

Abstract
Two-component regulatory systems require highly specific interactions between histidine kinase (transmitter) and response regulator (receiver) proteins. We have developed a novel genetic strategy that is based on tightly regulated synthesis of a given protein to identify domains and residues of an interacting protein that are critical for interactions between them. Using a reporter strain synthesizing the nonpartner kinase VanS under tight arabinose control and carrying a promoter-lacZ fusion activated by phospho-PhoB, we isolated altered recognition (AR) mutants of PhoB showing enhanced activation (phosphorylation) by VanS as arabinose-dependent Lac+ mutants. Changes in the PhoBAR mutants cluster in a “patch” near the proposed helix 4 of PhoB based on the CheY crystal structure (a homolog of the PhoB receiver domain) providing further evidence that helix 4 lies in the kinase-regulator interface. Based on the CheY structure, one mutant has an additional change in a region that may propagate a conformational change to helix 4. The overall genetic strategy described here may also be useful for studying interactions of other components of the vancomycin resistance and Pi signal transduction pathways, other two-component regulatory systems, and other interacting proteins. Conditionally replicative oriRR6Kγ attP “genome targeting” suicide plasmids carrying mutagenized phoB coding regions were integrated into the chromosome of a reporter strain to create mutant libraries; plasmids encoding mutant PhoB proteins were subsequently retrieved by P1-Int-Xis cloning. Finally, the use of similar genome targeting plasmids and P1-Int-Xis cloning should be generally useful for constructing genomic libraries from a wide array of organisms.
Keywords: P1-Int-Xis cloning, protein phosphorylation, two-component regulatory systems, vancomycin resistance
Protein–protein interactions are important in genetic regulatory responses in all cells. Identifying domains and amino acid residues that determine how a given protein recognizes an interacting partner protein(s) is therefore a fundamental problem. Many interacting proteins are members of gene families and yet interact only with specific proteins that are not (or only poorly) recognized by other members of the same family. Determination of the molecular basis of recognition between such proteins would provide new insights into the specific interactions that are critical in innumerable cellular signaling pathways.
Two-component regulatory systems are crucial in a variety of signal transduction pathways in bacteria (1). Most of these systems share a common biochemical signaling mechanism in which a histidine kinase (usually a membrane-associated sensor protein) autophosphorylates on a conserved histidine residue in response to an environmental stimulus and then transfers the phosphate group to a conserved aspartate residue on its partner response regulator (commonly a transcriptional activator). As many as 50 pairs of these kinase (transmitter) and response regulator (receiver) proteins may exist in a single bacterium such as Escherichia coli. Thus, interactions among these partner proteins must be highly specific to prevent undesirable interactions, or nonspecific cross talk (noise), between nonpartner proteins. Analogous signaling systems exist in eukaryotic cells including Saccharomyces cerevisiae (2), Arabidopsis thaliana (3), and others (4). In yeast, a two-component system (SLN1 and SSK1) provides signaling input to a mitogen-activating protein kinase cascade that is responsive to small molecules (5). Mitogen-activating protein kinase cascades are key integrators of convergent signal transduction pathways in mammalian cells (6).
The PhoR–PhoB two-component regulatory system is required for transcriptional activation of the phosphate (Pho) regulon of E. coli in response to environmental Pi levels (7). Pi control of the Pho regulon is a paradigm of a signal transduction pathway in which occupancy of a cell surface receptor(s) regulates gene expression in the cytoplasm. In response to Pi limitation, the sensor kinase PhoR is autophosphorylated by ATP, phospho-PhoR transfers the phosphate group to the response regulator PhoB, and phospho-PhoB activates transcription at Pho regulon promoters (the Pi signaling response). Deactivation (dephosphorylation) occurs upon growth shift to Pi excess conditions and requires in addition to PhoR, an intact Pst (Pi-specific transport) system and a protein called PhoU. Inhibition (prevention of phosphorylation) also requires PhoR, the Pst system, PhoU, and excess Pi. In the absence of PhoR, cross regulation of PhoB by two other signaling pathways—involving the catabolite sensor histidine kinase CreC (formerly called PhoM) or acetyl phosphate—result in high activation (phosphorylation) of PhoB in response to carbon sources (8–10).
The VanS–VanR two-component regulatory system is required for high-level inducible vancomycin resistance in the clinical isolate Enterococcus faecium BM4142 (11). The sensor kinase VanS is autophosphorylated in the presence of vancomycin (possibly due to accumulation of a cell wall precursor), phospho-VanS transfers the phosphate group to the response regulator VanR, and phospho-VanR activates transcription of the vanRSHAXYZ gene cluster required for vancomycin resistance (12).
VanS is also capable of high activation (phosphorylation) of PhoB due to cross talk when VanS is produced from a multicopy plasmid (13). Using cross talk as an in vivo assay, we previously identified VanS peptides capable of interfering with activation of PhoB by VanS; the peptides may correspond to a VanS domain for interaction with a response regulator. A similar approach had been used earlier to identify the phosphotransfer region of the kinase CheA for its partner response regulator CheY (14). We now describe a new strategy for studying sensor kinase-response regulator interactions; we used our new approach to isolate altered recognition (AR) PhoB mutants as gain-of-function mutants showing enhanced activation by VanS. We expect that methods developed in this study should be generally useful for studying other protein–protein interactions in vivo.
MATERIALS AND METHODS
Media and Reagents.
Luria–Bertani broth, MacConkey, 4-morpholinepropanesulfonic acid (Mops), and TYE media are described elsewhere (15). 5-bromo-4-chloro-3-indolyl β-d-galactopyranoside (X-Gal) was used at 40 μg per ml for detection of β-galactosidase. Ampicillin was added at 100 μg per ml; kanamycin, at 12.5 or 50 μg per ml to select kanamycin-resistant (KanR) integrants or maintain plasmids, respectively; and l-arabinose, at 0.13 mM for induction. Integrants were routinely grown without an antibiotic. Taq DNA polymerase (Promega) was used for polymerase chain reaction (PCR) mutagenesis and testing of single copy integration. Vent DNA polymerase (New England Biolabs) was used to generate DNAs for cloning.
Bacteria.
Unless noted otherwise, bacteria are derivatives of E. coli K-12 BW13711, which were constructed by P1kc transduction (15). Reporter strains have a PphnC−lacZ transcriptional fusion in single copy at the lac locus; they synthesize also the C-terminal cytoplasmic domain of PhoR (residues M63 to D431; denoted ′PhoR) or of VanS (residues M95 to S384; denoted ′VanS) under arabinose control from a ParaB−′phoR or ParaB−′vanS fusion in single copy at the araCBAD locus. These −lacZ and ParaB− fusions were constructed using methods similar to ones described elsewhere (16). In brief, the appropriate DNAs were made with the PCR, cloned, sequenced, and recombined onto the chromosome by allele replacement. The ParaB−′phoR strain BW23423 [lacIq PphnC−lacZWJ19 ΔphoBR580 ΔcreBCD153 Δ(pta ackA hisQ hisP)TA3516 phn(EcoB) ΔaraBADAH37::ParaB−NFLAG′phoRAH41] was used to test for activation of PhoB by ′PhoR; the ParaB−′vanS strains BW23425 (like BW23423, except ΔaraBADAH37::ParaB−′vanSAH49), BW23042 (like BW23425, except ΔphoR574), and BW23427 (like BW23425, except with the ′vanSH164Q allele), for activation by ′VanS or ′VanSH164Q (an inactive protein), and the ParaB−′vanS strain BW23562 (like BW23425, except ΔphoB578-phoR+), for inhibition by PhoR.
Plasmids.
A KanR, attP “genome targeting” suicide vector with the plasmid R6Kγ replication origin (oriRR6Kγ; unpublished data cited in ref. 17) and pINT-ts were from M. Koob (University of Minnesota School of Medicine). An oriRR6Kγ plasmid requires the transacting π protein (the pir gene product) for DNA replication. pINT-ts is an ampicillin-resistant, low copy, temperature-sensitive (Ts−) replicon plasmid synthesizing λ integrase (Int) under control of λcI857 (17). The same system for integration of similar plasmids into attλ has been used elsewhere (18). pAH57 is a derivative of pINT-ts that synthesizes both Int and excisionase (Xis); it was made by joining the EcoRI to BamHI backbone of pINT-ts with a 1.1-kbp EcoRI to BglII PCR fragment encoding Xis and N terminus of Int (restoring int+), which was PCR-generated using λDNA (from R. Somerville, Purdue University) as template. Our integration plasmids (Fig. 1) have an attP region in which a nearby NdeI site was destroyed; ones with Ptac contain the PstI to NdeI lacIq and Ptac segment of pASLR2 [from A. S. Lynch (Harvard Medical School) (18)]. oriRR6Kγ plasmids were maintained at different copy numbers in the pir+ host BW23473 (Δlac-169 robA1 creC510 hsdR514 endA recA1 ΔuidA::pir+), the isogenic pir-116 host BW23474, or similar hosts (16).
Figure 1.
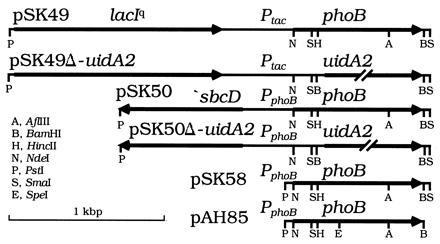
Plasmids. pSK49 and pSK49Δ-uidA2 express phoB or uidA from Ptac. pSK50 and pSK50Δ-uidA2 express phoB or uidA from PphoB. A 189-bp PphoB fragment in pSK58 and pAH85 was generated using PCR. An NdeI site coinciding with the phoB start codon and BamHI site immediately beyond the phoB stop codon were introduced by site-directed mutagenesis. pAH85 has a silent SpeI site at codon 112 of phoB that was introduced with PCR. Mutations were separated by subcloning the respective fragments into pSK58 or pAH85. Only relevant restriction sites are shown. All sites shown for pAH85 are unique. The 2.5-kbp vector backbone is not shown.
Mutagenesis, attP Plasmid Integration, and Mutant Selection.
Plasmid libraries were made by PCR mutagenesis (19) of phoB using pSK5 (20) as template, after which gel-purified NdeI to BamHI fragments were cloned into pSK49Δ-uidA2 or pSK50Δ-uidA2 (Fig. 1); plasmids were propagated in a pir+ host and then integrated into the chromosome of BW23042 or BW23425 to construct mutant libraries with phoB in single copy. Cells carrying pINT-ts were grown in Luria–Bertani broth with ampicillin at 30°C, transformed by electroporation, incubated at 37°C for 1 hr, 42°C for 30 min, spread onto glycerol Mops agar containing 2 mM Pi, arabinose, kanamycin, and X-Gal, and incubated at 37°C.
PCR Testing for Copy Number.
Two primers specific for the vector attP region and two specific for the chromosomal attλ region were used to verify strains with a single integrated plasmid. Details will be described elsewhere.
P1-Int-Xis (PIX) Cloning and DNA Sequencing.
We reasoned that we may be able to retrieve integrated oriRR6Kγ attP plasmids using a helper plasmid encoding both Int and Xis, which catalyze excision and recircularization of phage λ (21). Indeed, we showed that such plasmids are retrievable by P1kc transduction of a pir+ (or pir-116) host synthesizing Int and Xis, a procedure we named “PIX cloning” for P1-Int-Xis cloning. Accordingly, BW23473 carrying pAH57 was grown in Luria–Bertani broth with ampicillin at 30°C and, following infection and 2–3 hr incubation at 37°C, KanR colonies were selected. These were inoculated into Luria–Bertani–kanamycin broth to prepare plasmid DNAs, which were used for DNA sequencing and integration into new hosts. Both DNA strands were sequenced using an Applied Biosystems model 373A automated sequencer at the Dana–Farber Cancer Institute (Harvard Medical School) Core facility. Mutations were separated using a combination of cloning and PCR strategies with the plasmids shown in Fig. 1.
Enzyme Assays.
Measurements of bacterial alkaline phosphatase and β-galactosidase activities have been described (15).
RESULTS
Genetic Strategy for Identifying Mutants.
We considered that gain-of-function PhoB mutants isolated in a strain synthesizing the nonpartner kinase ′VanS may include altered recognition PhoBAR mutants that would be recognizable as ones requiring ′VanS for activation. We expected other gain-of-function mutants to include PhoBAR mutants requiring an endogenous kinase(s), as well as constitutively active (CA) mutants. We therefore developed reporter strains to facilitate the identification and characterization of ′VanS-activated PhoBAR mutants. With one exception these strains are ΔphoBR, ΔcreC, and Δ(pta ackA), so that production of activated PhoB depends upon the introduction of a phoB gene; activation of wild-type (wt) PhoB is disabled due to the combined absence of PhoR, the alternative kinase CreC, and acetyl phosphate (22). One otherwise isogenic ΔphoB–phoR+ strain was used to test the effects of PhoRwt. Many strains encode a mutant kinase (′VanS or ′PhoR) that behaves like a “constitutive kinase” because its N-terminal domain is absent. ′VanS (or ′PhoR) is synthesized from the tightly regulated ParaB promoter (23); this is done so that we can identify PhoBAR mutants activated by ‘VanS as ones showing activation only in the presence of arabinose. Strains with a single copy ParaB−′vanS (or −′phoR) fusion were used because preliminary experiments showed that only strains with a chromosomal (but not ones with a plasmid) ParaB−phoR fusion provided sufficiently tight arabinose control for activation of PhoBwt by PhoRwt (data not shown). In addition, strains carry two reporters for activation of PhoB, the PhoB-dependent PphnC−lacZ fusion (encoding β-galactosidase) and the phoA gene (encoding bacterial alkaline phosphatase). Thus, mutants are easily identified on indicator agar.
Mutant Isolation.
Our genetic strategy is illustrated in Fig. 2. We prepared several phoB plasmid libraries by random PCR mutagenesis followed by cloning the phoB coding region into a conditionally replicative oriRR6Kγ attP plasmid that requires the π replication protein (the pir gene product of the R6K plasmid) for DNA replication (Fig. 1). After propagation in a pir+ host, the plasmids were transformed into a (non-pir) PphnC−lacZ, ParaB−′vanS reporter strain carrying pINT-ts. Integrants resulting from site-specific recombination at attλ were selected on X-Gal agar containing kanamycin and arabinose. Lac+ colonies representing 0.1% or more of the total were screened for ones showing an arabinose-dependent Lac+ phenotype. Colonies showing arabinose dependence were candidates for VanS-activated PhoBAR mutants; others were candidates for PhoBCA mutants.
Figure 2.
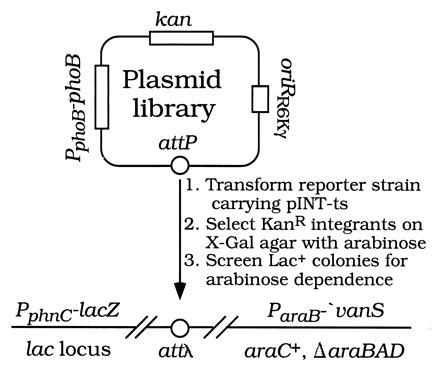
Genetic strategy for isolating PhoBAR mutants.
Mutant Characterization.
We characterized 11 mutants, including 6 candidates for PhoBAR mutants, 2 candidates for PhoBCA mutants, and 3 colonies showing no activation above background (PhoBwt or null mutants). PIX cloning was used to retrieve each integrated plasmid; the retrieved plasmids were again integrated into the same and different reporter strains to test each PhoB protein for activation by ′PhoR, ′VanS, ′VanSH164Q, or PhoRwt (under conditions of Pi limitation) and to test for deactivation or inhibition by PhoRwt. All PhoBAR mutants were activated by ′PhoR, ′VanS, and PhoRwt; none was activated by ′VanSH164Q, in which the histidine autophosphorylation site (residue 164) has been changed to a glutamine. In the presence of excess Pi, activation of all six PhoBAR mutants by ′VanS was inhibited by PhoRwt; in contrast, the two PhoBCA mutants were not deactivated by PhoRwt. Two PhoBwt plasmids served as controls because they showed normal regulation by ′PhoR and PhoRwt and no effect due to ′VanS. A single plasmid encoding a null PhoB protein that was not activated by any kinase served as another control.
DNA sequencing revealed that all 11 phoB coding regions had between 2 and 7 nucleotide changes, including several silent ones. Four PhoBAR mutants had the T97A amino acid change and different silent changes; one PhoBAR mutant had two residue changes (M17V and E87D); the sixth had three residue changes (Q28L, T83S, and S138R). The PhoBCA mutants had two (N40K and T76A) or five (M55V, T83S, E159G, T168S, and F229Y) residue changes. One PhoBwt had no amino acid change; the other had a single residue change (S175C). The PhoB null mutant had three changes (M17I, L95P, and D196G). The PhoBT97A and PhoBM17V,E87D PhoBAR mutants were isolated from pSK50Δ-uidA2 libraries; the PhoBQ28L,T83S,S138R PhoBAR mutant was isolated from a pSK49Δ-uidA2 library (Fig. 1).
Activation of PhoBAR Mutants.
In our reporter strains, activation of PhoBwt by ′PhoR occurs only in the presence of arabinose (Table 1). The low basal level seen in the absence of arabinose is similar to that seen in the absence of a phoR or phoB gene (data not shown). Also, in these reporter strains, no activation of PhoBwt by ′VanS is evident. In contrast, ′VanS activates PhoBM17V,E87D and PhoBT97A >200-fold and >400-fold, respectively. Furthermore, activation of these PhoB proteins by ′VanS occurs to levels similar to those when PhoBwt is activated by ′PhoR. Activation levels of PhoBT97A and PhoBwt by ′PhoR are nearly the same; activation of PhoBM17V,E87D by ′PhoR is enhanced ≈3- to 5-fold when compared with activation of PhoBwt by ′PhoR. Similar effects are seen when activation is compared using phoA or PphnC−lacZ as reporter.
Table 1.
In vivo activation of PhoB proteins by ′PhoR and ′VanS kinases
| Regulator | Kinase† | Bap sp
act*
|
β-galactosidase sp act†
|
||
|---|---|---|---|---|---|
| No arabinose | With arabinose | No arabinose | With arabinose | ||
| PhoBwt | None | 0.3 ± 0.1 | 0.3 ± 0.0 | 6.1 ± 0.2 | 6.5 ± 0.7 |
| PhoBwt | ′PhoR | 0.4 ± 0.1 | 185 ± 3 | 6.4 ± 0.2 | 194 ± 7 |
| PhoBwt | ′VanS | 0.4 ± 0.2 | 0.3 ± 0.0 | 6.1 ± 0.4 | 6.4 ± 0.1 |
| PhoBM17V,E87D | ′PhoR | 1.2 ± 1.5 | 516 ± 36 | 7.5 ± 2.2 | 968 ± 8 |
| PhoBM17V,E87D | ′VanS | 0.4 ± 0.2 | 63.5 ± 1.3 | 5.7 ± 0.5 | 53.1 ± 4.4 |
| PhoBM17V,E87D | ′VanSH164Q | 0.3 ± 0.0 | 0.7 ± 0.2 | 6.7 ± 0.2 | 4.7 ± 0.1 |
| PhoBT97A | ′PhoR | 0.6 ± 0.0 | 174 ± 8 | 6.7 ± 0.1 | 204 ± 10 |
| PhoBT97A | ′VanS | 0.4 ± 0.0 | 125 ± 9 | 6.4 ± 0.3 | 127 ± 7 |
Cells were assayed after growth in 0.06% glycerol Mops 2 mM Pi media without or with arabinose. Bap, bacterial alkaline phosphatase.
Specific activity (sp act) units are nanomoles of product formed per minute per cell optical density at 420 nm. Means of triplicate determinations with standard deviations are given.
Strains are integrants of BW23316 [like BW23423, except Δ(araBAD)AH37], BW23423, BW23425, or BW23427 with a pSK50 derivative encoding the respective PhoB protein.
Effects of Individual Residue Changes on Activation.
We assessed the contributions of individual changes by constructing integration plasmids and strains with each mutation alone. Our results showed that both M17V and E87D are required for the 200-fold activation of PhoBM17V,E87D by ′VanS, although a 5-fold activation of PhoBE87D is also apparent (Table 2). However, the M17V change appears to be solely responsible for the enhanced activation of PhoBM17V,E87D by ′PhoR. In contrast, T83S alone is sufficient for the 400-fold activation of PhoBQ28L,T83S,S138R by ′VanS. None of these residue changes alone showed a substantial effect on activation by ′PhoR (Table 2) or activation, deactivation, or inhibition by PhoRwt (data not shown).
Table 2.
In vivo activation of wild-type and mutant PhoB proteins
| Regulator | By ′PhoR*
|
By
′VanS†
|
||
|---|---|---|---|---|
| Bap sp act | Relative to PhoBwt | Bap sp act | Relative to PhoBwt | |
| PhoBwt | 186 ± 13 | 1.0 | 0.3 ± 0.0 | 1.0 |
| PhoBM17V,E87D | 469 ± 10 | 2.5 | 55.0 ± 15.9 | 167 |
| PhoBE87D | 207 ± 25 | 1.1 | 1.7 ± 0.2 | 5.2 |
| PhoBM17V | 410 ± 20 | 2.2 | 0.5 ± 0.1 | 1.6 |
| PhoBT97A | 182 ± 33 | 1.0 | 142 ± 4 | 432 |
| PhoBQ28L,T83S,S138R | 236 ± 2 | 1.3 | 128 ± 11 | 389 |
| PhoBT83S | 217 ± 21 | 1.2 | 137 ± 6 | 416 |
Cells were grown with arabinose and assayed as in Table 1. Each phoB allele is in single copy in a pSK58 or pAH85 derivative. Bap, bacterial alkaline phosphatase; sp act, specific activity.
Integrants of BW23423 were assayed. No arabinose values were 0.3 ± 0.0 or less.
Integrants of BW23425 were assayed. No arabinose values were 0.6 ± 0.0 or less.
DISCUSSION
We used a new strategy to identify the protein interface of PhoB that appears to be critical for interactions between kinase (transmitter) and response regulator (receiver) domains of two-component signal transduction proteins. It involves (i) tightly regulated synthesis of one component (the nonpartner ′VanS under tight arabinose control) and (ii) an in vivo assay system (a reporter strain with a PhoB-regulated promoter-lacZ fusion) to recognize PhoBAR mutants that can be activated by ′VanS. In preliminary experiments, we showed that strains with the homologous kinase PhoR under the same tight regulation reveal arabinose-dependent activation of PhoBwt. We therefore isolated gain-of-function mutants as Lac+ colonies in the presence of arabinose (for induction of ′VanS synthesis) and recognized PhoBAR mutants as ones requiring arabinose for the Lac+ phenotype. We anticipated being able to isolate PhoBAR mutants showing enhanced activation by the heterologous ′VanS kinase because of the initially weak interaction of ′VanS with PhoBwt; the reactivity between a maltose-binding protein–′VanS fusion protein (MBP-′VanS) and PhoBwt is 104-fold weaker than between MBP–′VanS and its partner response regulator VanR (20). Our use of a heterologous kinase to isolate PhoBAR mutants is in principle similar to the isolation of suppressor mutations as extragenic compensatory mutants. In similar ways, it should be possible to isolate response regulator or kinase mutants showing increased interaction with other heterologous partners.
The N-terminal receiver domain of PhoB is expected to share structural conservation with the chemotaxis response regulator CheY (24), the sporulation phosphorelay protein SpoOF (25), and the N-terminal domain of E. coli NarL (26) whose structures have been solved by x-ray crystallography. Based on the CheY structure, all six PhoBAR mutants have one residue changed (T83, E87, or T97) in the loop preceding helix 4 or in helix 4 (Fig. 3). The helix 4 region of CheY is thought to interact with the kinase CheA as changes in this region reduce affinity for CheA (30, 31). CheY changes reducing affinity are generally nonconservative ones, however. In contrast, PhoB changes showing enhanced activation by ′VanS are usually conservative ones (T83S, E87D, T97A). Conservative changes among closely related proteins may therefore be more significant than nonconserved residues for determining specificity of kinase-regulator interactions. Our results provide further evidence that the helix 4 region may be critical for interactions of response regulators and histidine kinases.
Figure 3.
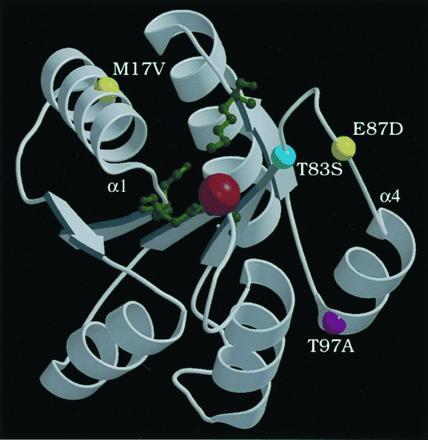
Ribbon diagram of Mg2+-bound CheY crystal structure from coordinates (27), drawn with molscript (28) and raster3d (29). CheY active site residues D12, D13, D57, and K109 are depicted as green ball-and-stick forms; a red ball shows Mg2+. The locations of the equivalent PhoB residues are shown as colored balls: T83 (cyan), M17 and E87 (yellow), and T97 (purple). The amino termini of helix 1 and helix 4 are labeled α1 and α4, respectively.
The PhoBAR mutants described here show nearly as high activation by ′VanS as by ′PhoR. Different classes of PhoBAR mutants may be revealed by using plasmids with more stringently controlled PhoB synthesis. No change leading to enhanced activation by ′VanS abolished activation by ′PhoR. Multiple changes may be necessary to gain recognition by ′VanS and to lose recognition by PhoR simultaneously. Mutants showing enhanced activation by ′VanS and reduced activation by ′PhoR may be found in a more exhaustive search. It is notable that no PhoBAR change makes PhoB more like the natural VanR sequence. On the contrary, T83 in PhoB and T81 in VanR are equivalent. Yet, the T83S change results in enhanced activation of PhoB by ′VanS. Clearly, the context of these residues is especially important for defining the recognition interface. In addition, the respective residue in SpoOF (S87) is structurally linked to the phosphorylation site (D59) via an intermediate residue (N61; ref. 25).
Our finding of one mutant with two distantly located changes (M17V and E87D), which are both required for activation by ′VanS, was at first unexpected. However, it has been recently shown that changes in helix 1 of CheY result in a conformational change (among others) in helix 4 that may mimic its molecular activation mechanism (32). Hence, M17V and E87D together may alter the helix 4 region resulting in enhanced activation by ′VanS. Changes in helix 1 of CheY and SpoOF also confer resistance to the phosphatase activity of CheZ (33) and Rap (25), respectively. Accordingly, M17V may act by abolishing interaction with a phospho-PhoB phosphatase, allowing more efficient activation of the PhoBM17V,E87D protein by ′VanS. In this regard, deactivation (dephosphorylation) of phospho-PhoB in vivo requires the Pst system and PhoU. While the Pst system and PhoU were always present in this study, no evidence indicates that a Pst component or PhoU acts on PhoB in the absence of PhoR (7).
The PhoBT97A and PhoBM17V,E87D proteins have now been purified to homogeneity. Preliminary kinetic studies of these PhoBAR proteins show that phosphotransfer from phospho-MBP–′VanS to PhoBT97A and PhoBM17V,E87D is 20-fold and 15-fold more rapid, respectively, than phosphotransfer to PhoBwt (20). The increased rates of phosphotransfer appear to be primarily due to changes in kcat without appreciable effects on KM values (unpublished data). An alteration in the PhoB interface may result in a different orientation of MBP–′VanS binding and thereby be responsible for such kinetic effects. More detailed studies are required to understand the biochemical basis of the enhanced activation of PhoBAR proteins by ′VanS.
Our approach for studying protein–protein interactions may have widespread application, not only for studying interactions between components of the vancomycin resistance and the Pi signal transduction pathways, but also for analyzing interactions between partner proteins of other two-component regulatory systems, as well as other interacting proteins involved in many diverse intracellular processes. For example, searching for mutants of PhoR capable of activating a nonpartner response regulator should allow one to define a domain(s) and residues of a signaling kinase for recognition of response regulators. Furthermore, when the activity of an expressed (given) protein depends on the presence (or absence) of a second protein, this indicates an interaction between the respective proteins. Therefore tightly regulated synthesis of other interacting proteins can be used to identify mutants affecting interactions with the partner protein(s) or to identify new interacting proteins.
Our searches for PhoBAR mutants involved use of a new cloning strategy that may be generally useful, not only for the isolation of mutants but also for the construction of genomic libraries. We made mutant libraries by cloning mutagenized phoB genes into conditionally replicative oriRR6Kγ attP plasmids, which were then integrated into the chromosome of a reporter strain by site-specific recombination. We were therefore able to screen mutants while avoiding problems often inherent in the use of multicopy plasmids. We subsequently retrieved the mutant plasmids by PIX cloning, a remarkably efficient process. Similar conditionally replicative oriRR6Kγ attP plasmids should also be useful as general cloning vectors for preparation of genomic libraries that can be maintained more stably as integrated plasmids and later retrieved individually or en masse by PIX cloning. Furthermore, the plasmid size of such genomic libraries should be limited only by the amount of DNA packageable by phage P1 (≈100 kbp).
Acknowledgments
We especially thank M. Koob and A. S. Lynch for providing detailed information on plasmids and for helpful discussions, J. T. Bolin and S. Han for assistance with structural data, and I. Tessman for reading the manuscript. This research was supported by National Institutes of Health Grants GM49338 and GM35392 to C.T.W. and B.L.W., respectively. S.L.F. was supported by National Institutes of Health Postdoctoral Fellowship GM16259. The Dana–Farber Cancer Institute Core facility is supported by National Institute of Health Grants CA06516 and AI28691.
Footnotes
Abbreviations: AR, altered recognition; CA, constitutively active; KanR, kanamycin resistant; Int, integrase; Xis, excisionase; PIX, P1-Int-Xis; wt, wild type; X-Gal, 5-bromo-4-chloro-3-indolyl β-d-galactopyranoside; MBP, maltose-binding protein.
References
- 1.Hoch J A, Silhavy T J. Two-Component Regulatory Systems. Washington, DC: Am. Soc. Microbiol.; 1995. [Google Scholar]
- 2.Ota I M, Varshavsky A. Science. 1993;262:566–569. doi: 10.1126/science.8211183. [DOI] [PubMed] [Google Scholar]
- 3.Chang C, Kwok S F, Bleecker A B, Meyerowitz E M. Science. 1993;262:539–544. doi: 10.1126/science.8211181. [DOI] [PubMed] [Google Scholar]
- 4.Alex L A, Borkovich K A, Simon M I. Proc Natl Acad Sci USA. 1996;93:3416–3421. doi: 10.1073/pnas.93.8.3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maeda T, Wurgler-Murphy S M, Saito H. Nature (London) 1994;369:242–245. doi: 10.1038/369242a0. [DOI] [PubMed] [Google Scholar]
- 6.Hill C S, Treisman R. Cell. 1995;80:199–211. doi: 10.1016/0092-8674(95)90403-4. [DOI] [PubMed] [Google Scholar]
- 7.Wanner B L. In: Escherichia coli and Salmonella typhimurium Cellular and Molecular Biology. Neidhardt F C, Curtiss R III, Ingraham J L, Lin E C C, Low K B Jr, Magasanik B, Reznikoff W, Riley M, Schaechter M, Umbarger H E, editors. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 1357–1381. [Google Scholar]
- 8.Wanner B L. J Bacteriol. 1992;174:2053–2058. doi: 10.1128/jb.174.7.2053-2058.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wanner B L, Jiang W, Kim S-K, Yamagata S, Haldimann A, Daniels L L. In: Regulation of Gene Expression in Escherichia coli. Lin E C C, Lynch A S, editors. Austin, TX: Landes; 1996. pp. 297–315. [Google Scholar]
- 10.Kim S-K, Wilmes-Riesenberg M R, Wanner B L. Mol Microbiol. 1996;21:135–147. doi: 10.1111/j.1365-2958.1996.tb02663.x. [DOI] [PubMed] [Google Scholar]
- 11.Arthur M, Depardieu F, Holman T, Wu Z, Wright G, Walsh C T, Courvalin P. In: Two-Component Signal Transduction. Hoch J A, Silhavy T J, editors. Washington, DC: Am. Soc. Microbiol.; 1995. pp. 387–391. [Google Scholar]
- 12.Walsh C T, Fisher S L, Park I-S, Prahalad M, Wu Z. Chem Biol. 1996;3:21–28. doi: 10.1016/s1074-5521(96)90079-4. [DOI] [PubMed] [Google Scholar]
- 13.Fisher S L, Jiang W, Wanner B L, Walsh C T. J Biol Chem. 1995;270:23143–23149. doi: 10.1074/jbc.270.39.23143. [DOI] [PubMed] [Google Scholar]
- 14.Morrison T B, Parkinson J S. Proc Natl Acad Sci USA. 1994;91:5485–5489. doi: 10.1073/pnas.91.12.5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wanner B L. Methods Mol Genet. 1994;3:291–310. [Google Scholar]
- 16.Metcalf W W, Jiang W, Daniels L L, Kim S-K, Haldimann A, Wanner B L. Plasmid. 1996;35:1–13. doi: 10.1006/plas.1996.0001. [DOI] [PubMed] [Google Scholar]
- 17.Hasan N, Koob M, Szybalski W. Gene. 1994;150:51–56. doi: 10.1016/0378-1119(94)90856-7. [DOI] [PubMed] [Google Scholar]
- 18.Lynch A S, Wang J C. Proc Natl Acad Sci USA. 1995;92:1896–1900. doi: 10.1073/pnas.92.6.1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cadwell R C, Joyce G F. PCR Methods Appl. 1992;2:28–33. doi: 10.1101/gr.2.1.28. [DOI] [PubMed] [Google Scholar]
- 20.Fisher S L, Kim S-K, Wanner B L, Walsh C T. Biochemistry. 1996;35:4732–4740. doi: 10.1021/bi9525435. [DOI] [PubMed] [Google Scholar]
- 21.Weisberg R A, Landy A. In: Lambda II. Hendrix R W, Roberts J W, Stahl F W, Weisberg R A, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1983. pp. 211–250. [Google Scholar]
- 22.Wanner B L, Wilmes-Riesenberg M R. J Bacteriol. 1992;174:2124–2130. doi: 10.1128/jb.174.7.2124-2130.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guzman L-M, Belin D, Carson M J, Beckwith J. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Volz K. Biochemistry. 1993;32:11741–11753. doi: 10.1021/bi00095a001. [DOI] [PubMed] [Google Scholar]
- 25.Madhusudan, Zapf J, Whiteley J M, Hoch J A, Xuong N H, Varughese K I. Structure (London) 1996;4:679–690. doi: 10.1016/s0969-2126(96)00074-3. [DOI] [PubMed] [Google Scholar]
- 26.Baikalov I, Shröder I, Kaczor-Grzeskowiak M, Grzeskowiak K, Gunsalus R P, Dickerson R E. Biochemistry. 1996;35:11053–11061. doi: 10.1021/bi960919o. [DOI] [PubMed] [Google Scholar]
- 27.Bellsolell L, Prieto J, Serrano L, Coll M. J Mol Biol. 1994;238:489–495. doi: 10.1006/jmbi.1994.1308. [DOI] [PubMed] [Google Scholar]
- 28.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 29.Merritt E A, Murphy M E P. Acta Crystallogr D. 1994;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]
- 30.Shukla D, Matsumura P. J Biol Chem. 1995;270:24414–24419. doi: 10.1074/jbc.270.41.24414. [DOI] [PubMed] [Google Scholar]
- 31.Swanson R V, Lowry D F, Matsumura P, McEvoy M M, Simon M I, Dahlquist F W. Nat Struct Biol. 1995;2:906–910. doi: 10.1038/nsb1095-906. [DOI] [PubMed] [Google Scholar]
- 32.Bellsolell L, Cronet P, Majolero M, Serrano L, Coll M. J Mol Biol. 1996;257:116–128. doi: 10.1006/jmbi.1996.0151. [DOI] [PubMed] [Google Scholar]
- 33.Sanna M G, Swanson R V, Bourret R B, Simon M I. Mol Microbiol. 1995;15:1069–1079. doi: 10.1111/j.1365-2958.1995.tb02282.x. [DOI] [PubMed] [Google Scholar]