenced by other genetic loci because mice with comparable—but not identical—genetic makeup with normal expression of TNFα show rapid and almost complete recovery of the brain following intracranial infection. Unexpectedly, this reparative function of TNFα in the CNS was dependent on TNFR1 (p55) for resolution of injury of the striatum but dependent on TNFR2 (p75) for resolution of injury of the hippocampus. The finding was not the result of different expressions of TNFR1 and TNFR2 in the various brain regions, as by immunocytochemistry, both the hippocampus and striatum showed staining for TNFR1 and TNFR2. A simple explanation for these observations could be that delayed resolution in the CNS pathology following encephalitis resulted from persistent virus antigen expression in areas of persistent morphologic injury. However, no virus antigen‐positive cells appeared chronically in any part of the brain in B6/129 TNF−/− mice despite the relative failure to resolve the pathology. In addition, quantitative RT‐PCR studies for viral RNA did not distinguish mice whose injuries resolved from mice with unresolved injuries. Therefore, the CNS virus infection was controlled normally despite the absence of TNFα. This implies that the delayed healing of the hippocampus and striatum was caused by an effect on CNS parenchymal cells independent of virus infection. The genetic experiments were corroborated by treatment of virus‐infected mice with recombinant mouse TNFα, which improved pathology in the hippocampus and striatum, and by treatment with a neutralizing mouse monoclonal antibody to mouse TNFα that worsened brain pathology. The fact that the protective effect of TNFα was seen in two different strains of mice argues that this is likely a robust biologic effect. Finally, we used serial MRI studies of individual TNF−/− mice or B6/129 F1 control mice to show that TNFα influenced the extent of CNS restoration longitudinally in individual mice. We are confident of the validity of our results because we did the appropriate control experiments on B6, 129/J, B6 129F1 and B6 129F2 mice, which did not show the changes in the hippocampus and striatum observed in TNF−/− mice. The total absence of hippocampal and striatal changes in our control mice eliminated any need for more laborious back‐cross experiments of B6/129F2 TNF−/− mice to B6 for multiple generations to obtain a line of TNF−/− mice without genetic drift. The experiments excluded for most unknown genetic factors outside of the TNF gene.
In our experiments, infecting over 250 TNF−/− mice resulted in no deaths following TMEV infection. This is in contrast to work using herpes simplex virus in which TNF−/− mice died of the infection (35). This implies that TMEV is intrinsically a much less virulent virus than herpes simplex. Even though we did not observe deaths in TNF−/−mice infected with TMEV, we did observe persistent injury in the hippocampus and striatum.
In most mouse strains studied to date, infection with TMEV results in two distinct phenotypes. In animals resistant to demyelination, such as prototypic C57BL/6 mice, virus replicates in the brain during the first 7 to 21 days of infection, resulting in encephalitis involving the hippocampus, striatum and cortex (7). Virus then rapidly clears from the brain in association with an intense inflammatory response (16) that protects against virus persistence and subsequent demyelination. This protective response mapped genetically to H‐2D genes of MHC by use of recombinant inbred strains (29) or transgenic mice (27). In resistant mice, the CD8+T cell response is directed against an immunodominant viral peptide (20), and both VP1 and VP2 capsid proteins are targets of cytotoxic lymphocytes in the brain(15). Our present results indicate that TNFα does not influence this protective immune response. Mice with genetic deletion of TNFα cleared virus antigen normally from neurons, oligodendrocytes and macrophages. In addition, genetic deletion of TNFα did not predispose these mice to a chronic demyelinating phenotype. As TNFα maps to the same chromosomal region or MHC, the absence of virus persistence or demyelination in TNF−/−mice indicates that the TNF deletion does not encroach on MHC function.
There is evidence that TNFα protects humans from CNS injury, especially with regard to demyelination. A clinical trial using a monoclonal antibody directed against TNFα was stopped prematurely in MS patients because of unexpected worsening of clinical deficits and MRI lesions (37). Of interest, this antibody was efficacious in rheumatoid arthritis patients. Unfortunately, cases of new‐onset CNS demyelination indistinguishable from MS have appeared in rheumatoid arthritis patients as a direct consequence of influencing TNFα with this monoclonal antibody. This provides the strongest human in vivo evidence that TNFα is one of a series of protective immune molecules for the CNS, along with interferon‐γ and interleukin 6 (IL‐6). One possibility for the change in phenotype observed in TNF−/− mice would be a change in astrocytes (GFAP‐positive cells), microglial cells (F480‐positive cells), or B cells (IgG‐ or IgM‐positive cells). We stained for these markers in the TMEV‐infected TNF−/− mice as well as the controls and saw no changes in their distribution (data not shown). Therefore, it was felt that any subtle effect of TNF on these cell types was overridden by the massive encephalitis observed in TMEV‐infected mice.
What are the mechanisms by which TNFα protects neurons following CNS viral injury? There are two general possibilities. First, TNFα may function directly on neurons or other CNS cells to enhance protection. Secondly, TNFα may indirectly induce restoration by stimulating other protective factors (growth factors, cytokines and interleukins). Mechanisms for the direct hypothesis include prevention of DNA fragmentation of injured neurons as demonstrated in ischemia of the CNS and in Alzheimer's disease. Anti‐apoptotic effects of TNFα have been shown to be the result of stimulating neuronal cells to express Bcl‐2. This has been described in both normal neurons and in prion protein‐deficient neuronal cells. TNFα also protects neurons from glutamate‐induced excitotoxicity (18) and from oxidative stress (5). This protection is dependent on TNFR2 and is enhanced by N‐methyl‐D‐aspartate receptor costimulation. The fact that TNFR2 was required for the healing of the hippocampus following virus infection is consistent with this finding. TNFα may also enhance the migration of neuronal precursor cells as has been shown in vitro using neurospheres, or TNFα may stabilize neuronal and mitochondrial membranes as has been shown in vitro in neurons and oligodendrocytes (6). TNFR2 has been found to be critical for oligodendrocyte regeneration (3). Mechanisms for the indirect hypothesis include activation of such factors as IL1‐β(7). TNFα acts via its receptors to stimulate synthesis of IL1‐β(12), which enhances CNS restoration in several models of demyelination (19). TNFα also stimulates epidermal growth factor expression in astrocytes (10), which may contribute to enhanced neuronal survival.
In this viral model system, TNFα protected different populations of neurons via either TNFR1 (p55) or TNFR2 (p75). Of interest, TNFR2 protected hippocampal neurons whereas TNFR1 protected striatal neurons. By immunocytochemistry, TNFR1 or TNFR2 were similarly expressed in the hippocampus and striatum. In contrast to our results, others propose that TNFR1 is important in TNFα‐mediated apoptosis whereas TNFR2 is important in mediating a TNFα anti‐apoptotic pathway (18). TNFR2‐mediated neuroprotection may work via a nuclear factor‐kappa B (NF‐kB) pathway (18). Of interest, NF‐kB activation via the TNFR1‐induced pathway does not protect cortical neurons from glutamate‐induced apoptosis, and in the retina, TNFR1 augments neuronal death whereas TNFR2 promotes neuroprotection (13). The reduction of neuronal loss in this system is associated with the presence of activated Akt/protein kinase B (13).
Deciphering the precise role of TNFα in the rapid resolution of injury of the hippocampus and the striatum following virus infection may explain other disease processes that specifically injure these parts of the brain (degenerative dementias, ischemia, anoxia and others). Ultimately, this family of factors or derivatives of these factors may potentially serve as therapeutic agents in a variety of CNS disease states.
ACKNOWLEDGMENTS
Grants from the National Institutes of Health (P01 NS 38468, R01 NS 32129) and the National MS Society Center Grant (CA 1011A8) supported this work. Lea Dacy helped edit the manuscript.
REFERENCES
- 1. Abrahamsohn IA, Coffman RL (1995) Cytokine and nitric oxide regulation of the immunosuppression in Trypanosoma cruzi infection. J Immunol 155:3955–3963. [PubMed] [Google Scholar]
- 2. Akassoglou K, Bauer J, Kassiotis G, Pasparakis M, Lassmann H, Kollias G, Probert L (1998) Oligodendrocyte apoptosis and primary demyelination induced by local TNF/p55TNF receptor signaling in the central nervous system of transgenic mice: models for multiple sclerosis with primary oligodendrogliopathy. Am J Pathol 153:801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP (2001) TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci 4:1116–1122. [DOI] [PubMed] [Google Scholar]
- 4. Bieber AJ, Kerr S, Rodriguez M (2003) Efficient central nervous system remyelination requires T cells. Ann Neurol 53:680–684. [DOI] [PubMed] [Google Scholar]
- 5. Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, et al (1996) Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med 2:788–794. [DOI] [PubMed] [Google Scholar]
- 6. Bruce‐Keller AJ, Geddes JW, Knapp PE, McFall RW, Keller JN, Holtsberg FW, et al (1999) Anti‐death properties of TNF against metabolic poisoning: mitochondrial stabilization by MnSOD. J Neuroimmunol 93:53–71. [DOI] [PubMed] [Google Scholar]
- 7. Buenz EJ, Howe CL (2006) Picornaviruses and cell death. Trends Microbiol 14:28–36. [DOI] [PubMed] [Google Scholar]
- 8. Buenz EJ, Rodriguez M, Howe CL (2006) Disrupted spatial memory is a consequence of picornavirus infection. Neurobiol Dis 24:266–273. [DOI] [PubMed] [Google Scholar]
- 9. Condorelli F, Sortino MA, Stella AM, Canonico PL (2000) Relative contribution of different receptor subtypes in the response of neuroblastoma cells to tumor necrosis factor‐alpha. J Neurochem 75:1172–1179. [DOI] [PubMed] [Google Scholar]
- 10. Donato NJ, Ince C, Rosenblum MG, Gallick GE (1989) Early events in the antiproliferative action of tumor necrosis factor are similar to the early events in epidermal growth factor growth stimulation. J Cell Biochem 41:139–157. [DOI] [PubMed] [Google Scholar]
- 11. Drescher KM, Murray PD, David CS, Pease LR, Rodriguez M (1999) CNS cell populations are protected from virus‐induced pathology by distinct arms of the immune system. Brain Pathol 9:21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Feuerstein GZ, Liu T, Barone FC (1994) Cytokines inflammation and brain injury: role of tumor necrosis factor‐alpha. Cerebrovasc Brain Metab Rev 6:341–360. [PubMed] [Google Scholar]
- 13. Fontaine V, Mohand‐Said S, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U (2002) Neurodegenerative and neuroprotective effects of tumor necrosis factor (TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2. J Neurosci 22:rC216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Korner H, Riminton DS, Strickland DH, Lemckert FA, Pollard JD, Sedgwick JD (1997) Critical points of tumor necrosis factor action in central nervous system autoimmune inflammation defined by gene targeting. J Exp Med 186:1585–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lin X, Thiemann NR, Pease LR, Rodriguez M (1995) VP1 and VP2 capsid proteins of Theiler's virus are targets of H‐2D‐restricted cytotoxic lymphocytes in the central nervous system of B10 mice. Virology 214:91–99. [DOI] [PubMed] [Google Scholar]
- 16. Lindsley MD, Rodriguez M (1989) Characterization of the inflammatory response in the central nervous system of mice susceptible or resistant to demyelination by Theiler's virus. J Immunol 142:2677–2682. [PubMed] [Google Scholar]
- 17. Lipton HL (1975) Theiler's virus infection in mice: an unusual biphasic disease process leading to demyelination. Infect Immun 11:1147–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marchetti L, Klein M, Schlett K, Pfizenmaier K, Eisel UL (2004) Tumor necrosis factor (TNF)‐mediated neuroprotection against glutamate‐induced excitotoxicity is enhanced by N‐methyl‐D‐aspartate receptor activation. Essential role of a TNF receptor 2‐mediated phosphatidylinositol 3‐kinase‐dependent NF‐kappa B pathway. J Biol Chem 279:32869–32881. [DOI] [PubMed] [Google Scholar]
- 19. Mason JL, Suzuki K, Chaplin DD, Matsushima GK (2001) Interleukin‐1beta promotes repair of the CNS. J Neurosci 21:7046–7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mendez‐Fernandez YV, Johnson AJ, Rodriguez M, Pease LR (2003) Clearance of Theiler's virus infection depends on the ability to generate a CD8+ T cell response against a single immunodominant viral peptide. Eur J Immunol 33:2501–2510. [DOI] [PubMed] [Google Scholar]
- 21. Milani D, Zauli G, Rimondi E, Celeghini C, Marmiroli S, Narducci P, et al (2003) Tumour necrosis factor‐related apoptosis‐inducing ligand sequentially activates pro‐survival and pro‐apoptotic pathways in SK‐N‐MC neuronal cells. J Neurochem 86:126–135. [DOI] [PubMed] [Google Scholar]
- 22. Paya CV, Patick AK, Leibson PJ, Rodriguez M (1989) Role of natural killer cells as immune effectors in encephalitis and demyelination induced by Theiler's virus. J Immunol 143:95–102. [PubMed] [Google Scholar]
- 23. Paya CV, Leibson PJ, Patick AK, Rodriguez M (1990) Inhibition of Theiler's virus‐induced demyelination in vivo by tumor necrosis factor alpha. Int Immunol 2:909–913. [DOI] [PubMed] [Google Scholar]
- 24. Pierce ML, Rodriguez M (1989) Erichrome stain for myelin on osmicated tissue embedded in glycol methacrylate plastic. J Histotechnol 12:35–36. [Google Scholar]
- 25. Pirko I, Johnson A, Gamez J, Macura SI, Rodriguez M (2004) Disappearing “T1 black holes” in an animal model of multiple sclerosis. Front Biosci 9:1222–1227. [DOI] [PubMed] [Google Scholar]
- 26. Powrie F, Leach MW, Mauze S, Menon S, Caddle LB, Coffman RL (1994) Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity 1:553–562. [DOI] [PubMed] [Google Scholar]
- 27. Rodriguez M, David CS (1995) H‐2 Dd transgene suppresses Theiler's virus‐induced demyelination in susceptible strains of mice. J Neurovirol 1:111–117. [DOI] [PubMed] [Google Scholar]
- 28. Rodriguez M, Buchmeier MJ, Oldstone MB, Lampert PW (1983) Ultrastructural localization of viral antigens in the CNS of mice persistently infected with lymphocytic choriomeningitis virus (LCMV). Am J Pathol 110:95–100. [PMC free article] [PubMed] [Google Scholar]
- 29. Rodriguez M, Leibowitz J, David CS (1986) Susceptibility to Theiler's virus‐induced demyelination. Mapping of the gene within the H‐2D region. J Exp Med 163:620–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rodriguez M, Patick AK, Pease LR (1990) Abrogation of resistance to Theiler's virus‐induced demyelination in C57BL mice by total body irradiation. J Neuroimmunol 26:189–199. [DOI] [PubMed] [Google Scholar]
- 31. Rodriguez M, Zoecklein LJ, Howe CL, Pavelko KD, Gamez JD, Nakane S, Papke LM (2003) Gamma interferon is critical for neuronal viral clearance and protection in a susceptible mouse strain following early intracranial Theiler's murine encephalomyelitis virus infection. J Virol 77:12252–12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Scherbel U, Raghupathi R, Nakamura M, Saatman KE, Trojanowski JQ, Neugebauer E, et al (1999) Differential acute and chronic responses of tumor necrosis factor‐deficient mice to experimental brain injury. Proc Natl Acad Sci USA 96:8721–8726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schwartz M, Hauben E (2002) T cell‐based therapeutic vaccination for spinal cord injury. Prog Brain Res 137:401–406. [DOI] [PubMed] [Google Scholar]
- 34. Sedel F, Bechade C, Vyas S, Triller A (2004) Macrophage‐derived tumor necrosis factor alpha, an early developmental signal for motoneuron death. J Neurosci 24:2236–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sergerie Y, Rivest S, Boivin G (2007) Tumor necrosis factor‐alpha and interleukin‐1 beta play a critical role in the resistance against lethal herpes simplex virus encephalitis. J Infect Dis 196:853–860. [DOI] [PubMed] [Google Scholar]
- 36. Sidman RL, Angevine JB, Pierce ET (1971) Atlas of the Mouse Brain and Spinal Cord. Harvard University Press: Cambridge, MA, USA. [Google Scholar]
- 37. Van Oosten BW, Barkhof F, Truyen L, Boringa JB, Bertelsmann FW, Von Blomberg BM, et al (1996) Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti‐tumor necrosis factor antibody cA2. Neurology 47:1531–1534. [DOI] [PubMed] [Google Scholar]
- 38. Viel JJ, McManus DQ, Smith SS, Brewer GJ (2001) Age‐ and concentration‐dependent neuroprotection and toxicity by TNF in cortical neurons from beta‐amyloid. J Neurosci Res 64:454–465. [DOI] [PubMed] [Google Scholar]
- 39. Warrington AE, Asakura K, Bieber AJ, Ciric B, Van Keulen V, Kaveri SV, et al (2000) Human monoclonal antibodies reactive to oligodendrocytes promote remyelination in a model of multiple sclerosis. Proc Natl Acad Sci USA 97:6820–6825. [DOI] [PMC free article] [PubMed] [Google Scholar]