

Abstract
The posterior paraventricular nucleus of the thalamus (THPVP) has been identified as a forebrain region that modulates the central nervous system (CNS) response to recurrent experiences of stressors. The THPVP is activated in response to a single (SH) or recurrent (RH) experience of the metabolic stress of hypoglycemia. In this study, we evaluated whether temporary experimental inactivation of the THPVP would modify the neuroendocrine response to SH or RH. Infusion of lidocaine (LIDO) or vehicle had no effect on the neuroendocrine response to SH, comparable to findings with other stressors. THPVP vehicle infusion concomitant with RH resulted in a prevention of the expected impairment of neuroendocrine responses, relative to SH. LIDO infusion with RH resulted in significantly decreased glucagon and sympathoadrenal responses, relative to SH. These results suggest that the THPVP may contribute to the sympathoadrenal stimulation induced by hypoglycemia; and emphasizes that the THPVP is a forebrain region that may contribute to the coordinated CNS response to metabolic stressors.
Keywords: Rat, Posterior paraventricular nucleus of the thalamus (THPVP), Hypoglycemia, Catecholamine, Stress, Glucagon
1. Introduction
With repeated exposure to hypoglycemic stress in a time frame of hours to days, the CNS-initiated neuroendocrine counterregulatory response becomes impaired. The impairment is global and represents the efferent component of the clinical syndrome, hypoglycemia-associated autonomic failure or HAAF (Cryer, 1993). We have established a model of this impaired response, in which rats experience three bouts of insulin-induced hypoglycemia within a 24-h period. In our model, there are impairments of the sympathoadrenal (adrenal medulla) projection of the sympathetic nervous system and the pancreatic islet α-cell, with modest impairment of the hypothalamic–pituitary–adrenal (HPA) axis. This results in decreased epinephrine (EPI), glucagon, ACTH, and corticosterone, respectively (Evans et al., 2001). In our model, we have mapped forebrain expression of the immediate early gene product, c-fos, as an indicator of the pattern of forebrain activation in association with a single, vs. multiple, bouts of hypoglycemia. We observed decreased activation of the paraventricular nucleus of the hypothalamus (PVN) and dorsomedial hypothalamus (DMH) in rats that experienced multiple, vs. a single bout of hypoglycemia. In subsequent studies, we have demonstrated that reversible inactivation of these regions during a single bout of hypoglycemia (SH) resulted in impairments of the sympathoadrenal and HPA axis responses (Evans et al., 2003, 2004). Thus, decreased medial hypothalamic activation may contribute to the impaired counterregulatory response of recurrent hypoglycemia (RH).
In addition to decreased medial hypothalamic activation with multiple bouts of hypoglycemia, we observed maintained activation of the posterior paraventricular nucleus of the thalamus (THPVP), such that c-fos expression was comparable between rats that experienced one, vs. three, bouts of hypoglycemia (Evans et al., 2001). Importantly, functional imaging studies have now identified the thalamus, including the THPVP, as a region of increased neuronal activity during SH and RH in non-diabetic and diabetic human subjects (Teh et al., 2007; Teves et al., 2004 and 2007 meeting of the American Diabetes Association, with permission from A.M. Arbelaez). Activity of the THPVP has been implicated in the altered neuroendocrine response to the repeated experience of other stressors, including the phenomenon of stress habituation (decreased HPA activation): the THPVP contributes to feedback inhibition by glucocorticoids on the HPA axis, in chronically but not acutely stressed rats (Jaferi et al., 2003). Bhatnagar and others have demonstrated that permanent inactivation of the THPVP, via electrolytic lesion, prevents the blunted ACTH response that occurs with multiple experience of restraint stress, whereas the HPA response to a novel, heterotypic, stressor is not altered by THPVP lesion (Bhatnagar et al., 2000, 2002; Jaferi et al., 2003; Jaferi and Bhatnagar, 2006; Ottenweller et al., 1989). These data suggest that the THPVP is linked to the CNS ‘memory’ of a stressor experience. In addition to its involvement in the response to a psychological stressor such as restraint (Bhatnagar et al., 2000, 2002), the THPVP is activated by the chronic physiological stressor of intermittent hypoxia (Sica et al., 2000).
Afferent inputs and efferent projections from the THPVP support a functional role in the integrated response to stressors. Connectivity has been reported between the THPVP and key brainstem and hypothalamic sites including the NTS, parabrachial nucleus, and dorsal raphe, as well as the lateral, DMH, suprachiasmatic, and arcuate nuclei of the hypothalamus (Bhatnagar et al., 2000; Moga et al., 1995; Otake et al., 1994; Phillipson and Bohn, 1994). Connections with limbic areas such as the bed nucleus of the stria teminalis, amygdala, and medial prefrontal cortex are likewise consistent with a role in the modulation of the stress response. From these regions, the THPVP receives adrenergic, noradrenergic, CRHergic, and dopaminergic inputs (Freedman and Cassell, 1994; Otake and Nakamura, 1995, 1998; Otake and Ruggiero, 1995; Otake et al., 1995; Phillipson and Bohn, 1994). Despite the evidence of the functional neuroanatomy, which would implicate it in the response to metabolic stressors, the role of the THPVP in the neuroendocrine response to hypoglycemia has not been evaluated. In the present study, we tested whether reversible inactivation of the THPVP altered the neuroendocrine response to a single (SH), or multiple (RH), bouts of hypoglycemia. We hypothesized that if THPVP activation contributed to the impaired glucagon, sympathoadrenal, or HPA responses to a third bout of hypoglycemia, then those responses might be enhanced by reversible inactivation of the THPVP. Our findings suggest that THPVP activation makes a complex contribution to the response to hypoglycemia, facilitating the sympathoadrenal response, but with no effect on the HPA response to hypoglycemia. Because the glucagon and sympathoadrenal – and not the HPA – responses are the most critical for counter-regulation to hypoglycemia, our findings suggest that the THPVP has the capacity to qualitatively modulate stress responses, depending upon the specific nature of the stressor.
2. Results
Neuroendocrine data for the experimental groups are provided in Table 1. Baseline (t.0) plasma glucose levels and t.120 min glucose nadirs are matched between the two experimental conditions (SH vs. RH). All baseline (t.0) neuroendocrine parameters are matched as well. We observed two types of effects of our experimental procedures. First, there was a differential effect of PBS or LIDO infusion in RH rats compared with SH rats (Fig. 1). Second, there was an effect of LIDO (vs. PBS) infusion in RH rats. Similar to previous studies in our hypoglycemia model, these differential effects were observed for glucagon and sympathoadrenal responses.
Table 1. Glucose and neuroendocrine data with SH and RH.
| 0 min
(baseline) |
30 min | 60 min | 90 min | 120 min | |
|---|---|---|---|---|---|
| SH (single bout of hypoglycemia) | |||||
| Plasma glucose (mg/dl) | |||||
| PBS | 106±2 | 52±5 | 37±3 | 33±2 | 31±2 |
| LIDO | 103±3 | 52±4 | 39±2 | 31±2 | 32±2 |
| Glucagon (pg/ml) | |||||
| PBS | 124±26 | 224±21 | 262±46 | 248±42 | 307±32 |
| LIDO | 127±9 | 248±25 | 351±36 | 299±34 | 384±37 |
| ACTH (pg/ml) | |||||
| PBS | 25±7 | 62±14 | 208±38 | 307±54 | 319±56 |
| LIDO | 17±4 | 78±19 | 250±49 | 274±50 | 288±53 |
| CORT (μg/dl) | |||||
| PBS | 6.3±1 | 11.6±1.8 | 19.5±1.7 | 22.5±1.3 | 24.2±1.3 |
| LIDO | 6.1±1.1 | 12.7±1.6 | 20.2±1.9 | 23.4±1.9 | 22.9±1.6 |
| EPI (ng/ml) | |||||
| PBS | 94±10 | 412±93 | 2010±455 | 2618±489 | 3308±467 |
| LIDO | 93±11 | 620±181 | 2233±687 | 3113±425 | 2460±228 |
| NEPI (ng/ml) | |||||
| PBS | 242±17 | 332±45 | 670±99 | 756±65 | 918±126 |
| LIDO | 257±21 | 428±49 | 706±134 | 881±85 | 761±40 |
| RH (third bout of hypoglycemia) | |||||
| Plasma glucose (mg/dl) | |||||
| PBS | 110±1 | 50±2 | 39±2 | 35±2 | 35±3 |
| LIDO | 109±3 | 51±2 | 41±2 | 39±2 | 36±2 |
| Glucagon (pg/ml) | |||||
| PBS | 164±22 | 291±22 | 244±25 | 291±37 | 289±39 |
| LIDO | 142±3 | 250±18 | 230±21 | 238±16 | 415±49 |
| ACTH (pg/ml) | |||||
| PBS | 11±2 | 70±19 | 133±20 | 322±55 | 284±27 |
| LIDO | 20±6 | 55±12 | 87±23 | 230±53 | 302±44 |
| CORT (μg/dl) | |||||
| PBS | 5.7±0.8 | 10.1±1.1 | 17.6±1.1 | 21.4±0.8 | 23.4±1.1 |
| LIDO | 8.3±0.9 | 11.9±1.1 | 15.2±1.5 | 19.8±1.2 | 21.4±0.9 |
| EPI (ng/ml) | |||||
| PBS | 93±20 | 530±127 | 1412±272 | 2634±346 | 3341±531 |
| LIDO | 114±15 | 441±56 | 986±181 | 1942±282 | 2370±347 |
| NEPI (ng/ml) | |||||
| PBS | 206±15 | 353±33 | 572±65 | 794±77 | 971±115 |
| LIDO | 258±24 | 314±28 | 449±40 | 664±66 | 769±76 |
Plasma glucose, glucagon, adrenocorticotropic hormone (ACTH), corticosterone (CORT), epinephrine (EPI), and norepinephrine (NE) measures taken at baseline (t.0 min) and at 30 min intervals throughout a 2-h bout of hypoglycemia. PBS (phosphate buffered saline) and LIDO (lidocaine) infusions were made into the THPVP. Values are represented as the mean±SEM.
Fig. 1.
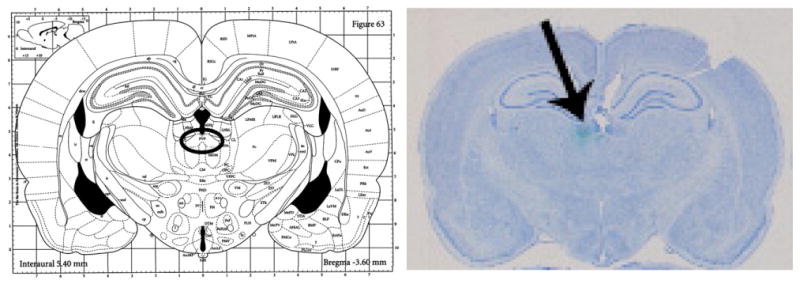
Bilateral guide cannula placement. Left, figure from Paxinos and Watson, 1986; the circle indicates the target area of the THPVP. Right, actual bilateral guide cannula placement. Cresyl violet stained coronal section showing cannula track (black arrow) and cannula end (green dye) marking the target area of the THPVP. (The specific placement coordinates for this micrograph correspond to −3.3 mm AP from bregma; ±0.2 mm ML from midline; −5.4 mm DV from skull.)
Somewhat surprisingly, we did not observe the expected blunting of glucagon or sympathoadrenal responses to RH (HAAF), in rats receiving (control) PBS infusions into the THPVP (see t.120 min data for SH-PBS vs. RH-PBS, Table 1). Glucagon, EPI, and NEPI data are depicted in Fig. 1 (Upper) as responses in RH rats that are normalized, compared to time-matched responses in SH rats (for which the value at each timepoint would be 100%). There was no overall effect of hypoglycemia experience (RH vs. SH) across the 2-h infusion period for any of the three key counterregulatory neuroendocrine responses.
An effect of recurrent hypoglycemia on the neuroendocrine counterregulatory response was manifest in rats infused with LIDO. Fig. 1 (Lower) depicts glucagon, EPI, and NEPI responses in RH/LIDO rats that are normalized in comparison with responses from SH/LIDO rats. When LIDO was infused into the THPVP, there was a significant overall effect for glucagon (p=0.038) and EPI (p=0.022), and a trend for NEPI (p=0.06), to be decreased in RH rats compared with SH rats. This effect in LIDO-infused rats was observed in the middle of the bout of hypoglycemia, such that glucagon responses were suppressed at 60 min (p=0.005) and 90 min (p=0.036); EPI responses were suppressed at 60 min (p=0.05) and 90 min (p=0.038); and NEPI responses were suppressed at 30 min (p=0.044), in RH rats vs. SH rats. All responses at t.120 min were comparable between SH/LIDO and RH/LIDO rats (see also Table 1, t.120 min). Thus the prevention of the impaired counterregulatory response in the PBS-infused rats might have been due to activation of some forebrain neuroendocrine pathways, which drive the response to hypoglycemia, but which are blunted when LIDO is infused into the THPVP (see Discussion). LIDO infusion into the THPVP did not produce any noticeable behavioral effects.
We also observed differences in the counterregulatory response between PBS- and LIDO-treated RH rats. Infusion of LIDO (compared with PBS) had differential effects on sympathoadrenal and glucagon responses to hypoglycemia. Specifically, LIDO (vs. PBS) had no overall effect on glucagon responses in either SH or RH rats, although there was a trend in both groups for the glucagon response to be amplified by LIDO at t.120 min (see Table 1, t.120 min, SH/PBS vs. SH/LIDO and RH/PBS vs. RH/LIDO). However, in RH but not SH rats, LIDO (vs. PBS) infusion significantly suppressed the NEPI response (overall, F(1,22)=6.483, p=0.02; t.30 p=0.018; t.60 p=0.035). There was a trend for LIDO infusion to suppress the overall EPI response (F(1,22)=3.861, p=0.06). This effect of LIDO is illustrated in Fig. 2, which shows glucagon, E, and NE responses in RH/LIDO normalized to corresponding, timepoint-matched, RH/PBS values set to 100%.
Fig. 2.
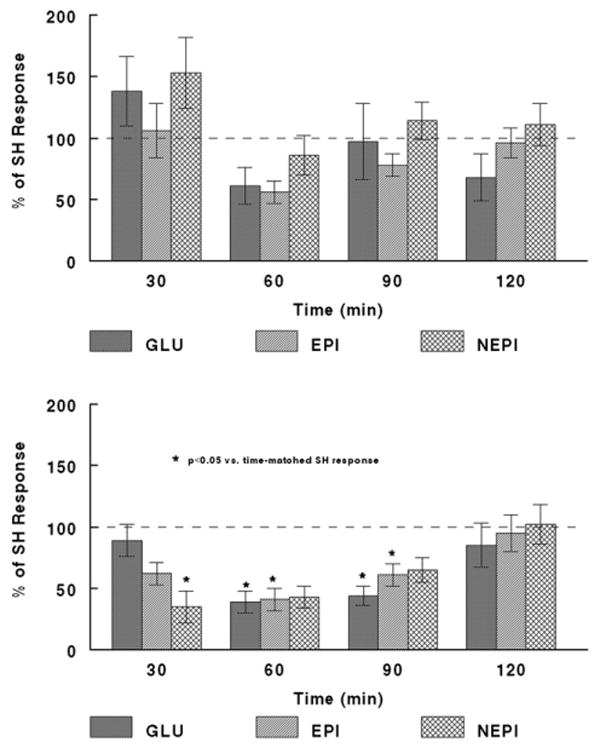
Comparison of Glucagon, EPI, and NEPI responses to single or recurrent hypoglycemia with PBS or LIDO infusion into the THPVP. Upper: Glucagon, EPI, and NEPI responses to recurrent hypoglycemia (RH) are comparable to those in rats experiencing a single (naive) bout of hypoglycemia (SH), when PBS is infused into the THPVP. Data for RH subjects are expressed as a % of the identical, time-matched responses for SH subjects (all mean SH responses being set to ‘100%’), and are shown as mean±standard error of the mean. See Table 1 and Results for group sizes. Lower: Glucagon, EPI, and NEPI responses to RH are decreased relative to those of rats experiencing a single (naive) bout of hypoglycemia (SH), when LIDO is infused into the THPVP. Data for RH subjects are expressed as a % of the identical, time-matched responses for SH subjects (all mean SH responses being set to ‘100%’), and are shown as mean±standard error of the mean. See Table 1 for group sizes. ‘*’ indicates p<0.05 vs. SH response, at the timepoint indicated.
Finally, as shown in Table 1, infusion of LIDO into the THPVP during a 2-h bout of hypoglycemia had no effect on the ACTH or CORT responses, in SH or RH rats. Baseline ACTH and CORT levels were low, and comparable to those we have previously reported in non-cannulated rats (Evans et al., 2001). Although permanent THPVP lesion has been shown to modulate the HPA response to other stressors, the ACTH/CORT response is not the most rapid or critical response for acute correction of blood glucose levels. Therefore, it is perhaps not surprising that THPVP inactivation had no effect on the HPA response to hypoglycemia.
3. Discussion
The goal of this study was to test the hypothesis that the THPVP contributes to the impaired neuroendocrine counterregulatory response that occurs with multiple experiences of the metabolic stressor, hypoglycemia (HAAF). In fact, our studies suggest that the THPVP does have a role in the impaired counterregulatory response. The results presented here in our animal model are consistent with recent work by Cryer and Amiel and colleagues in human subjects, demonstrating increased activity in the THPVP induced by recent antecedent recurrent hypoglycemia (Teh et al., 2007; Teves et al., 2004 and 2007 meeting of the American Diabetes Association, with permission from A.M. Arbelaez). However, the role of the THPVP may be more complex than originally hypothesized. First, control PBS infusions in SH rats experiencing a single (naive) bout of hypoglycemia had no effect on the neuroendocrine responses, when compared with other data from our laboratory in this model, in subjects with no CNS cannulas (Evans et al., 2001), or identical experimental protocols where cannulas were aimed at the paraventricular or dorsomedial nuclei of the hypothalamus (Evans et al., 2003, 2004). Likewise, acute LIDO infusions in SH rats had no effect on the neuroendocrine responses. This is consistent with findings in other stress models, i.e., THPVP inactivation does not impair the response to a naive experience of a stressor.
Our results in rats experiencing a third bout of hypoglycemia (RH), however, were quite surprising. Acute THPVP infusion of PBS vehicle in RH rats resulted in essentially normal counterregulatory responses (that is, comparable to those of the SH rats; Fig. 1, upper), whereas we would have expected blunted glucagon and EPI levels. This suggests that some aspect of the preparation resulted in an excitatory drive on some stress-responsive outflow pathway(s) leading to enhanced glucagon and sympathoadrenal responses. Note that this cannot be ascribed to a non-specific stress response since, as mentioned above, all of the responses to a naive experience of hypoglycemia were normal. Therefore the ‘normalized’ counterregulatory response of the RH/PBS rats is somehow tied to the repeated experience of the metabolic stressor.
Also unexpected was the response to LIDO infusion in the RH rats. As shown in Fig. 1, lower, LIDO infusion resulted in a suppression of glucagon and sympathoadrenal responses. We would have hypothesized that this treatment would simulate the effect of a permanent, physical, THPVP lesion, and in this circumstance, the counterregulatory responses would have been normalized. However, the pattern of blunted responsiveness (compared with SH/LIDO) nonetheless does not imitate the impaired counterregulatory response that we normally observe with a third bout of hypoglycemia (Evans et al., 2001): the typical impaired response pattern becomes manifest at or after 1 h and is most apparent at 120 min. However, in the THPVP RH/LIDO rats (compared to SH/LIDO rats), glucagon and sympathoadrenal responses were restored by 120 min, and were most prominently impaired at 60 and 90 min. This complex result might be interpreted as a composite of THPVP cannulation-induced enhanced neuroendocrine responding (as seen in RH/PBS rats), and the temporary local inactivation of the THPVP or immediate surrounding area by LIDO, which is overridden by continued drive on stress pathways by the maintained hypoglycemic stimulus. Importantly, the observation of LIDO effects in RH, but not SH rats, suggests that the prior experience of hypoglycemia alters neural activity, priming the THPVP, such that responsivity to additional hypoglycemic challenges is changed. Indeed, measures of regional cerebral blood flow, which reflect increased neural activity, in human subjects during hypoglycemia are found to be increased in the THPVP (Teh et al., 2007; Teves et al., 2004 and 2007 meeting of the American Diabetes Association, with permission from A.M. Arbelaez).
Neither ACTH nor CORT responses were altered by infusion of saline or LIDO in the RH rats. This result might seem contrary to those from studies using other stressors in which the THPVP appears to be play a role in the habituation of the ACTH response to repeated experience of the homotypic stressor (Bhatnagar et al., 2000, 2002; Jaferi et al., 2003; Jaferi and Bhatnagar, 2006; Ottenweller et al., 1989). However, we do not observe consistent habituation of the HPA response in our recurrent hypoglycemia model, with only modest decrements of ACTH and CORT. Since the most rapid, hence critical, endocrine responses to hypoglycemia are the release of glucagon and epinephrine, adrenal glucocorticoids are secondary and it is possible that THPVP involvement in the CNS response to stressors may be linked to stressor-specific efferent responses, which differ in the case of psychological and metabolic challenges.
As mentioned above, neither PBS nor LIDO infusion altered the neuroendocrine response in the SH rats. However, there was a significant suppression of the NEPI response in RH rats infused with LIDO vs. PBS. This suggests that LIDO infusion into the THPVP decreased sympathetic nervous system activation, as plasma NEPI reflects spillover from peripheral sympathetic nerve terminals. Conversely, this would suggest that the activation (c-fos expression) of the THPVP that we have documented in both SH and RH (Evans et al., 2001) contributes to the activation of the sympathetic nervous system. A very speculative interpretation of our data emerges from this finding combined with the observation of low plasma NEPI at 30 min in RH/LIDO rats (Fig. 1, lower): that a primary contribution of the THPVP in the CNS response to hypoglycemia may be the support of the sympathetic nervous system. This in turn can contribute to adrenal activation and EPI release, and both EPI and the sympathetic nervous system stimulate glucagon release. As shown in Fig. 1, lower (compare neuroendocrine response at t.30 and t.60 min), decreased low plasma NEPI precedes decreased EPI and glucagon, thus, suppressed sympathetic nervous system activation may be responsible for the subsequently decreased EPI and glucagon.
The connectivity of the THPVP makes it a strong candidate for modulation of the counterregulatory response to hypoglycemia, as numerous synaptic connections exist through the extent of the neuroaxis. For example, direct synaptic afferent input comes from the hindbrain (Otake et al., 1995; Phillipson and Bohn, 1994). The hindbrain contains glucoreceptive cells capable of detecting and initiating the full spectrum of counterregulatory responses (Ritter et al., 2000; Ritter, 2003). Specifically, hindbrain catecholamine neurons have been shown to be necessary neural substrates that transmit glucoprivic signals to forebrain or spinal cord sites (Ritter et al., 2001; Ritter, 2003) that are critical for the expression of neuroendocrine, autonomic, and behavioral counterregulatory responses to glucoprivation. Hypothalamic activation, as determined by c-fos-immunoreactive neurons (Ritter et al., 2001), and upregulation of arcuate nucleus NPY and AGRP mRNA expression in response to glucoprivation (Fraley and Ritter, 2003), are absent when rostrally projecting hindbrain catecholamine neurons are permanently lesioned, or in models of repeated glucoprivation. Hindbrain neural substrates involved in glucoprivic detection and/or initiation of counterregulatory responses may influence the activation of the THPVP, in addition to the hypothalamus, in response to hypoglycemia. Additionally, mono- and di-synaptic connections exist between the THPVP, limbic circuitry (BNST, amygdala, LH, medial prefrontal cortex), and the medial hypothalamus that modulates medial hypothalamic responsivity to stressors (Bhatnagar et al., 2000; Freedman and Cassell, 1994; Jaferi and Bhatnagar, 2006; Moga et al., 1995; Otake et al., 1994, 1995; Otake and Nakamura, 1995, 1998; Otake and Ruggiero, 1995; Phillipson and Bohn, 1994).
One limitation of our studies is our inability to precisely localize the anatomical site(s) that are contributing to the effects we report here. Our intermittent infusion paradigm was designed to provide continuous action of LIDO (which does not require continuous infusion) and it is estimated that the radius of spread of the infusion is <1 mm3 (Albert and Madryga, 1980; Martin, 1991; Seamans et al., 1995; Tehovnik and Sommer, 1997). Thus, while we aimed to target the THPVP, it is possible that other CNS structures are contributing to the effects we observe here, such as the habenula or the dentate gyrus. We are currently investigating the potential roles of these brain regions in the response to hypoglycemia. Nonetheless, based on our experimental design, it is clear that the THPVP would have been the major, if not the exclusive, target for the LIDO infusion.
In conclusion, our findings support a role of the THPVP in modulation of the neuroendocrine counterregulatory response to hypoglycemia. This finding contributes to the evidence obtained with other types of stressors, implicating the THPVP in stress regulation. However, the pattern of effects of THPVP inactivation suggests that hypoglycemia is a unique type of physical stressor that elicits THPVP modulation of the most critical and acute components of the counterregulatory response, and not simply HPA axis activation. Finally, our study emphasizes the role of forebrain stress regulatory circuitry in the response to hypoglycemia, in addition to hypothalamic and hindbrain sites that have served as the focus for most studies to date. Our findings support clinical evidence and demonstrate the potential for significant modulation of metabolic homeostasis by brain limbic circuitry.
4. Experimental procedure
4.1. Subjects
Male Wistar rats (Animal Technologies Limited, Kent, WA; 350–400 g) were maintained on a 12–12 h light–dark schedule (lights on at 6 AM, off at 6 PM), with ad libitum access to food and water and were studied during the lights-on portion of the light cycle. All procedures were approved by the Animal Studies Subcommittee of the VA Puget Sound Health Care System Research and Development Committee, and adhere to the guiding principles for research set forth by the American Physiological Society (2002).
4.2. Surgery
All animals underwent bilateral implantation of intravenous (IV) silastic catheters under ketamine/xylazine anesthesia (60 mg/kg ketamine (KetaFlo™, Abbott Laboratories, Chicago, IL), 7.8 mg/kg xylazine (Xyla-Ject®, Phoenix Pharmaceutical, St. Joseph, MO)) as in our previous studies (Figlewicz et al., 2002). For each rat, the right jugular vein and the left submaxilary vein were catheterized. Catheters were tunneled subcutaneously and exteriorized through a midline incision in the scalp. Each rat also received bilateral 26 gauge stainless steel guide cannulae (Plastics One, Roanoke, Virginia) inserted at an angle and aimed at the THPVP using the stereotaxic coordinates 3.5 mm AP, ±0.8 mm ML, −4.5 mm DV from bregma, according to the atlas of Paxinos and Watson (1986). Acceptable placement ranges were: −3.2 to −3.8 AP, 0.0 to +0.8 mm ML, and −4.2 to −5.0 mm DV. For experimental infusions into the THPVP, obturators were removed from the guide cannulae, and infusion cannulae were inserted that extended 1 mm beyond the tip of the guide cannula. The intracranial cannulae and IV catheters were held in place by acrylic cement (Lang Dental, Wheeling, IL) and skull screws (Small Parts, Miami Lakes, FL). Animals received subcutaneous 3 cm3 Lactated Ringers solution (Baxter Pharmaceutical Products, New Providence, NJ) and intramuscular 0.2 cm3 Gentamicin antibiotic (Bayer AG, Leverkusen, Germany), and were maintained on a circulating-water heating pad until recovery from anesthesia. Catheter lines were filled with 25–60% polyvinylpyrrolidone (PVP10, Sigma, St. Louis, MO)/heparin (1000 units/ml; Elkins-Sinn, Cherry Hill, NJ), and kept patent by a heparin (100 units/ml) flush every 3 days. All animals were allowed to recover at least one week and had to have regained weight to at least the presurgical level, and were on a positive weight gain trajectory, before undergoing the experimental procedure. THPVP cannula placements were verified at the end of the experiment and only data from animals with correct placements (approximately 85% successful placements) are included in the final analysis. Fig. 3 shows the location of the THPVP and placement of guide cannulas.
Fig. 3.
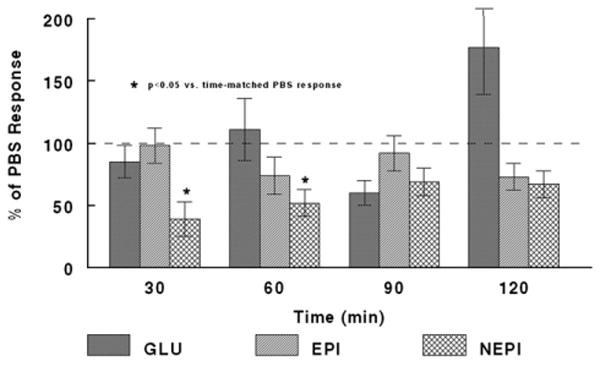
Glucagon, EPI, and NEPI responses to recurrent hypoglycemia with PBS or LIDO infusion into the THPVP. THPVP LIDO suppresses the NEPI response to a third bout of hypoglycemia (RH), relative to THPVP PBS-infused RH subjects. Data for LIDO-infused subjects are expressed as a % of the identical, time-matched responses for PBS-infused subjects (all mean PBS responses being set to ‘100%’), and are shown as mean±standard error of the mean. See Table 1 for group sizes. ‘*’ indicates p<0.05 vs. PBS response, at the timepoint indicated.
4.3. Experiments
Prior to performing experiments, animals were familiarized with square acrylic test chambers (∼30 cm×30 cm×30 cm) and the microinjection procedure as described by Evans et al. (2003, 2004). Subsequent to this, the animals were placed in the test chambers for at least 2 h before experimental procedures began. Injection and withdrawal cannulae were connected, and the experiment began once the animals were observed to be calm (∼10–15 min after connecting cannulae). Rats either received two 120-min insulin infusions separated by a 60-min break on Day 1, followed by one 120-min insulin infusion on Day 2 (recurrent hypoglycemia or RH); or they received only a single 120-min insulin infusion (single hypoglycemia or SH). Animals received insulin (Novolin® R, regular human insulin, recombinant DNA origin, Novo Nordisk, Princeton, NJ; 0.5 U/100 g body weight/h) IV over 120 min infused at a rate of 1.14 ml/h. In combination with the insulin infusions, THPVP infusions of LIDO (2%; Sigma, St. Louis, MO) or PBS vehicle (0.1 M phosphate-buffered saline, pH 7.4; Oxoid, Bastingstoke, England) were carried out by programmable syringe pumps (SP101i, World Precision Instruments, Sarasota, FL) (Evans et al., 2003). Rats either received LIDO infusion together with a single bout of hypoglycemia (SH); or they received LIDO infusion on Day 2 during a third bout of hypoglycemia (RH). As described in our previous study (Evans et al., 2003), 10 min neuropil infusions of LIDO or PBS were made every 25 min, at a rate of 100 nl/min. This dose regimen of LIDO is based on functional pharmacokinetics of LIDO in brain tissue for establishing a continuous block of neuronal activity (Martin, 1991; Tehovnik and Sommer, 1997). Furthermore, the infusion paradigm has been shown to cause no discernible tissue damage on histological evaluation in our previous studies (Evans et al., 2003, 2004), and has an estimated volume of functional spread of <1 mm3 (Albert and Madryga, 1980; Jaferi and Bhatnagar, 2006; Ritter, 2003, and see Evans et al., 2003 for additional references). Blood samples (1.5 ml) were drawn every 30 min and immediately replaced with equivalent volumes of donor blood drawn from unstressed rats (donor rats were routinely handled such that they were habituated to the blood draws and unstressed on the morning of the experiments). Plasma samples were stored frozen for subsequent use in assays.
4.4. Histology
Following the termination of infusions, each animal was overdosed with sodium pentobarbital (Nembutal®, Abbott Laboratories, Chicago, IL). Fast green dye (0.3 μl; VWR, Westchester, PA) was infused into the THPVP to mark the guide cannula track, and then animals were perfused transcardially with 0.9% saline followed by 4% paraformaldehyde. Brains were removed and placed in 4% paraformaldehyde at 4 °C for 3 days. Brains were submersed in 30% sucrose followed by freezing at −80 °C in embedding media (Fisher, Pittsburgh, PA), until sectioning at 40 μm. Tissue sections were mounted on slides and viewed under low power magnification to verify cannula placement.
4.5. Plasma assays
Plasma from blood samples were obtained for the measurement of neuroendocrine counterregulatory responses to hypoglycemia and stored at −80 °C until assayed. Blood for the catecholamine assays was collected on EGTA:glutathione (2.3 mg/ml:1.5 mg/ml; Sigma, St. Louis, MO). Tubes for glucagon assays contained 10 μl of 1 M benzamidine (Sigma, St. Louis, MO) and 1 U heparin. Blood for glucose, corticosterone, and ACTH assays was collected on EDTA and aprotinin (0.7 TIU; Sigma, St. Louis, MO). The assays have been described previously (Evans et al., 2001). Briefly, a radioenzymatic method as described in Evans et al. (1978) was used for determination of plasma EPI and NEPI. A radioimmunoassay procedure was used for plasma corticosterone measurement as described in van Dijk et al. (1997). Plasma glucose was measured spectrophotometrically, after a glucose oxidase reaction, with a Dynatech MR5000 microplate reader connected to a PC computer running Dynatech Biolinx software (Dyantech Laboratories, Chantilly, VA). Glucagon was assayed by the Linco glucagon RIA kit (Linco Research, St. Charles, MO). Measurements of ACTH were made using the Nichols Institute Diagnostics immunoradiometric assay kit (Nichols Institute Diagnostics, San Juan Capistrano, CA).
4.6. Statistical analysis
Data from the plasma assays were analyzed using two-factor ANOVA (time×hypoglycemia experience [SH or RH] or time×treatment [LIDO or PBS]) for overall (integrated) effects. Specific post hoc comparisons were carried out using Student's t-test. Significance for all tests was taken as p≤0.05.
Acknowledgments
These studies were supported by the Department of Veterans Affairs (Merit Review Program and Research Career Scientist support for D. Figlewicz Lattemann); the National Institutes of Health (NIDDK R21-062446 and DK40963 for D. Figlewicz Lattemann); and the American Diabetes Association (N. Sanders, S. Al-Noori). We thank Libby Colasurdo, Jen Bennett, Aryana Zavosh, Connie Holmes West, Pam Gronbeck, and the Metabolism Lab for excellent technical support with assays, histology, and surgical preparations.
Abbreviations
- RH
recurrent hypoglycemia
- SH
single hypoglycemia
- THPVP
posterior paraventricular nucleus of the thalamus
- LIDO
lidocaine
- EPI
epinephrine
- NEPI
norepinephrine
- PBS
phosphate buffered saline
References
- Albert DJ, Madryga FJ. An examination of the functionally effective spread of 4 microliters of slowly infused lidocaine. Behav Neural Biol. 1980;29:378–384. doi: 10.1016/s0163-1047(80)90335-0. [DOI] [PubMed] [Google Scholar]
- American Physiological Society. Guiding principles for research involving animals and human beings. Am J Physiol, Regul Integr Comp Physiol. 2002;283:R281–R283. doi: 10.1152/ajpregu.00279.2002. [DOI] [PubMed] [Google Scholar]
- Bhatnagar S, Viau V, Chu A, Soriano L, Meijer OC, Dallman MF. A cholecystokinin-mediated pathway to the paraventricular thalamus is recruited in chronically stressed rats and regulates hypothalamic–pituitary–adrenal function. J Neurosci. 2000;20:5564–5573. doi: 10.1523/JNEUROSCI.20-14-05564.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar S, Huber R, Nowak N, Trotter P. Lesions of the posterior paraventricular thalamus block habituation of hypothalamic–pituitary–adrenal responses to repeated restraint. J Neuroendocrinol. 2002;14:403–410. doi: 10.1046/j.0007-1331.2002.00792.x. [DOI] [PubMed] [Google Scholar]
- Cryer PE. Hypoglycemia unawareness in IDDM. Diabetes Care. 1993;16:40–47. doi: 10.2337/diacare.16.3.40. [DOI] [PubMed] [Google Scholar]
- Evans MI, Halter JB, Porte D., Jr Comparison of double- and single-isotope enzymatic derivative methods for measuring catecholamines in human plasma. Clin Chem. 1978;24:567–570. [PubMed] [Google Scholar]
- Evans SB, Wilkinson CW, Bentson K, Gronbeck P, Zavosh A, Figlewicz DP. PVN activation is suppressed by repeated hypoglycemia but not antecedent corticosterone in the rat. Am J Physiol, Regul Integr Comp Physiol. 2001;281:R1426–R1436. doi: 10.1152/ajpregu.2001.281.5.R1426. [DOI] [PubMed] [Google Scholar]
- Evans SB, Wilkinson CW, Gronbeck P, Bennett JL, Taborsky GJ, Jr, Figlewicz DP. Inactivation of the PVN during hypoglycemia partially simulates hypoglycemia-associated autonomic failure. Am J Physiol, Regul Integr Comp Physiol. 2003;284:R57–R65. doi: 10.1152/ajpregu.00439.2002. [DOI] [PubMed] [Google Scholar]
- Evans SB, Wilkinson CW, Gronbeck P, Bennett JL, Zavosh A, Taborsky GJ, Jr, Figlewicz DP. Inactivation of the dorsomedial nucleus of the hypothalamus selectively inhibits the ACTH and corticosterone responses to hypoglycemia. Am J Physiol. 2004;286:R123–R128. doi: 10.1152/ajpregu.00328.2003. [DOI] [PubMed] [Google Scholar]
- Figlewicz DP, Van Dijk G, Wilkinson CW, Gronbeck P, Zavosh A. Effects of repetitive hypoglycemia on neuroendocrine response and brain tyrosine hydroxylase activity in the rat. Stress. 2002;5:217–226. doi: 10.1080/1025389021000010558. [DOI] [PubMed] [Google Scholar]
- Fraley GS, Ritter S. Immunolesion of norepinephrine and epinephrine afferents to medial hypothalamus alters basal and 2-deoxy-d-glucose-induced neuropeptide Y and agouti gene-related protein messenger ribonucleic acid expression in the arcuate nucleus. Endocrinology. 2003;144:75–83. doi: 10.1210/en.2002-220659. [DOI] [PubMed] [Google Scholar]
- Freedman LJ, Cassell MD. Relationship of thalamic basal forebrain projection neurons to the peptidergic innervation of the midline thalamus. J Comp Neurol. 1994;348:321–342. doi: 10.1002/cne.903480302. [DOI] [PubMed] [Google Scholar]
- Jaferi A, Bhatnagar S. Corticosterone can act at the posterior paraventricular thalamus to inhibit hypothalamic–pituitary–adrenal activity in animals that habituate to repeated stress. Endocrinology. 2006;147:4917–4930. doi: 10.1210/en.2005-1393. [DOI] [PubMed] [Google Scholar]
- Jaferi A, Nowak N, Bhatnagar S. Negative feedback functions in chronically stressed rats: role of the posterior paraventricular thalamus. Physiol Behav. 2003;78:365–373. doi: 10.1016/s0031-9384(03)00014-3. [DOI] [PubMed] [Google Scholar]
- Martin JH. Autoradiographic estimation of the extent of reversible inactivation produced by microinjection of lidocaine and muscimol in the rat. Neurosci Lett. 1991;127:160–164. doi: 10.1016/0304-3940(91)90784-q. [DOI] [PubMed] [Google Scholar]
- Moga MM, Weis RP, Moore RY. Efferent projections of the paraventricular thalamic nucleus in the rat. J Comp Neurol. 1995;359:221–238. doi: 10.1002/cne.903590204. [DOI] [PubMed] [Google Scholar]
- Otake K, Nakamura Y. Sites of origin of corticotropin-releasing factor-like immunoreactive projection fibers to the paraventricular thalamic nucleus in the rat. Neurosci Lett. 1995;201:84–86. doi: 10.1016/0304-3940(95)12148-w. [DOI] [PubMed] [Google Scholar]
- Otake K, Nakamura Y. Single midline thalamic neurons projecting to both the ventral striatum and the prefrontal cortex in the rat. Neuroscience. 1998;86:635–649. doi: 10.1016/s0306-4522(98)00062-1. [DOI] [PubMed] [Google Scholar]
- Otake K, Ruggiero DA. Monoamines and nitric oxide are employed by afferents engaged in midline thalamic regulation. J Neurosci. 1995;15:1891–1911. doi: 10.1523/JNEUROSCI.15-03-01891.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otake K, Reis DJ, Ruggiero DA. Afferents to the midline thalamus issue collaterals to the nucleus tractus solitarii: an anatomical basis for thalamic and visceral reflex integration. J Neurosci. 1994;14:5694–5707. doi: 10.1523/JNEUROSCI.14-09-05694.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otake K, Ruggiero DA, Nakamura Y. Adrenergic innervation of forebrain neurons that project to the paraventricular thalamic nucleus in the rat. Brain Res. 1995;697:17–26. doi: 10.1016/0006-8993(95)00749-g. [DOI] [PubMed] [Google Scholar]
- Ottenweller JE, Natelson BH, Pitman DL, Drastal SD. Adrenocortical and behavioral responses to repeated stressors: toward an animal model of chronic stress and stress-related mental illness. Biol Psychiatry. 1989;26:829–842. doi: 10.1016/0006-3223(89)90123-6. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. Atlas of the Rat Brain in Stereotaxic Coordinates. 4th. Academic Press; San Diego, CA: 1986. [Google Scholar]
- Phillipson OT, Bohn MC. C1–3 adrenergic medullary neurones project to the paraventricular thalamic nucleus in the rat. Neurosci Lett. 1994;176:67–70. doi: 10.1016/0304-3940(94)90873-7. [DOI] [PubMed] [Google Scholar]
- Ritter SA. Localized glucoprivation of hindbrain but not hypothalamic sites stimulates corticosterone and glucagon secretion. Appetite. 2003;4:315. [Google Scholar]
- Ritter S, Dinh TT, Zhang Y. Localization of hindbrain glucoreceptive sites controlling food intake and blood glucose. Brain Res. 2000;856:37–47. doi: 10.1016/s0006-8993(99)02327-6. [DOI] [PubMed] [Google Scholar]
- Ritter S, Bugarith K, Dinh TT. Immunotoxic destruction of distinct catecholamine subgroups produces selective impairment of glucoregulatory responses and neuronal activation. J Comp Neurol. 2001;432:197–216. doi: 10.1002/cne.1097. [DOI] [PubMed] [Google Scholar]
- Seamans JK, Floresco SB, Phillips AG. Functional differences between the prelimbic and anterior cingulate regions of the rat prefrontal cortex. Behav Neurosci. 1995;109:1063–1073. doi: 10.1037//0735-7044.109.6.1063. [DOI] [PubMed] [Google Scholar]
- Sica AL, Greenberg HE, Scharf SM, Ruggiero DA. Chronic-intermittent hypoxia induces immediate early gene expression in the midline thalamus and epithalamus. Brain Res. 2000;883:224–228. doi: 10.1016/s0006-8993(00)02800-6. [DOI] [PubMed] [Google Scholar]
- Teh MM, Dunn J, Samarasinghe Y, Choudhary P, Pernet A, Wilson B, Marsden P, Reed L, Amiel S. Reduced activation of lateral orbitofrontal cortex in hypoglycemia associated with hypoglycemia unawareness in type 1 diabetes: a [15O]-H2O positron emission tomography study. Diabetes. 2007;56 1:A44. [Google Scholar]
- Tehovnik EJ, Sommer MA. Effective spread and time course of neural inactivation caused by lidocaine injection in monkey cerebral cortex. J Neurosci Methods. 1997;74:17–26. doi: 10.1016/s0165-0270(97)02229-2. [DOI] [PubMed] [Google Scholar]
- Teves D, Videen TO, Cryer PE, Powers WJ. Activation of human medial prefrontal cortex during autonomic responses to hypoglycemia. PNAS. 2004;101:6217–6221. doi: 10.1073/pnas.0307048101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk G, Donahey JC, Thiele TE, Scheurink AJ, Steffens AB, Wilkinson CW, Tenenbaum R, Campfield LA, Burn P, Seeley RJ, Woods SC. Central leptin stimulates corticosterone secretion at the onset of the dark phase. Diabetes. 1997;46:1911–1914. doi: 10.2337/diabetes.46.11.1911. [DOI] [PubMed] [Google Scholar]