

Abstract
Protein kinase D (PKD) is a novel protein serine kinase which has recently been implicated in diverse cellular functions, including apoptosis and cell proliferation. The purpose of our present study was: (i) to define the activation of PKD in intestinal epithelial cells treated with hydrogen peroxide (H2O2), an agent which induces oxidative stress, and (ii) to delineate the upstream signaling mechanisms mediating the activation of PKD. We found that the activation of PKD is induced by H2O2 in both a dose- and time-dependent fashion. PKD phosphorylation was attenuated by rottlerin, a selective protein kinase C (PKC)-δ inhibitor, and small interfering RNA (siRNA) directed against PKC-δ, suggesting the regulation of PKD activity by upstream PKC-δ. Activation of PKD was also blocked by a Rho kinase (ROK) specific inhibitor, Y27632, as well as C3, a Rho protein inhibitor, demonstrating that the Rho/ROK pathway also mediates PKD activity in intestinal cells. In addition, H2O2-induced PKC-δ phosphorylation was inhibited by C3 treatment, further suggesting that PKC-δ is downstream of Rho/ROK. Interestingly, H2O2-induced intestinal cell apoptosis was enhanced by PKD siRNA. Taken together, these results clearly demonstrate that oxidative stress induces PKD activation in intestinal epithelial cells, and this activation is regulated by upstream PKC-δ and Rho/ROK pathways. Importantly, our findings suggest that PKD activation protects intestinal epithelial cells from oxidative stress-induced apoptosis. These findings have potential clinical implications to intestinal injury associated with oxidative stress (e.g., necrotizing enterocolitis in infants).
Keywords: Intestinal epithelial injury, oxidative stress, PKD, PKC-δ, Rho/ROK
INTRODUCTION
Oxidative stress is noted in cells when concentrations of reactive oxygen species (ROS) exceed the capacity of cellular antioxidant protective mechanisms. ROS-mediated damage to cellular components is thought to contribute to the pathophysiology of numerous diseases that affect tissues of various organ system including the gastrointestinal (GI) tract (4, 41). In addition, many types of mammalian cells rapidly produce ROS in response to various stimuli, leading to the hypothesis that ROS constitute biologically important signaling molecules. Cells exposed to ROS at high concentrations or for extended periods undergo cellular DNA damage, which is widely known to induce cell death via either apoptosis and/or necrosis (11). Oxidative stress is known to contribute to various inflammatory conditions of the GI tract (4, 41). In particular, ROS has been implicated in the pathogenesis of necrotizing enterocolitis (NEC), a devastating disease in premature infants which can lead to sepsis and death. Although numerous risk factors for this disease process have been identified, the exact cellular mechanisms involved in its pathogenesis are largely unknown.
Protein kinase D (PKD), initially referred to as protein kinase C (PKC)-μ(14), is a serine-threonine kinase with a distinctive structure and substrate specificity different from the PKC family members (34). PKD has been implicated in many important intracellular signal transduction pathways via PKC-dependent mechanisms. Importantly, PKD is activated by oxidative stress and induces survival pathways in fibroblast cells (33, 36, 37). However, its potential role in the regulation of oxidative stress-induced intestinal epithelial cell injury is not known. PKC-δ, a novel PKC isoform, is activated by a variety of apoptotic stimuli in different cellular systems. Although the majority of the studies indicate that PKC-δ is involved in the induction of cell apoptosis, there are other reports indicating a role of PKC-δ in cell survival and in anti-apoptotic responses (6).
The Rho family of small GTPases, including well-known Rho proteins (RhoA and RhoB), Rac1 and Cdc42, are members of the Ras superfamily of GTPases. The Rho family regulates many biological processes, including cytoskeletal regulation, membrane trafficking, cell adhesion, cell polarization and transcriptional activity (1, 7). The Rho family has been implicated in cell growth and apoptosis in response to diverse stimuli (2, 18). The Rho kinase (ROK), including isoform α and β, is an effector of Rho proteins. It has been shown that membrane blebbing during apoptosis is mediated through a caspase-mediated activation of ROKβ (9). However, the exact role of these cellular signaling kinases in oxidative stress-induced intestinal epithelial cell injury is not known.
In the present study, we show activation of PKD by hydrogen peroxide (H2O2) treatment in intestinal epithelial cells. This activation of PKD is regulated by PKC-δ and Rho/ROK pathways, which also play a critical anti-apoptosis role during the oxidative stress-mediated intestinal epithelial cell injury. Using small interfering (si) RNA targeting PKD, we also demonstrate the protective role of PKD during H2O2-induced intestinal epithelial cell apoptosis. Together, our findings suggest an important protective role for PKD in intestinal cells in response to oxidative damage. These results have potential clinical implications for the management of diseases (such as NEC) which are related to ROS-mediated injury.
MATERIALS AND METHODS
Reagents and antibodies
PKD and PKC-δ siRNA were synthesized by Custom SMARTPool siRNA Design Service of Dharmacon, Inc. (Lafayette, CO). The non-specific control siRNA was also obtained from Dharmacon. Recombinant GST-C3 and GST control proteins were purified from E. coli using constructs provided by Dr. Keith Burridge (University of North Carolina, Chapel Hill, NC). GF109203X (GFX), Ro31-8220, rottlerin and Y27632 were from BIOMOL Research Laboratories Inc. (Plymouth Meeting, PA). Syntide-2 and Gö6983 were from CALBIOCHEM (La Jolla, CA). 2′,7′-dichlorofluorescein diacetate (DCFH-DA) was from Sigma Chemical Co. (St. Louis, MO). PKD, PKC-δ, poly (ADP-ribose) polymerase (PARP), and caspase-3 polyclonal antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). The anti-phospho-PKD (Ser744/748) antibody was from Cell Signaling Technology (Beverly, MA). The anti-phospho-PKC-δ (Tyr311) antibody was from Stressgen Biotechnologies (San Diego, CA). The secondary antibodies were from Pierce (Rockford, IL). Alexa Fluor 488 antibody for fluorescent staining was from Molecular Probes (Eugene, OR). The enhanced chemiluminescence (ECL) system for Western immunoblot analysis was from Amersham (Arlington Heights, IL). The concentrated protein assay dye reagent was from Bio-Rad (Hercules, CA). Tissue culture media and reagents were from GIBCO-BRL (Grand Island, NY). All other reagents were of molecular biology grade and purchased from Sigma Chemical Co. (St. Louis, MO).
Cell culture and transfection
The RIE-1 cell line (a generous gift from Dr. Kenneth D. Brown; Cambridge Research Station, Babraham, Cambridge, U.K.) is a diploid, nontransformed, crypt-like cell line derived from rat small intestine (5). IEC-6 cell line (purchased from American Type Culture Collection; Manassas, VA) was derived from normal rat intestinal crypt cells and was developed and characterized by Quaroni et al (26). For all experiments, RIE-1 cells were used between passages 18–29, and IEC-6 cells were used between passages 23–31. Both cell lines were found to be free of Mycoplasma contamination by polymerase chain reaction method. Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum (FBS) in 5% CO2 at 37°C. For experimental purposes, cells were plated in 100-mm dishes and grown to near confluence. Cells were treated with the indicated concentrations of H2O2 at 37°C. For inhibitor studies, cells were pretreated with inhibitors for 30 min and then treated with H2O2 in combination with inhibitors for another 30 min. siRNA or GST-C3 protein was transfected by electroporation (400V, 500 μF for siRNA; 450V, 25 μF for GST-C3 protein) using GenePulser XCell (Bio-Rad, Hercules, CA).
Immunoprecipitation, in vitro kinase assays and Western blotting
Immunoprecipitation and in vitro kinase assays were performed as described previously (21). In brief, proteins (50 μg) were incubated with PKD antibodies (1:50) on a shaker for 2 h at 4°C followed by another 2 h incubation with 30 μl of protein A-Sepharose beads at 4°C. The immunocomplexes were suspended in 20 μl of kinase buffer and kinase reaction, with or without 2.5 μg of syntide-2 as a substrate, was started by adding 5 μCi of [γ-32P]ATP and incubated for 10 min at 30°C. Reactions were stopped by the addition of 2x Tris-glycine sample buffer. Samples were denatured by boiling for 5 min and separated by NuPAGE 4–12% Bis-Tris gels. Gels were incubated in Gel-Dry drying solution (Invitrogen) for 5 min and dried at 60°C for 60 min followed by exposure to x-ray film. For Western blotting, equal amounts of protein were resolved on NuPAGE Bis-Tris gels and electrophoretically transferred to polyvinylidene difluoride membranes; the membranes were incubated with primary antibodies overnight at 4°C followed by secondary antibodies conjugated with horseradish peroxidase. Membranes were developed using the ECL detection system.
Immunofluorescent staining and fluorescent microscopy
Cells were grown in chamber slides. Three days after seeding, cells were treated either with or without H2O2 (500 μM) for 15 min. After treatment, cells were fixed with 4% paraformaldehyde for 20 min at 37°C. After three washes with phosphate buffered saline (PBS), the cells were permeabilized with 0.3% Triton X-100 for 15 min at 37°C and blocked with 1% bovine serum albumin (BSA)-PBS for 20 min. The cells were incubated with rabbit polyclonal anti-PKD antibody diluted 1:100 with 1% BSA-PBS for 1 h at room temperature or overnight at 4°C. Cells were washed three times with PBS and incubated with Alexa 488-conjugated anti-rabbit secondary antibody diluted 1:500 in 1% BSA-PBS. The fluorescence of PKD immunoreactivity was observed under a fluorescent microscope.
Cell death assay and measurement of reactive oxygen species
RIE-1 cells (4x104 cells/well) were seeded in 24-well plates for 24 h. Cells were transfected with siRNA and then grown for an additional 72 h. Cells were treated with various doses of H2O2 in growth medium for 3h or H2O2 (500 μM) over a time course. Both adherent and floating cells were collected and washed 3 times using cold PBS. Quantitation of apoptosis was performed using a Cell Death Detection ELISAplus kit (Roche Applied Science; Indianapolis, IN) following the manufacturer’s instructions. ROS was measured using a modification of methods described by Wang et al (39). In brief, 6.4x103 cells were plated in 96-well plates. After 24 h, cells were incubated with 50 μM of the fluorescent probe 2′,7′-dichlorofluorescein diacetate (DCFH-DA) for 30 min. DCFH-DA was removed and cells were washed twice with PBS and incubated with 500 μM of H2O2 in DMEM containing 5% FBS for 3 h. DCFH-DA fluorescence was determined at an excitation of 485 nm and an emission of 520 nm by a fluorescent microplate-reader (FLUOstar Optima; BMG Labtech Inc., Durham, NC).
Statistical analysis
All experiments were repeated at least three times and data are reported as mean ± SEM. Data were analyzed using the Kruskal-Wallis test due to heterogeneous variability in each group. All tests were assessed at the 0.05 level of significance. All statistical computations were conducted using the SAS® system, Release 8.2 (SAS Insitute, Inc., Cary, NC).
RESULTS
H2O2 induces PKD activation in a dose- and time-dependent fashion
H2O2 has been implicated in the activation of PKD in Swiss 3T3 fibroblasts, COS-7 cells, HeLa cells and NIH 3T3 fibroblasts (33, 36, 37). In order to determine whether an activation of PKD occurs during oxidative stress injury in intestinal epithelial cells, RIE-1 cells and another normal rat intestinal cell line, IEC-6, were treated with or without H2O2 and PKD activation determined by Western blot using anti-phospho-PKD (Ser744/748) antibody. First, RIE-1 cells were treated with or without various concentrations of H2O2 (10 to 500 μM) for 15 min.
PKD phosphorylation was noted using 250 μM of H2O2 with increased phosphorylation at 500 μM (Fig. 1A, upper panel). Next, RIE-1 cells were treated with 500 μM of H2O2 over a time course. PKD activation occurred rapidly at 5 min with a significant increase at 10 min after treatment with H2O2 (Fig. 1A, lower panel). Similar results in IEC-6 cells confirmed that PKD activation, in response to H2O2, was not limited to RIE-1 cells (Fig. 1B), suggesting that the activation of PKD by H2O2 occurs in intestinal epithelial cells, consistent with previous studies in other cell types (33, 36, 37).
Figure 1. H2O2 induced PKD activation in RIE-1 and IEC-6 cells.
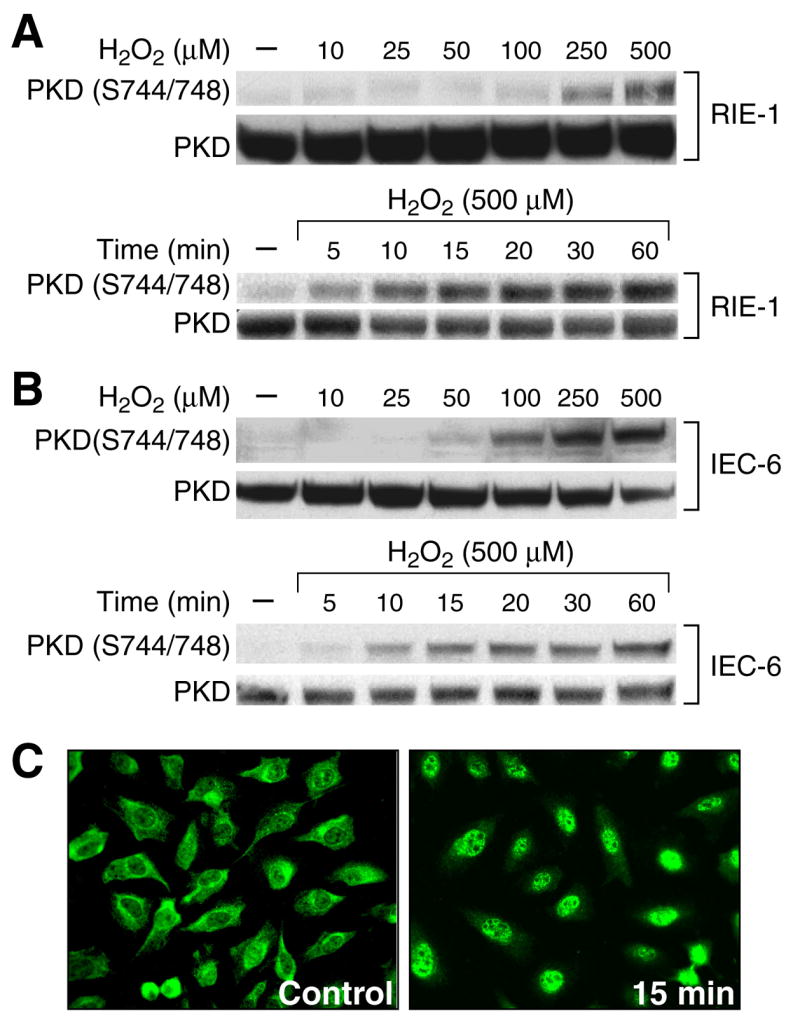
(A) RIE-1 cells were treated with or without various concentrations of H2O2 for 15 min. PKD phosphorylation was detected by Western blotting using anti-phospho-PKD (Ser744/748) antibody. The membrane was stripped and reprobed with anti-PKD antibody as a loading control. RIE-1 cells were treated with 500 μM of H2O2 over a time course study. (B) Similar experiments were performed using IEC-6 cells. (C) RIE-1 cells were grown on chamber slides for 3 days and then treated with H2O2 (500 μM) or vehicle (control) for 15 min. A representative photomicrograph is shown demonstrating PKD localization in the nucleus using immunofluorescent imaging 15 min after H2O2 treatment. (Representative photomicrograph from 3 separate experiments).
To further demonstrate the activation of PKD by H2O2 treatment, we performed PKD immunofluorescent staining (Fig. 1C). In untreated control RIE-1 cells, PKD was noted in Golgi-like structures in the cytosol. In contrast, PKD localization to the nucleus was noted in RIE-1 cells after treatment with 500 μM of H2O2 for 15min (Fig. 1C, right panel). These data indicate that oxidative stress stimulates PKD nuclear accumulation in RIE-1 cells.
H2O2 stimulates apoptosis in RIE-1 cells
H2O2 is a known intracellular stress agent which induces apoptosis (19). We next examined whether H2O2 increases apoptosis in RIE-1 cells. Cells were treated with various dosages of H2O2 for 3 h. Then, using the dose which produces significant cell apoptosis (i.e., 500 μM), we also performed a time course study. As shown in figure 2A, treatment of RIE-1 cells with 500 μM of H2O2 did not induce cell death until 3 h after treatment. Next, RIE-1 cells were treated with different doses of H2O2 for 3 h (Fig. 2B). The results demonstrate that H2O2 induces intestinal epithelial cell apoptosis in a dose-dependent manner.
Figure 2. H2O2 induced apoptosis in RIE-1 cells.
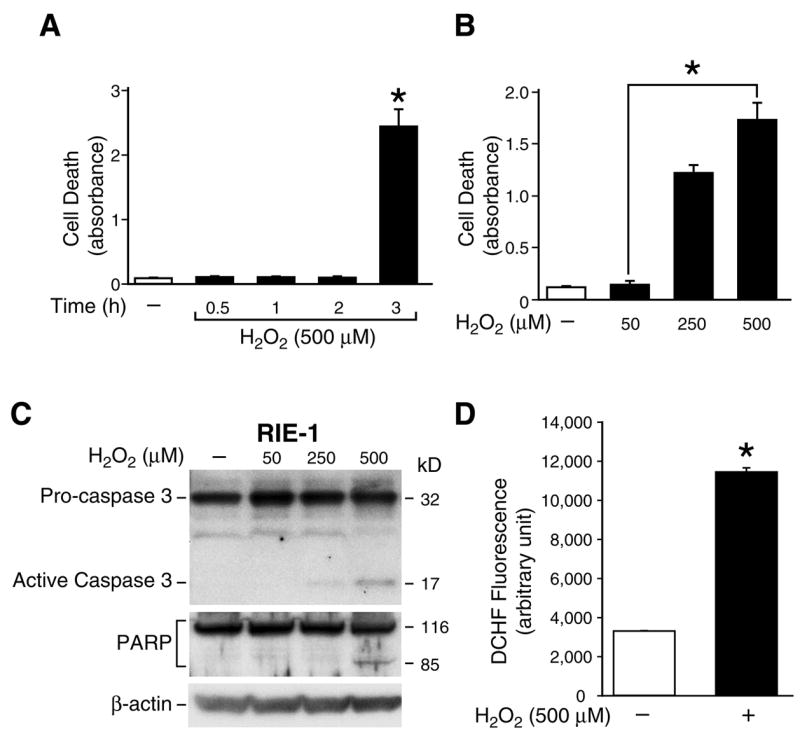
(A) RIE-1 cells were treated with or without H2O2 (500 μM) in normal growth medium over a time course and cell death assays performed. Experiments were performed in triplicate and results are expressed as mean ± SEM (n=3); * p< 0.05 vs. control (−). (B) RIE-1 cells were treated with or without various doses of H2O2 in normal growth medium for 3 h and cell death assays performed. Experiments were performed in triplicate and results are expressed as mean ± SEM (n=3); * p< 0.05 vs. control (−). (C) RIE-1 cells were treated with or without various doses of H2O2 in normal growth medium for 3 h. Both attached and floating cells were collected, lysed and Western blot analysis was performed for the expression of caspase 3 or PARP cleavage products; β-actin protein expression was detected to monitor equal loading. (D) RIE-1 cells were loaded with DCFH/DA (50 μM) for 30 min and were subsequently exposed to H2O2 (500 μM) for 3 h. Cellular fluorescence in each well was measured and immediately recorded. The DCFH fluorescence increased in H2O2-exposed cells. Results are expressed as mean ± SEM (n=3); * p< 0.05 vs. control (−).
In many cell types, apoptosis involves the activation of cysteine-dependent aspartate-directed proteases known as caspases (43). Therefore, we measured the protein levels of cleaved caspase 3 as an apoptotic marker. In addition, poly (ADP-ribose) polymerase (PARP), a nuclear zinc finger DNA-binding protein, is a caspase 3 target and cleavage of PARP has also been used extensively as a marker of apoptosis (23). Cells were treated with or without different doses of H2O2 (50, 250, 500 μM) for 3 h. Western blots were performed using anti-caspase 3 or anti-PARP-1 antibodies. Treatment of RIE-1 cells with 500 μM of H2O2 for 3 h induced caspase-3 activation and PARP cleavage (Fig. 2C). To further confirm that the H2O2-induced apoptosis was a result of increased cellular ROS, the generation of ROS in RIE-1 cells was measured. Treatment with 500 μM of H2O2 for 3 h caused a significant increase in intracellular ROS accumulation in RIE-1 cells when compared to vehicle-treated (control) cells (Fig. 2D).
PKD siRNA enhances H2O2-induced cell death
PKD activation in HeLa cells was shown to protect oxidative stress-induced cell death (33), but whether PKD activation also protects intestinal epithelial cells from oxidative stress induced-cell apoptosis is unknown. To further determine if PKD plays a role in the protection of RIE-1 cells from H2O2-induced apoptosis, we transfected PKD siRNA into RIE-1 cells and, 3 days later, cells were treated with 500 μM of H2O2 for 3 h. Western blotting demonstrated that the inhibition of PKD expression by siRNA significantly decreased endogenous PKD levels and concurrently PKD phosphorylation when compared to control siRNA (Fig. 3A). The cell death assay demonstrated that H2O2 treatment enhanced intestinal epithelial cell apoptosis by approximately 1.5 fold compared to cells without H2O2 treatment in control siRNA transfected cells (Fig. 3B). More importantly, H2O2 treatment induced cell death up to 5 fold when compared to cells in the absence of H2O2 in PKD siRNA transfected cells, strongly suggesting that the suppression of endogenous PKD enhances H2O2-induced intestinal epithelial cell apoptosis. Additionally, this increase in H2O2-induced apoptosis in PKD siRNA transfected RIE-1 cells was confirmed by increased activation of caspase 3 and PARP cleavage (Fig. 3C).
Figure 3. PKD siRNA promoted H2O2-induced cell death.

(A) RIE-1 cells were transfected with PKD siRNA or the control siRNA. After 3 days, cells were treated with H2O2 (500 μM) for 3 h and protein extracted and analyzed by Western blot. Inhibition of PKD expression by PKD siRNA was shown using anti-PKD antibody (upper panel). PKD phosphorylation was determined using anti-phospho-PKD (Ser744/748) antibody (middle panel). β-actin was used as a loading control (bottom panel). A representative result from 3 experiments is shown. (B) RIE-1 cells were transfected with PKD siRNA or the control siRNA. After 3 days, cells were treated with 500 μM of H2O2 for 3 h and cell death assays performed. Results from 3 experiments are expressed as mean ± SEM (n=3); * p< 0.05 vs. control siRNA without H2O2; † p< 0.05 vs. control siRNA and H2O2. (C) RIE-1 cells were transfected with PKD or control siRNA for 72 h, and then treated with H2O2 (500 μM) for 3 h. Both attached and floating cells were collected, lysed and analyzed by Western blot for the expression of caspase 3 or PARP cleavage products; β-actin was used as a protein loading control.
PKC and ROK pathway regulation of H2O2-induced PKD activation
Phosphorylation of serines 744/748 in PKD has previously been associated with a dependence on PKC activity (36, 37) and the Rho/ROK pathway (22, 42) in certain cell types. To investigate whether H2O2-induced PKD activation in intestinal epithelial cells is mediated by PKC or Rho/ROK, RIE-1 and IEC-6 cells were treated with three well-characterized PKC inhibitors (Gö6983, GFX, Ro31-8220), which inhibit activity of classic and novel PKC isoforms but not PKD (12, 29), and the PKC-δ selective inhibitor, rottlerin (13), as well as the ROK inhibitor Y27632 (10). H2O2-induced PKD activation in RIE-1 cells was significantly attenuated in the presence of all of these inhibitors (Fig. 4A, upper panel). The membrane was stripped and reprobed with total PKD antibody to monitor the loading equality (Fig. 4A, middle panel). Similar results were obtained in IEC-6 cells (Fig. 4B, upper and middle panels). Quantitative analysis confirmed that H2O2 treatment increases PKD phosphorylation by 40.3% and 42.9% in both RIE-1 and IEC-6 cells, respectively; these increases were attenuated to baseline level by all of PKC inhibitors and ROK inhibitor (Fig. 4A,B; bottom panels). These data further demonstrate that the activation of PKD is PKC or Rho/ROK pathway-dependent in intestinal epithelial cells.
Figure 4. Regulation of PKD activity by PKC and ROK inhibitors.

(A) RIE-1 cells were pretreated with or without inhibitors [Gö6983 (1 μM), GF109203X (1 μM), Ro31-8220 (1 μM), Rottlerin (5 μM), Y27632 (30 μM)] as indicated for 30 min and then treated with H2O2 (250 μM) in combination with inhibitors for another 15 min. PKD phosphorylation was determined by Western blot analysis using the anti-phospho-PKD (Ser744/748) antibody (upper panel). PKD expression was assessed as a loading control (middle panel). Densitometric analysis of protein level from corresponding treatment lanes [n=3 immunoblots; * p< 0.05 vs. H2O2 only (lane 2)] (bottom panel). (B) Similar experiments were performed using IEC-6 cells. A representative Western blot (upper and middle panels) and corresponding densitometric analysis are shown [n=3 immunoblots; * p< 0.05 vs. only H2O2 (lane 2)] (bottom panel).
Oxidative stress-mediated PKD activation is regulated by PKC-δ
PKC-δ has been implicated in both pro- and anti-apoptosis (15). Based on significant inhibition of PKD activation by rottlerin, a PKC-δ selective inhibitor, we further wanted to confirm the involvement of PKC-δ in oxidative stress-mediated PKD activation in intestinal epithelial cells. Rottlerin effectively inhibits PKC-δ kinase activity at a concentration of 5–10 μM (13). To examine the sensitivity of PKD phosphorylation to rottlerin, RIE-1 cells were pretreated with various doses of rottlerin for 30 min and, then, in combination with H2O2 and rottlerin for another 30 min. Western blots were performed using anti-phospho-PKD (Ser744/748) antibody. H2O2-induced PKD phosphorylation was decreased with the 2.5 μM dosage of rottlerin and markedly decreased using 5 μM (Fig. 5A). This result was confirmed in IEC-6 cells (Fig. 5B). To further demonstrate a specific role of PKC-δ in the regulation of PKD activity in H2O2-induced cell apoptosis, siRNA-targeting PKC-δ was transfected into RIE-1 cells for 3 days. The expression of PKC-δ protein was significantly decreased by PKC-δ siRNA compared to control siRNA (Fig. 5C). Furthermore, PKD activation was significantly inhibited by PKC-δ siRNA either in the presence or absence of H2O2 compared with the control siRNA (Fig. 5C). To further support the specific nature of PKC-δ regulation of PKD activation, we next performed an in vitro PKD kinase assay using syntide-2, a specific PKD substrate. Consistent with the PKD protein expression level, PKC-δ siRNA also significantly decreased PKD kinase activity (Fig. 5C).
Figure 5. PKC-δ regulation of PKD phosphorylation.

(A) RIE-1 cells were pretreated with or without rottlerin for 30 min and then treated with H2O2 (250 μM) in combination with inhibitors for another 15 min. PKD phosphorylation was determined by Western blot analysis using the anti-phospho-PKD (Ser744/748) antibody. PKD expression was assessed as a loading control. (B) Similar experiments as above were performed using IEC-6 cells. A representative result from 3 experiments is shown. (C) RIE-1 cells were transfected with control siRNA or PKC-δ siRNA. After 3 days, cells were treated with or without H2O2 (500 μM) for 3 h and Western blotting performed using total cell lysates. PKC-δ expression was examined using anti-PKC-δ antibody (top panel). The membranes were reprobed with anti-phospho-PKD (Ser744/748) antibody (2nd panel) and β-actin was utilized as a loading control (3rd panel). PKD kinase assay in vitro was performed in the presence of syntide-2 using the same lysates (bottom panel). (D) RIE-1 cells were transfected with control siRNA or PKC-δ siRNA. After 3 days, cells were treated with or without H2O2 (500 μM) for 3 h and cell death assays performed. Results are expressed as mean ± SEM (n = 3). * p< 0.05 vs. control; † p< 0.05 vs. control siRNA and H2O2 (500 μM). (E) RIE-1 cells were transfected with PKC-δ or control siRNA. After 72 h, cells were treated with or without H2O2 (500 μM) for 3 h. Both attached and floating cells were collected, lysed and analyzed by Western blot for the expression of caspase 3 or PARP cleavage products; β-actin was used as a loading control.
To examine the role PKC-δ in H2O2-induced intestinal cell apoptosis, cell death assay was performed in RIE-1 cells transfected with either PKC-δ or control siRNA (Fig. 5D). In cells transfected with control siRNA, H2O2 treatment enhanced cell death approximately 2-fold compared to cells in the absence of H2O2. In contrast, H2O2 treatment induced cell death up to 4-fold when compared to cells in the absence of H2O2 and transfected with PKC-δ siRNA (Fig. 5D). We also determined caspase 3 and PARP cleavage in RIE-1 cells transfected with PKC-δ siRNA and found that PKC-δ siRNA resulted in increased active procaspase 3 as well as PARP cleavage when compared to control siRNA (Fig. 5E). Taken together, these results strongly suggest the regulation of PKC-δ on PKD activity in H2O2-induced cell apoptosis in intestinal epithelial cells and, moreover, demonstrate a specific role of PKC-δ in the regulation of PKD activation during H2O2-induced intestinal epithelial cell apoptosis.
Rho/ROK pathway regulation of PKD activity during H2O2-induced apoptosis
ROK has been implicated in cell membrane blebbing, which often occurs when cells become committed to apoptosis (27). However, the importance of ROK in apoptosis remains to be elucidated (17). Based on the inhibition of PKD phosphorylation by the ROK inhibitor Y27632 (Fig. 4), we next examined whether the Rho/ROK pathway is indeed involved in H2O2-induced PKD activation. For this purpose, both RIE-1 and IEC-6 cells were treated with various concentrations of Y27632 to examine the sensitivity of PKD activity and Western blots performed using anti-phospho-PKD (Ser744/748) antibody to detect PKD activation. PKD phosphorylation was inhibited slightly by 5 μM of Y27632 but significantly at doses of both 15 μM and 30 μM, suggesting the relatively specific regulation of PKD activation by ROK (Fig. 6A). Similar results were obtained in IEC-6 cells (Fig. 6B). Next, RIE-1 cells were treated with 500 μM of H2O2, alone or in combination with Y27632, and apoptosis measured (Fig. 6C). Treatment with Y27632 significantly enhanced H2O2-induced cell death, which is consistent with the protective role of PKD in H2O2-induced apoptosis.
Figure 6. H2O2-mediated PKD activation was attenuated by Rho/ROK inhibitors.

(A) RIE-1 cells were pretreated with or without Y27632 for 30 min and then treated with H2O2 (250 μM) in combination with inhibitors for another 15 min. PKD phosphorylation was determined by Western blot analysis using the anti-phospho-PKD (Ser744/748) antibody. PKD expression was determined as a loading control. (B) Similar experiments as above were performed using IEC-6 cells. A representative result from 3 experiments is shown. (C) RIE-1 cells were pretreated with Y27632 for 30 min and then with 500 μM of H2O2 in combination with Y27632 and cell death assay performed. Results are expressed as mean ± SEM (n = 3). * p< 0.05 vs. control (−); † p< 0.05 vs. control and H2O2 (500 μM). (D) RIE-1 cells were transfected with GST-C3 or GST proteins (as a control) overnight. Cells were treated with or without H2O2 (250 μM) for 15 min. Cells were lysed and protein extracted for Western blots. PKD phosphorylation was monitored using anti-phospho-PKD (Ser744/748) antibody. β-actin was used as a loading control.
ROK serves as an effector of Rho proteins (27). C3 toxin is a Rho protein inhibitor (27). To further determine whether Rho proteins are also involved in this process, RIE-1 cells were transfected with GST-C3 as well as GST as control (Fig. 6D). PKD phosphorylation was totally blocked by C3 toxin treatment (Fig. 6D, upper panel), further demonstrating the involvement ofthe Rho/ROK pathway in the regulation of PKD activity. PKC-δ was shown to be a regulator of PKD phosphorylation, downstream of the Rho/ROK pathway, in the human pancreatic carcinoid cell line BON (22). Based on the findings presented here that PKC-δ plays an anti-apoptotic role in H2O2-induced cell death and regulation of PKD phosphorylation, we further determined the sequential regulation of PKD phosphorylation by PKC-δ and Rho/ROK pathway. The membrane was reprobed with anti-phospho-PKC-δ (Tyr 311) antibody to detect PKC-δ tyrosine phosphorylation, which is often noted in apoptosis (Fig. 6D, middle). As expected, PKC-δ tyrosine phosphorylation at 311 was also decreased by C3 treatment. This result demonstrated that PKD phosphorylation was regulated by PKC-δ downstream of the Rho/ROK pathway, consistent with previous results by Li et al (22). Taken together, these results provide evidence that the signaling pathway of H2O2-induced PKD activation requires the Rho/ROK pathway, and PKC-δ is downstream of this pathway in intestinal epithelial cells.
DISCUSSION
ROS has been implicated in the pathogenesis of various inflammatory conditions of the GI tract, including NEC in premature infants; however, the cellular mechanisms of action in the intestinal epithelial cells are not yet clearly defined. In this study, we show that H2O2 treatment generates ROS and results in significant intestinal epithelial cell apoptosis. Moreover, we demonstrate, for the first time, that H2O2 stimulates PKD phosphorylation via PKC-δ and Rho/ROK-dependent mechanisms to function as an important anti-apoptotic pathway during oxidative stress-mediated intestinal epithelial cell injury.
ROS include a group of molecules such as hydrogen peroxide (H2O2), superoxide anion (O2−), singlet oxygen (O2), and hydroxyl radicals (OH). Cells possess endogenous antioxidant enzyme systems to protect against the deleterious effects of free oxygen radicals. Therefore, cells undergo oxidative stress when levels of ROS exceed the counter-regulatory antioxidant capacity. The exposure of cells to oxidative stress requires cellular responses to avoid lethal damage through induction of necrosis or apoptosis (3). Furthermore, oxidative stress can concurrently stimulate protective cellular functions as a compensatory response to stressful stimuli. The PKD enzymes have recently been implicated in diverse cellular functions, including Golgi organization and plasma membrane directed transport, metastasis, immune responses, apoptosis and cell proliferation (28). Activation of PKD by oxidative stress has been recently reported in a variety of cell types (30–33, 36, 37, 40). Using HeLa cells as a model, Storz et al (33), recently showed that oxidative stress induces both tyrosine phosphorylation in the PH domain and in the activation loop serine residues leading to PKD activation (30–33). PKD phosphorylation at Ser 744/748 was induced in oxidative stress mediated by menadione, a superoxide generator, in hepatocytes as a protective cell survival response (40). However, oxidative stress-induced activation of PKD has never been reported in intestinal epithelial cells.
In our present study, we demonstrate activation of PKD in H2O2-induced oxidative stress in RIE-1 and IEC-6 cells, and that the activation of PKD protects from H2O2-induced cell apoptosis. Consistent with previous reports in Swiss 3T3 fibroblasts (36, 37) and HeLa cells (30–33), we also noted phosphorylation of PKD at the activation loop Ser744/748 induced by H2O2 treatment in intestinal epithelial cells. More importantly, we found that suppression of PKD by RNAi rendered RIE-1 cells more sensitive to oxidative stress-induced cell apoptosis. Therefore, our study demonstrates an important role of PKD in the protection of oxidative stress-induced intestinal epithelial cell injury.
The function of PKC-δ, a member of the novel PKC family, depends on the cell type and specific stimulus. PKC-δ has been considered as a redox-sensitive kinase and known to play a key role in promoting apoptotic cell death in various cell types (6). However, more recent studies have demonstrated a role of PKC-δ either in cell survival or in anti-apoptosis (16, 20, 24, 38). In human neutrophils, PKC-δ is a critical factor of tumor necrosis factor (TNF)-related antiapoptotic signaling. Inhibition of PKC-δ resulted in the inhibition of TNF-mediated inhibition of apoptosis (16). Using a colon cancer cell line, COLO 205, Lewis et al. (20) demonstrated that PKC-δ inhibition is sufficient for caspase-3-independent apoptosis. Similarly, PKC-δ plays a critical role in the anti-apoptotic effect of basic fibroblast growth factor (FGF) in granulosa cells (24). Storz et al. (33) proposed a model in which two distinct signaling pathways converge at the level of PKD; the Src-Abl pathway, which controls tyrosine phosphorylation and activation loop phosphorylation of PKD in response to oxidative stress, is mediated by the PKC pathway, specifically, the PKC-δ isoform (30–32). On the other hand, tyrosine-phosphorylated PKC-δ was required for H2O2-induced phosphorylation of vascular endothelial growth factor receptor-3, which is activated in response to H2O2 and promotes endothelial cell survival (38). In our present study, we utilized the PKC-δ selective inhibitor, rottlerin, as well as PKC-δ siRNA to examine the role of PKC-δ in the regulation of oxidative stress-induced PKD activity in intestinal epithelial cells. We found that PKC-δ siRNA promoted H2O2-induced cell death, suggesting an anti-apoptotic role of PKC-δ in intestinal epithelial cells. We also showed that the PKD phosphorylation at the activation loop Ser744/748 was significantly inhibited by rottlerin and PKC-δ siRNA, demonstrating that PKC-δ is an upstream kinase of PKD, consistent with the findings in HeLa cells.
The Rho family and its effector ROK have been implicated in the regulation of apoptosis (2, 8). The human RhoA, CDC42, and Rac-1 proteins and their guanine nucleotide exchange factor Dbl efficiently induce the transcriptional activity of nuclear factor kappa B (NF-κB) (25), which is clearly capable of controlling the expression of several anti-apoptotic factors to enhance cell survival (35). This indirect evidence suggests that Rho family proteins play a role in the cellular survival pathway. In this study, we showed that exposure of RIE-1 or IEC-6 cells to Y27632, a selective ROK inhibitor, induces the inhibition of H2O2-induced PKD phosphorylation. To our knowledge, this is the first report describing H2O2-induced PKD activation is regulated by the Rho/ROK pathway. Furthermore, treatment of RIE-1 cells with Y27632 enhanced H2O2-induced cell death, suggesting a role of Rho/ROK pathway in the protective effect of PKD against H2O2-induced cell apoptosis. Moreover, PKD phosphorylation was completely blocked by C3 toxin, a specific Rho protein inhibitor, and the PKC-δ phosphorylation at tyrosine 311 was also decreased by C3 treatment, suggesting the regulation of PKD activity by PKC-δ downstream of Rho/ROK pathway.
In summary, the results presented in this report demonstrate a critical role of PKD activation in oxidative stress-induced intestinal epithelial cell injury. The results also demonstrate that the PKD activity is regulated by PKC-δ downstream of the Rho/ROK pathway. Here, we propose a model for PKD-mediated cell survival pathway in oxidative stress-induced intestinal epithelial cell injury. Oxidative stress is strongly implicated in various diseases, including NEC; the potent activation of PKD may provide new mechanistic insights into both physiological and pathological processes in intestinal epithelial cells.
Acknowledgments
The authors thank Tatsuo Uchida for statistical analysis and Karen Martin for manuscript preparation. This work was supported by the grants RO1 DK61470, R37AG10885, RO1 DK48498 and PO1 DK35608 from the National Institutes of Health. This paper was presented, in part, at the annual meeting of the American Gastroenterological Association (May 18–24, 2004, New Orleans, LA) and was previously published in abstract form (Gastroenterology 126:A147, 2004).
References
- 1.Aspenstrom P, Fransson A, Saras J. Rho GTPases have diverse effects on the organization of the actin filament system. Biochem J. 2004;377:327–337. doi: 10.1042/BJ20031041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aznar S, Lacal JC. Rho signals to cell growth and apoptosis. Cancer Lett. 2001;165:1–10. doi: 10.1016/s0304-3835(01)00412-8. [DOI] [PubMed] [Google Scholar]
- 3.Bergamini CM, Gambetti S, Dondi A, Cervellati C. Oxygen, reactive oxygen species and tissue damage. Curr Pharm Des. 2004;10:1611–1626. doi: 10.2174/1381612043384664. [DOI] [PubMed] [Google Scholar]
- 4.Bernotti S, Seidman E, Sinnett D, Brunet S, Dionne S, Delvin E, Levy E. Inflammatory reaction without endogenous antioxidant response in Caco-2 cells exposed to iron/ascorbate-mediated lipid peroxidation. Am J Physiol Gastrointest Liver Physiol. 2003;285:G898–906. doi: 10.1152/ajpgi.00042.2003. [DOI] [PubMed] [Google Scholar]
- 5.Blay J, Brown KD. Characterization of an epithelioid cell line derived from rat small intestine: demonstration of cytokeratin filaments. Cell Biol Int Rep. 1984;8:551–560. doi: 10.1016/0309-1651(84)90054-7. [DOI] [PubMed] [Google Scholar]
- 6.Brodie C, Blumberg PM. Regulation of cell apoptosis by protein kinase c δ. Apoptosis. 2003;8:19–27. doi: 10.1023/a:1021640817208. [DOI] [PubMed] [Google Scholar]
- 7.Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 8.Coleman ML, Olson MF. Rho GTPase signalling pathways in the morphological changes associated with apoptosis. Cell Death Differ. 2002;9:493–504. doi: 10.1038/sj.cdd.4400987. [DOI] [PubMed] [Google Scholar]
- 9.Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol. 2001;3:339–345. doi: 10.1038/35070009. [DOI] [PubMed] [Google Scholar]
- 10.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dypbukt JM, Ankarcrona M, Burkitt M, Sjoholm A, Strom K, Orrenius S, Nicotera P. Different prooxidant levels stimulate growth, trigger apoptosis, or produce necrosis of insulin-secreting RINm5F cells. The role of intracellular polyamines. J Biol Chem. 1994;269:30553–30560. [PubMed] [Google Scholar]
- 12.Gschwendt M, Dieterich S, Rennecke J, Kittstein W, Mueller HJ, Johannes FJ. Inhibition of protein kinase C μ by various inhibitors. Differentiation from protein kinase c isoenzymes. FEBS Lett. 1996;392:77–80. doi: 10.1016/0014-5793(96)00785-5. [DOI] [PubMed] [Google Scholar]
- 13.Gschwendt M, Muller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, Marks F. Rottlerin, a novel protein kinase inhibitor. Biochem Biophys Res Commun. 1994;199:93–98. doi: 10.1006/bbrc.1994.1199. [DOI] [PubMed] [Google Scholar]
- 14.Johannes FJ, Prestle J, Eis S, Oberhagemann P, Pfizenmaier K. PKCμ is a novel, atypical member of the protein kinase C family. J Biol Chem. 1994;269:6140–6148. [PubMed] [Google Scholar]
- 15.Kanthasamy AG, Kitazawa M, Kanthasamy A, Anantharam V. Role of proteolytic activation of protein kinase Cδ in oxidative stress-induced apoptosis. Antioxid Redox Signal. 2003;5:609–620. doi: 10.1089/152308603770310275. [DOI] [PubMed] [Google Scholar]
- 16.Kilpatrick LE, Sun S, Korchak HM. Selective regulation by δ-PKC and PI 3-kinase in the assembly of the antiapoptotic TNFR-1 signaling complex in neutrophils. Am J Physiol Cell Physiol. 2004;287:C633–642. doi: 10.1152/ajpcell.00486.2003. [DOI] [PubMed] [Google Scholar]
- 17.Kobayashi K, Takahashi M, Matsushita N, Miyazaki J, Koike M, Yaginuma H, Osumi N, Kaibuchi K, Kobayashi K. Survival of developing motor neurons mediated by Rho GTPase signaling pathway through Rho-kinase. J Neurosci. 2004;24:3480–3488. doi: 10.1523/JNEUROSCI.0295-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lacal JC. Regulation of proliferation and apoptosis by Ras and Rho GTPases through specific phospholipid-dependent signaling. FEBS Lett. 1997;410:73–77. doi: 10.1016/s0014-5793(97)00444-4. [DOI] [PubMed] [Google Scholar]
- 19.Lee WC, Choi CH, Cha SH, Oh HL, Kim YK. Role of ERK in hydrogen peroxide-induced cell death of human glioma cells. Neurochem Res. 2005;30:263–270. doi: 10.1007/s11064-005-2449-y. [DOI] [PubMed] [Google Scholar]
- 20.Lewis AE, Susarla R, Wong BC, Langman MJ, Eggo MC. Protein kinase C δ is not activated by caspase-3 and its inhibition is sufficient to induce apoptosis in the colon cancer line, COLO 205. Cell Signal. 2005;17:253–262. doi: 10.1016/j.cellsig.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 21.Li J, Hellmich MR, Greeley GH, Jr, Townsend CM, Jr, Evers BM. Phorbol ester-mediated neurotensin secretion is dependent on the PKC-α and -δ isoforms. Am J Physiol Gastrointest Liver Physiol. 2002;283:G1197–1206. doi: 10.1152/ajpgi.00177.2002. [DOI] [PubMed] [Google Scholar]
- 22.Li J, O’Connor KL, Hellmich MR, Greeley GH, Jr, Townsend CM, Jr, Evers BM. The role of protein kinase D in neurotensin secretion mediated by protein kinase C-α/-δ and Rho/Rho kinase. J Biol Chem. 2004;279:28466–28474. doi: 10.1074/jbc.M314307200. [DOI] [PubMed] [Google Scholar]
- 23.Oliver FJ, de la Rubia G, Rolli V, Ruiz-Ruiz MC, de Murcia G, Murcia JM. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J Biol Chem. 1998;273:33533–33539. doi: 10.1074/jbc.273.50.33533. [DOI] [PubMed] [Google Scholar]
- 24.Peluso JJ, Pappalardo A, Fernandez G. Basic fibroblast growth factor maintains calcium homeostasis and granulosa cell viability by stimulating calcium efflux via a PKC δ-dependent pathway. Endocrinology. 2001;142:4203–4211. doi: 10.1210/endo.142.10.8460. [DOI] [PubMed] [Google Scholar]
- 25.Perona R, Montaner S, Saniger L, Sanchez-Perez I, Bravo R, Lacal JC. Activation of the nuclear factor-κB by Rho, CDC42, and Rac-1 proteins. Genes Dev. 1997;11:463–475. doi: 10.1101/gad.11.4.463. [DOI] [PubMed] [Google Scholar]
- 26.Quaroni A, Wands J, Trelstad RL, Isselbacher KJ. Epithelioid cell cultures from rat small intestine. Characterization by morphologic and immunologic criteria. J Cell Biol. 1979;80:248–265. doi: 10.1083/jcb.80.2.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4:446–456. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- 28.Rykx A, De Kimpe L, Mikhalap S, Vantus T, Seufferlein T, Vandenheede JR, Van Lint J. Protein kinase D: a family affair. FEBS Lett. 2003;546:81–86. doi: 10.1016/s0014-5793(03)00487-3. [DOI] [PubMed] [Google Scholar]
- 29.Shirai Y, Saito N. Activation mechanisms of protein kinase C: maturation, catalytic activation, and targeting. J Biochem (Tokyo) 2002;132:663–668. doi: 10.1093/oxfordjournals.jbchem.a003271. [DOI] [PubMed] [Google Scholar]
- 30.Storz P, Doppler H, Johannes FJ, Toker A. Tyrosine phosphorylation of protein kinase D in the pleckstrin homology domain leads to activation. J Biol Chem. 2003;278:17969–17976. doi: 10.1074/jbc.M213224200. [DOI] [PubMed] [Google Scholar]
- 31.Storz P, Doppler H, Toker A. Activation loop phosphorylation controls protein kinase D-dependent activation of nuclear factor kappaB. Mol Pharmacol. 2004;66:870–879. doi: 10.1124/mol.104.000687. [DOI] [PubMed] [Google Scholar]
- 32.Storz P, Doppler H, Toker A. Protein kinase Cδ selectively regulates protein kinase D-dependent activation of NF-κB in oxidative stress signaling. Mol Cell Biol. 2004;24:2614–2626. doi: 10.1128/MCB.24.7.2614-2626.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Storz P, Toker A. Protein kinase D mediates a stress-induced NF-κB activation and survival pathway. Embo J. 2003;22:109–120. doi: 10.1093/emboj/cdg009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Valverde AM, Sinnett-Smith J, Van Lint J, Rozengurt E. Molecular cloning and characterization of protein kinase D: a target for diacylglycerol and phorbol esters with a distinctive catalytic domain. Proc Natl Acad Sci U S A. 1994;91:8572–8576. doi: 10.1073/pnas.91.18.8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van der Woude CJ, Kleibeuker JH, Jansen PL, Moshage H. Chronic inflammation, apoptosis and (pre-)malignant lesions in the gastro-intestinal tract. Apoptosis. 2004;9:123–130. doi: 10.1023/B:APPT.0000018794.26438.22. [DOI] [PubMed] [Google Scholar]
- 36.Waldron RT, Rey O, Zhukova E, Rozengurt E. Oxidative stress induces protein kinase C-mediated activation loop phosphorylation and nuclear redistribution of protein kinase D. J Biol Chem. 2004;279:27482–27493. doi: 10.1074/jbc.M402875200. [DOI] [PubMed] [Google Scholar]
- 37.Waldron RT, Rozengurt E. Oxidative stress induces protein kinase D activation in intact cells. Involvement of Src and dependence on protein kinase C. J Biol Chem. 2000;275:17114–17121. doi: 10.1074/jbc.M908959199. [DOI] [PubMed] [Google Scholar]
- 38.Wang JF, Zhang X, Groopman JE. Activation of vascular endothelial growth factor receptor-3 and its downstream signaling promote cell survival under oxidative stress. J Biol Chem. 2004;279:27088–27097. doi: 10.1074/jbc.M314015200. [DOI] [PubMed] [Google Scholar]
- 39.Wang X, Simpkins JW, Dykens JA, Cammarata PR. Oxidative damage to human lens epithelial cells in culture: estrogen protection of mitochondrial potential, ATP, and cell viability. Invest Ophthalmol Vis Sci. 2003;44:2067–2075. doi: 10.1167/iovs.02-0841. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y, Schattenberg JM, Rigoli RM, Storz P, Czaja MJ. Hepatocyte resistance to oxidative stress is dependent on protein kinase C-mediated down-regulation of c-Jun/AP-1. J Biol Chem. 2004;279:31089–31097. doi: 10.1074/jbc.M404170200. [DOI] [PubMed] [Google Scholar]
- 41.Yamamoto K, Kushima R, Kisaki O, Fujiyama Y, Okabe H. Combined effect of hydrogen peroxide induced oxidative stress and IL-1α on IL-8 production in CaCo-2 cells (a human colon carcinoma cell line) and normal intestinal epithelial cells. Inflammation. 2003;27:123–128. doi: 10.1023/a:1023813710941. [DOI] [PubMed] [Google Scholar]
- 42.Yuan J, Slice LW, Rozengurt E. Activation of protein kinase D by signaling through Rho and the α subunit of the heterotrimeric G protein G13. J Biol Chem. 2001;276:38619–38627. doi: 10.1074/jbc.M105530200. [DOI] [PubMed] [Google Scholar]
- 43.Zimmermann KC, Bonzon C, Green DR. The machinery of programmed cell death. Pharmacol Ther. 2001;92:57–70. doi: 10.1016/s0163-7258(01)00159-0. [DOI] [PubMed] [Google Scholar]