

Abstract
Parasitic nematodes that cause elephantiasis and river blindness threaten hundreds of millions of people in the developing world. We have sequenced the ~90 megabase (Mb) genome of the human filarial parasite Brugia malayi and predict ~11,500 protein coding genes in 71 Mb of robustly assembled sequence. Comparative analysis with the free-living, model nematode Caenorhabditis elegans revealed that, despite these genes having maintained little conservation of local synteny during ~350 million years of evolution, they largely remain in linkage on chromosomal units. More than 100 conserved operons were identified. Analysis of the predicted proteome provides evidence for adaptations of B. malayi to niches in its human and vector hosts and insights into the molecular basis of a mutualistic relationship with its Wolbachia endosymbiont. These findings offer a foundation for rational drug design.
The phylum Nematoda is speciose and abundant and, although most species are free-living, many are parasitic. Over one-third of all humans, mainly in the developing world, carry a nematode infection. Parasitic worms typically cause chronic, debilitating infections that are often difficult to treat and that, despite the high cost to human health, have been neglected in biomedical research. Current knowledge of nematode molecular genetics and developmental biology is largely based on extensive studies of the free-living, bacteriovorous species Caenorhabditis elegans. Here, we present the initial analysis of the genome of the human filarial parasite Brugia malayi.
Brugia malayi is endemic in Southeast Asia and Indonesia. Like other filarial nematodes, B. malayi develops through four larval stages into an adult male or female (fig. S1), entirely within one of two host species—a mosquito vector (Culex, Aedes, and Anopheles) and humans, where adult worms can live for more than a decade. B. malayi was chosen for whole-genome sequencing (1) because it is the only major human filarial pathogen that can be maintained in small laboratory animals. Most filarial nematodes, including B. malayi, carry three genomes: nuclear, mitochondrial (available at GenBank, accession no. AF538716), and that of an alphaproteobacterial endosymbiont, Wolbachia. We present here the draft assembly and annotated genome of the TRS strain of B. malayi. We provide comparative analyses with Caenorhabditis and another well-annotated member of the superphylum Ecdysozoa, Drosophila melanogaster, to further illuminate the origins of novelty and loss of ancestral characters in the model species and the parasite. Comparative genome analysis reveals key features of Nematoda that define the scope of molecular diversity that has contributed to the success of the phylum. The analysis also uncovers adaptations that appear to have evolved in the B. malayi genome in response to the pressures of parasitism and to the presence of the parasite’s Wolbachia endosymbiont, wBm.
The B. malayi nuclear genome is organized as five chromosomes (2), including an XY sex-determination pair, and has been estimated to be 80 to 100 megabases (Mb) (3, 4). The sequence of the B. malayi nuclear genome was obtained to ~9× coverage with the use of whole-genome shotgun (WGS) sequencing (1, 5). The sequences were assembled into scaffolds totaling ~71 Mb of data with a further ~17.5 Mb of contigs not integrated into any scaffold (orphan contigs). The repeat content of the B. malayi genome, estimated at ~15% (1), may have contributed significantly to assembly difficulties (5, 6). From these sequence data, we estimate that the B. malayi genome is 90 to 95 Mb (Table 1) (5). In comparison, the C. elegans genome is 100 Mb and the Caenorhabditis briggsae genome 104 Mb. The overall G + C content (30.5%) is lower than that of C. elegans (35.4%) or C. briggsae (37.4%) (6).
Table 1.
General features and genome content of B. malayi, with C. elegans values for comparison. Scaffolds are also called super-contigs, they are assembled into large regions of ordered sequence fragments separated by gaps. Orphan-contigs are sequence fragments (contigs) that do not have information allowing them to be linked to other fragments and into scaffolds. Singletons are sequences that cannot be assembled into sequence fragments (contigs). N50 is a length-weighted average of contig or scaffold size, such that the average nucleotide in an assembly will appear in a contig (or scaffold) of N50 size or greater.
| Features | B. malayi | C. elegans |
|---|---|---|
| Overall | ||
| Estimated size of genome (Mb) | 90–95 | |
| Total number of bp of assembled sequence (bp) | 88,363,057 | |
| Number of scaffolds | 8,180 | |
| N50 of scaffolds (bp) | 93,771 | |
| Maximum length of scaffold (bp) | 6,534,162 | |
| Number of bp assembled into scaffolds (bp) | 70,837,048 | |
| Number of orphan contigs | 18,868 | |
| Number of bp assembled into orphan contigs (bp) | 17,526,009 | |
| Number of singletons | 176,099 | |
| Number of bp in singletons (bp) | 108,289,205 | |
| Protein-coding regions | ||
| Percent of genome containing protein-coding sequence (%) | 17.84 | |
| Number of gene models | 11,515* | |
| Number of proteins | 11,508 (9,839)† | |
| Max/average protein length (amino acids) | 9,445/371 | 18,563/440 |
| Gene density (genes per Mb) | 162 | 228 |
| Number of exons | 83,672 | |
| Mean/median exon size (bp) | 159/140 | 307/147 |
| Mean/median number of exons per gene | 7.27/5 | 6.38/6 |
| Number of bp included in exons | 13,282,846 | |
| Number of introns | 72,157 | |
| Mean/median intron size (bp) | 311/219 | 320/68 |
| Number of bp included in introns | 22,512,502 | |
| Mean length of intergenic region (bp) | 3,783 | 2218 |
| Overall G + C content (%) | 30.5 | 35.4 |
| Exons, G + C content (%) | 39.6 | 42.9 |
| Introns, G + C content (%) | 27.6 | 29.1 |
| Intergenic regions, G + C content (%) | 30.9 | 32.5 |
| Non–protein coding genes | ||
| Transfer RNA (tRNA) genes (+ tRNA pseudogenes) | ~233 (+26) | |
| 5S ribosomal RNA (found in scaffolds and orphan contigs) | ~400 |
This number includes seven pseudogenes.
The number of proteins 100 amino acids long or larger.
The complement of protein-coding genes was derived by automated gene prediction from the ~71-Mb assembly and by manual annotation of selected gene families (table S1). The 11,515 robustly predicted gene-coding regions occupy ~32% of the sequence at an average density of 162 genes/Mb (Table 1).
After inclusion of genes estimated to be found in the unannotated portion of the genomic sequence (5), we infer that B. malayi has between 14,500 and 17,800 protein-coding genes, agreeing with previous estimates (7). Even the higher estimate is lower than the 19,762 (WormBase data release WS133) and 19,507 (6) genes reported for C. elegans and C. briggsae, respectively, which suggests that parasitic nematode genomes have fewer genes than their free-living counterparts, echoing a pattern observed in bacterial pathogens.
For the six scaffolds longer than 1 Mb, totaling ~25 Mb of the genome, the arrangement of B. malayi genes was compared with that of their C. elegans orthologs (Fig. 1). Linkage is in general conserved: For large regions of the B. malayi genome, orthologs map predominantly to one (or, in the case of scaffold 14972, two) C. elegans chromosome(s) (Fig. 1, A to C), which indicates maintenance of linkage of these genes despite ~350 million years of separation (8). However, local gene order is not conserved (Fig. 1D). The largest, 6.5-Mb scaffold contains interdigitating blocks of genes that map to chromosomes 4 and X of C. elegans, which suggests there were ancient breakage and fusion events between linkage groups. These data support a model where within-linkage group rearrangements have been many times more common than between-linkage group translocations (7, 9), a pattern that may be typical of nematode genomes (6, 10).
Fig. 1.
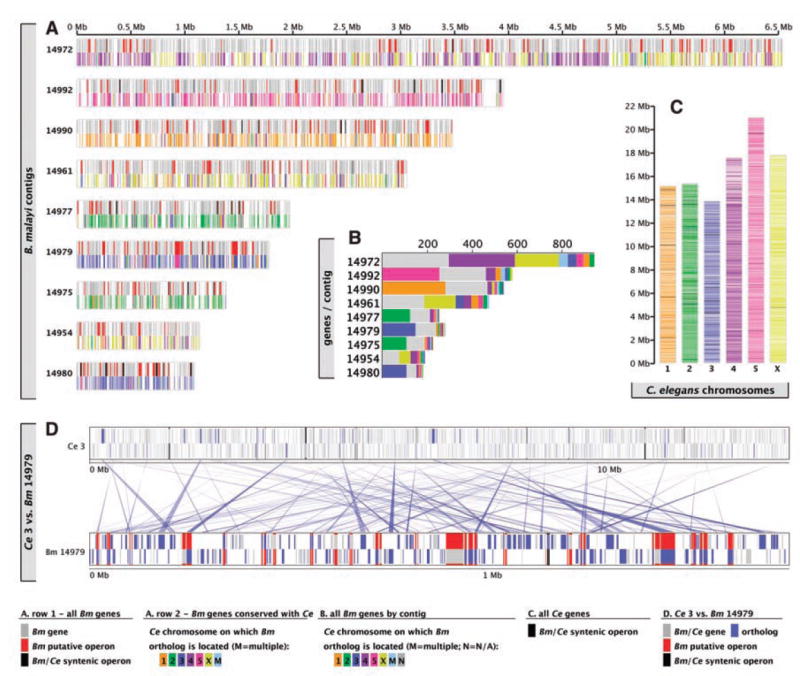
Genome organization of B. malayi in comparison with C. elegans. (A) Genes on the eight largest B. malayi scaffolds. The upper part of each scaffold cartoon represents genes in conserved operons (black) or B. malayi–specific operons (red) or genes not in operons (gray). The lower part indicates genes with orthologs in C. elegans, colored to indicate the C. elegans chromosome on which the ortholog is located. (B) Numbers of genes on the longest B. malayi scaffolds that have orthologs on each of the six C. elegans chromosomes. (C) Distribution on the six chromosomes of C. elegans of orthologs of B. malayi genes. Orthologs map to both the conserved, central cores of the autosomes and to the less well conserved genes on the autosome arms. (D) Relative arrangements of the genes on B. malayi scaffold 14979 and their orthologs on C. elegans chromosome 3. Forward-and reverse-strand genes are distinguished (forward on top, reverse below). Red rectangles correspond to putative B. malayi operons, and black rectangles correspond to conserved operon structures.
Operons are a common form of gene organization in bacteria and some protozoa, but in Metazoa, operons have been identified only in nematodes, platyhelminths, and urochordates (11, 12). Using 1000 base pairs (bp) as the upper limit of intergenic spacing, 838 potential operons (5), containing ~1800 genes (16% of the total; 2 to 5 genes per operon), were found in the assembled genome (Fig. 2 and fig. S2). Of these putative operons, only 10% of the gene pairs were also in operons in C. elegans (table S2).
Fig. 2.
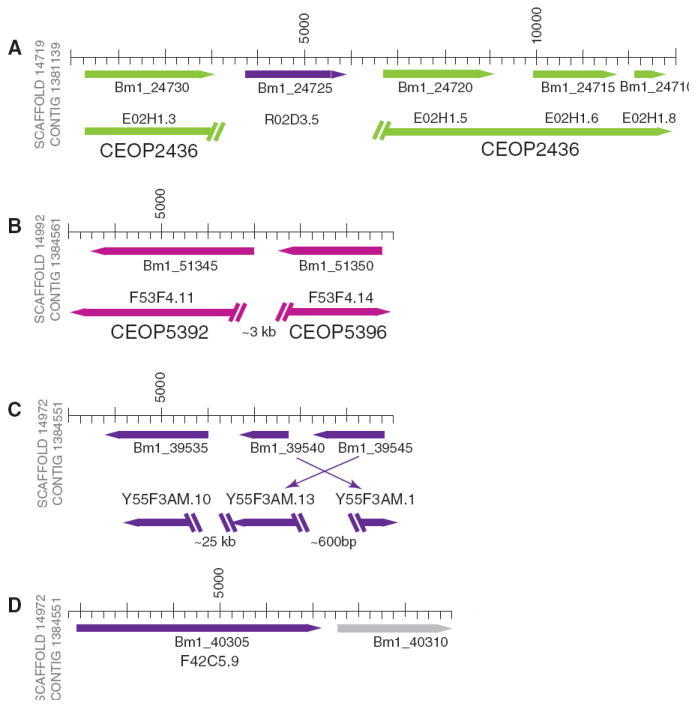
Operon diversity in the genome of Brugia malayi. The cartoon shows the structure of four putative B. malayi operons (scale is shown in base pairs). Gene transcriptional orientations are shown with colored arrows, and the C. elegans chromosome on which the ortholog is contained is indicated (II, green; IV, dark purple; V, light purple, or none, grey), along with the peptide identification number (table S2). C. elegans orthologs are aligned below the B. malayi contigs, and their gene names and operon identifiers are shown. The angled double bars show gaps in the C. elegans genomic DNA; the relative size of the gap is indicated. (A) Four predicted B. malayi genes, annotated as “hypothetical,” have orthologs in a C. elegans operon. The C. elegans ortholog of the second gene is found on a different chromosome; the ortholog of E02H1.4 has not been identified within the B. malayi genome sequence. (B) In C. elegans, the orthologs of these genes are found in operons adjacent to each other but in opposing transcriptional orientations. (C) The C. elegans orthologs of genes found in this operon are relatively close to each other but are not in an operon. (D) The C. elegans ortholog of the first gene is not in the operon while the downstream gene (independently verified GenBank accession CAD22104) has no identifiable C. elegans ortholog.
To obtain an estimate of the core complement of proteins that defines the phylum Nematoda, we compared the proteomes of B. malayi, C. elegans, and C. briggsae. Comparisons with the arthropod D. melanogaster were also made to help define a list of lineage-restricted genes. We identified 3979 sets of orthologs with representatives in all four species and 1726 sets of orthologs limited to the three nematode species (fig. S3A; tables S3 to S8). The average pairwise identity of B. malayi proteins with orthologs from either caenorhabditid species is ~ 48%. The genes conserved in nematodes but absent from the fly include cathepsin Z–like cysteine proteases, major sperm proteins, and cuticle collagens, as well as several families of unknown function. In addition, these orthologs were significantly enriched (2.4- to 4.4-fold; P > 0.0017) for genes with RNA interference (RNAi) phenotypes in C. elegans (fig. S4), which is consistent with a gene set essential to the core of nematode biochemisty and cell biology. These lineage-restricted families may define a molecular “bauplan” of Nematoda.
As noted above, the B. malayi genome appears to have fewer genes than C. elegans. On examination, much of the disparity in gene numbers can be accounted for by the extent to which gene families in Brugia and Caenorhabditis have undergone lineage-specific expansion. More than 8% of the 5780 B. malayi–C. elegans ortholog clusters were expanded in C. elegans (fig. S3C).
Comparing the occurrence of protein domains in B. malayi, C. elegans, C. briggsae, and D. melanogaster (figs. S3B and S5 and table S9) revealed, to our surprise, that B. malayi is in some ways more similar to the fly than to the model worms. For example, B. malayi and D. melanogaster have similar numbers of genes of the most abundant domains, whereas several of the most abundant domains in the caenorhabditid nematodes rank much lower in or are absent from the filarial or fly genomes (fig. S5). For domains with high abundance in all four species, C. elegans tends to have 1.5- to 2-fold as many instances as do B. malayi or D. melanogaster (fig. S5).
The distinctive biology of B. malayi is likely to be underpinned by novel proteins with unique functions. After extensive comparative analyses, 20% of the predicted proteins were found to be B. malayi-specific (fig. S6 and table S10). More than one-third of the 1977 hypothetical proteins found only in B. malayi were confirmed by B. malayi expressed sequence tags. These genes constitute an interesting list of initial candidates for functional studies of putatively filaria-specific gene products.
The drugs used for treatment of filarial parasites, although effective in the short-term control of worm burden and transmission, require extended courses of treatment that have traditionally compromised their long-term effectiveness. Recently, issues of the emergence of drug resistance have become a concern (13, 14). From the genome sequence we can identify several systems likely to be fruitful targets for the discovery of additional drug targets. (i) Molting: The B. malayi genome contains many homologs of genes that encode molecules required for molting in C. elegans (15) including proteases, protease inhibitors, nuclear hormone receptors (NRs), cuticular collagens, and chitinases (table S11). (ii) Nuclear receptors: Twenty-seven members of the NR family were identified in the B. malayi genome including orthologs of Ecr (not present in the caenorhabditids) and other NRs acting in the D. melanogaster ecdysone-response cascade (table S12). (iii) Collagens and collagen processing: B. malayi has ~82 genes that encode for a collagen repeat (including cuticular collagens and basement membrane collagens) (table S13), which is less than half the number of collagens found in the C. elegans genome (~180). It also encodes enzymes important for cuticular collagen processing such as blisterase-like proteases, protease inhibitors, tyrosinases, mixed-function oxidases, and peptidyl-prolyl isomerase (table S1). (iv) Neuronal signaling: Seven putative biogenic amine heterotrimeric guanosine 5’-triphosphate–binding protein (G protein)–coupled receptors, 44 Cys-loop receptors, and 36 genes encoding potassium channels (table S14) were identified in B. malayi, a number of which are orthologs of C. elegans genes that can be mutated to give paralytic or uncoordinated phenotypes. (v) The B. malayi kinome: The B. malayi genome encodes ~205 conventional and ~10 atypical protein kinases (Table 2), of which 142 appear to be of fundamental importance based on the severity of their RNAi phenotypes in C. elegans (table S15). (vi) Reliance on host and endosymbiont metabolism: As 9 of 10 enzymes required for de novo purine synthesis, 6 of 7 genes required for heme biosynthesis, and all 5 enzymes required for de novo riboflavin biosynthesis are absent from the B. malayi genome, the worm may be forced to meet requirements for these key metabolic factors by active uptake of host-supplied molecules (16) or through reliance on wBm, which has complete purine, heme, and riboflavin synthesis pathways (17).
Table 2.
The kinomes of the nematodes B. malayi, C. elegans, and C. briggsae in comparison with that of Homo sapiens. Eukaryotic protein kinases are mainly defined on the basis of sequence similarity of their catalytic domains, plus knowledge of accessory domains and any known modes of regulation. Conventional protein kinases (EPKs) include AGC [adenosine 3′,5′-monophosphate–dependent protein kinase/protein kinase G/protein kinase C] kinases regulated by cyclic-nucleotide and calcium-phospholipid binding; casein kinase 1 and close relatives (CK1); calmodulin-dependent kinases (CaMK), cyclin-dependent kinases (CDK)/mitogen-activated protein kinases/glycogen synthase kinases/CDK-like kinases (CMGC); receptor guanylate cyclase (RGC); a group including many kinases functioning in MAP kinase cascades (STE); tyrosine kinases (TK); tyrosine kinase-like kinases (TKL). Atypical protein kinases (APKs) include Alpha (exemplified by myosin heavy chain kinase of Dictyostelium discoideum); phosphatidylinositol 3-kinase–related kinases (PIKK); pyruvate dehydrogenase kinases (PHDK); “right open reading frame” (RIO) [named as such as it was one of two adjacent genes that were found to be transcribed divergently from the same intergenic region].
| Organism
|
|||||
|---|---|---|---|---|---|
| Protein kinases | H. sapiens | C. elegans | C. briggsae | B. malayi | Kinases shared by all 3 nematodes |
| EPKs | |||||
| AGC | 84 | 35 | 46 | 22 | 19 |
| CAMK | 98 | 63 | 69 | 41 | 23 |
| CK1 | 12 | 91 | 77 | 31 | 13 |
| CMGC | 70 | 56 | 60 | 33 | 28 |
| RGC | 5 | 27 | 24 | 4 | 4 |
| STE | 61 | 35 | 27 | 27 | 16 |
| TK | 93 | 96 | 73 | 35 | 21 |
| TKL | 55 | 22 | 21 | 12 | 9 |
| Total | 478 | 425 | 397 | 205 | 133 |
| APKs | |||||
| PIKK | 6 | 5 | 4 | 5 | 4 |
| Alpha | 6 | 1 | 1 | 1 | 1 |
| PDHK | 5 | 1 | 1 | 1 | 1 |
| RIO | 3 | 3 | 3 | 3 | 3 |
| Total | 20 | 10 | 9 | 10 | 9 |
Effective RNAi by soaking worms in double-stranded RNA(dsRNA) has been demonstrated in B. malayi adults (18). We therefore expected to find components of the RNAi pathway in the genome (table S16). However, some genes necessary for systemic RNAi in C. elegans appear to be absent from B. malayi, including sid-1, a membrane channel that transfers dsRNA molecules from a source cell to neighboring cells (19); sid-2; sid-3; and rsd-6. The presence of a putative drsh-1 ortholog suggests that B. malayi is also capable of microRNA processing. The effectiveness of RNAi in B. malayi implies either that these genes are rapidly evolving or are not required in B. malayi or that alternate pathways for siRNA transfer exist. Improvement of RNAi protocols for filarial nematodes would offer an attractive testing platform for verifying candidate drug targets.
Mapping B. malayi genes onto the C. elegans protein-protein interaction network (20) reveals an interesting pattern of evolutionarily conserved relations within the context of interconnected functional modules (figs. S7 and S8). Of 957 B. malayi genes that could be mapped, only 30 were found to be nematode-specific (supporting online text), revealing the overall conserved nature of the protein interaction network (21). Given the low level of sequence similarity between the two nematodes, the identification of conserved functional modules indicates that results from investigations of these complexes within C. elegans may be effectively translated to B. malayi.
B. malayi interacts with two hosts during its life cycle and is thought to have evolved mechanisms to suppress, subvert, or exploit host defense systems (22). Comparison of sequences of predicted proteins of B. malayi to that of interleukins, chemokines, and other signaling molecules from humans identified intriguing candidates including two genes encoding members of the macrophage migration inhibition (MIF) family of signaling molecules (23), transforming growth factor beta (TGFβ) homologs (including Bm-tgh-1) (24), and a member of the PDZ domain/interleukin 16 family (table S17). These proteins may be immune modulators that promote parasite survival or growth and differentiation factors important in parasite development. In addition, members of the ALT (abundant larval transcript) family of proteins have been implicated as virulence factors through their ability to modulate macrophage function (25). The B. malayi genome sequence revealed an unexpected diversity of 13 ALT genes (table S17), most of which are expressed by adult parasites. Note that the ALTs represent one of the few gene families that are expanded in B. malayi but not in C. elegans (which has only one member).
The innate immune systems encoded in the B. malayi genome have a complexity comparable to those of C. elegans (26) and include thioester proteins, scavenger receptors, C-type lectins, and galectins. However, both nematodes lack the peptidoglycan-recognition and lipopolysaccharide-binding proteins found in arthropods. Although there are orthologs of some components of the DAF-2, TGFβ, and p38 mitogen-activated protein (MAP) kinase signaling cascades in B. malayi and C. elegans, there is no evidence for the nuclear factor κB and Dif pathways. None of the small antibacterial peptides described from C. elegans and Ascaris suum (27) were identified in B. malayi, which suggests that the parasite might have a unique set of small peptide effectors or may lack this effector arm altogether. B. malayi gene products implicated in defense against and interaction with mammalian and insect immune systems were found, including seven genes encoding antioxidants deployed at the cuticle surface, where they may protect against oxyradical attack (28) (table S17).
Four representative organisms involved in the maintenance of Brugian filariasis have now been sequenced: the nematode parasite; its Wolbachia endosymbiont, wBm; mosquito vectors (Aedes and Anopheles); and the human host. Together these present opportunities for a systems-based approach to understanding the molecular basis of parasitism and for identification of targets for intervention. In addition, defining the molecular mechanisms that allow filarial worms to persist for decades in an immunologically competent host may yield new strategies for the control of autoimmunity and the management of transplanted tissues.
The differences in genome content and organization between Caenorhabditis and B. malayi underscore the importance of obtaining additional genome data from representative species from across the diversity of the Nematoda (29). The ability to carry out large-scale comparative genomics within Nematoda will be key in defining molecules and pathways unique to nematode development and parasitism that can serve as the targets for the next generation of antinematode drugs and vaccines.
Acknowledgments
GenBank (EF588824 to EF588901); the Brugia malayi contigs are available in GenBank (DS237653, DS238272, DS238705, DS239028, DS239057, DS239291, DS239377, and DS239315); the Brugia malayi scaffolds are available in GenBank (AAQA01000958, AAQA01000097, AAQA01001500, AAQA01000425, AAQA01001819, AAQA01001498, AAQA01000384, AAQA01001952, AAQA01000736, AAQA01000571, and AAQA01000369); and the microarray primers sequences that were used for RT-PCR are deposited in ArrayExpress (A-TIGR-28). This work was supported by an NSF grant to J.H.W. and H.T., a National Institute of Allergy and Infectious Diseases grant to E.G., and New England Biolabs Incorporated support to B.E.S.
Footnotes
Supporting Online Material
www.sciencemag.org/cgi/content/full/317/5845/1756/DC1
Materials and Methods
SOM Text
Figs. S1 to S8
Tables S1 to S17
References
References and Notes
- 1.Ghedin E, Wang S, Foster JM, Slatko BE. Trends Parasitol. 2004;20:151. doi: 10.1016/j.pt.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 2.Sakaguchi Y, Tada I, Ash LR, Aoki Y. J Parasitol. 1983;69:1090. [PubMed] [Google Scholar]
- 3.Sim BK, Shah J, Wirth DF, Piessens WF. Ciba Found Symp. 1987;127:107. doi: 10.1002/9780470513446.ch8. [DOI] [PubMed] [Google Scholar]
- 4.McReynolds LA, DeSimone SM, Williams SA. Proc Natl Acad Sci USA. 1986;83:797. doi: 10.1073/pnas.83.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Materials and methods are available as supporting material on Science Online.
- 6.Stein LD, et al. PLoS Biol. 2003;1:E45. doi: 10.1371/journal.pbio.0000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitton C, Daub J, Thompson M, Blaxter M. Methods Mol Biol. 2004;270:75. doi: 10.1385/1-59259-793-9:075. [DOI] [PubMed] [Google Scholar]
- 8.Vanfleteren JR, et al. Mol Phylogenet Evol. 1994;3:92. doi: 10.1006/mpev.1994.1012. [DOI] [PubMed] [Google Scholar]
- 9.Guiliano DB, et al. Genome Biol. 2002;3:RESEARCH0057.1. doi: 10.1186/gb-2002-3-10-research0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee KZ, Eizinger A, Nandakumar R, Schuster SC, Sommer RJ. Nucleic Acids Res. 2003;31:2553. doi: 10.1093/nar/gkg359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blumenthal T. Brief Funct Genomic Proteomic. 2004;3:199. doi: 10.1093/bfgp/3.3.199. [DOI] [PubMed] [Google Scholar]
- 12.Guiliano DB, Blaxter ML. PLOS Genetics. 2006;2:e198. doi: 10.1371/journal.pgen.0020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Awadzi K, et al. Ann Trop Med Parasitol. 2004;98:231. doi: 10.1179/000349804225003253. [DOI] [PubMed] [Google Scholar]
- 14.Schwab AE, Boakye DA, Kyelem D, Prichard RK. Am J Trop Med Hyg. 2005;73:234. [PubMed] [Google Scholar]
- 15.Frand AR, Russel S, Ruvkun G. PLoS Biol. 2005;3:e312. doi: 10.1371/journal.pbio.0030312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen SN, Howells RE. Exp Parasitol. 1981;51:296. doi: 10.1016/0014-4894(81)90117-x. [DOI] [PubMed] [Google Scholar]
- 17.Foster J, et al. PLoS Biol. 2005;3:e121. doi: 10.1371/journal.pbio.0030121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aboobaker AA, Blaxter ML. Mol Biochem Parasitol. 2003;129:41. doi: 10.1016/s0166-6851(03)00092-6. [DOI] [PubMed] [Google Scholar]
- 19.Winston WM, Molodowitch C, Hunter CP. Science. 2002;295:2456. doi: 10.1126/science.1068836. [DOI] [PubMed] [Google Scholar]
- 20.Li S, et al. Science. 2004;303:540. doi: 10.1126/science.1091403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharan R, et al. Proc Natl Acad Sci USA. 2005;102:1974. [Google Scholar]
- 22.Murray J, Gregory WF, Gomez-Escobar N, Atmadja AK, Maizels RM. Mol Biochem Parasitol. 2001;118:89. doi: 10.1016/s0166-6851(01)00374-7. [DOI] [PubMed] [Google Scholar]
- 23.Pastrana DV, et al. Infect Immun. 1998;66:5955. doi: 10.1128/iai.66.12.5955-5963.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gomez-Escobar N, Lewis E, Maizels RM. Exp Parasitol. 1998;88:200. doi: 10.1006/expr.1998.4248. [DOI] [PubMed] [Google Scholar]
- 25.Gomez-Escobar N, et al. BMC Biol. 2005;3:8. doi: 10.1186/1741-7007-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alper S, McBride SJ, Lackford B, Freedman JH, Schwartz DA. Mol Cell Biol. 2007;27:5544. doi: 10.1128/MCB.02070-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kato Y, Komatsu S. J Biol Chem. 1996;271:30493. doi: 10.1074/jbc.271.48.30493. [DOI] [PubMed] [Google Scholar]
- 28.Ou X, Tang L, McCrossan M, Henkle-Duhrsen K, Selkirk ME. Exp Parasitol. 1995;80:515. doi: 10.1006/expr.1995.1064. [DOI] [PubMed] [Google Scholar]
- 29.Blaxter ML. Adv Parasitol. 2003;54:101. doi: 10.1016/s0065-308x(03)54003-9. [DOI] [PubMed] [Google Scholar]
- 30.Funding for this project was provided by a grant from the National Institute for Allergy and Infectious Diseases, NIH (NIAID/NIH U01-AI50903) awarded to E.G. and A.L.S. We would like to acknowledge our colleagues in the Filarial Genome Consortium and the filarial research community for their continued support and encouragement. The Filarial Genome Consortium was initiated by grants from the United Nations Special Programme for Research and Training in Tropical Diseases (TDR), which is cosponsored by the U.N. Children’s Fund (UNICEF), U.N. Development Programme (UNDP), World Bank, and World Health Organization (WHO) (T23/79/152 to A.L.S.; T23/79/153 to B.S.; and T23/79/157 to S.A.W.). This whole-genome shotgun project has been deposited at the DNA Databank of Japan (DDBJ), European Molecular Biology Laboratory (EMBL), and GenBank under the project accession AAQA00000000. The version described in this paper is the first version AAQA01000000. The data are also available in WormBase release WS175.