

Abstract
Vanilloid agonists such as capsaicin activate ion flux through the TRPV1 channel, a heat- and ligand-gated cation channel that transduces painful chemical or thermal stimuli applied to peripheral nerve endings in skin or deep tissues. We have probed the SAR of a variety of 1,4-dihydropyridine (DHP) derivatives as novel "enhancers" of TRPV1 activity by examining changes in capsaicin-induced elevations in 45Ca2+-uptake in either cells ectopically expressing TRPV1 or in cultured dorsal root ganglion (DRG) neurons. The enhancers increased the maximal capsaicin effect on 45Ca2+-uptake by typically 2 – 3 fold without producing an action when used alone. The DHP enhancers contained 6-aryl substitution and small alkyl groups at the 1 and 4 positions, and a 3-phenylalkylthioester was tolerated. Levels of free intracellular Ca2+, as measured by calcium imaging, were also increased in DRG neurons when exposed to the combination of capsaicin and the most efficacious enhancer 23 compared to capsaicin alone. Thus, DHPs can modulate TRPV1 channels in a positive fashion.
Introduction
Capsaicin 1 is the neuroactive component of hot chili peppers, paprika and related spices, which stimulates peripheral terminals of nociceptive sensory neurons to produce a sensation that mimics painful heat. The receptor for capsaicin is the vanilloid receptor 1 (TRPV1/VR1) located on the nerve endings, axon and soma of a subpopulation of nociceptive (pain-sensing) sensory ganglion neurons.1 TRPV1 is a member of the transient receptor potential (TRP) ion channel family, named for its homology with drosophila TRP channels and its ability to recognize ligands containing a vanillin moiety, such as capsaicin 1 (Chart 1).2 The ion flux induced upon activation of TRPV1 depolarizes the afferent nerve ending, transmitting an action potential from the skin to synaptic contacts with spinal cord second order neurons, which then transmit the information to the brain where the perception of pain is registered.3 Because of its critical role in acute and chronic inflammatory pain, antagonists of TRPV1 are sought as new analgesic agents.4–10 Selected non-vanilloid antagonists of TRPV1 (the first antagonist 2 and clinical candidates SB-705498 3 and AMG 517 4) and representative activators (an endogenous lipid mediator 5, and the potent agonist 6) are shown in Chart 1.2 The effect of heat on TRPV1 was also reversibly reduced by micromolar nifedipine, a typical dihydropyridine (DHP) that is used clinically as an L-type calcium channel antagonist.11 A number of compounds have been found that can either specifically block access of putative endovanilloids12 to the binding site or, by an allosteric effect, prevent opening of the pore-loop (P-loop) domain of the channel.2 The organometallic derivative ruthenium red is a pore-loop blocker of TRPV1.8,13 More recently, an ATP binding region within the large intracellular ankyrin repeat amino terminal domain was identified that appears to regulate receptor desensitization,14 which contrasts functionally with an ATP interaction at the Walker A motif in the C-terminal region that increases TRPV1 activity.15 Thus, multiple auxiliary, pharmacologically tractable domains are present in TRPV1 that can be targeted to block or modulate ion channel function.
Chart 1.

Selected known activating (1, 5, 6) and inhibiting (2 – 4) ligands of TRPV1.
In addition to blocking, the stimulation of TRPV1 has also been utilized therapeutically. Chronic topical application of high concentration capsaicin, which deadens pain sensation locally, has been available as an over-the-counter treatment for arthritic pain for many years, and higher dose formulations are also being explored.16 Resiniferatoxin (RTX) 6, one of the most potent vanilloid agonists, is being used in animals to treat intractable pain conditions resulting from advanced cancer, and potentially in human patients.17,18 The therapeutic effect in this application results from Ca2+ excitotoxicity that occurs selectively in TRPV1-expressing, primary afferent nociceptive neurons when RTX is administered in the vicinity of the cell body or the peripheral or central nerve terminals.19–21
While TRPV1 is obviously located on the plasma membrane, another receptor pool (TPRV1ER) also occurs intracellularly, where it is localized on the endoplasmic reticulum (ER).13,19,22–26 Several studies have demonstrated that TPRV1ER is capable of being stimulated by exogenously applied vanilloids, since they readily cross cell membranes.13,23–26 We have hypothesized that activation of TPRV1ER can contribute to nociceptive signaling due to de novo, intracellularly produced endovanilloids that release Ca2+ from the ER following stimulation of TPRV1ER.19,22 The multiplicity of environmental and intra- and intercellular stimuli that are capable of activating TRPV1 suggest that additional insights might be obtained by further pharmacological evaluation of the vanilloid binding site, various sites on the ion channel pore, or other non-vanilloid sites on this receptor/ion channel.8–10,14,26
We have discovered that a class of 1,4-dihydropyridine (DHP) derivatives with 6-aryl substitution has a novel action as enhancers of TRPV1 activity in several experimental models. They greatly increase the maximal ion flux stimulated by capsaicin, but exhibit minimal or no intrinsic agonist activity of their own. It is possible that these enhancers, in the presence of endovanilloids, would exhibit agonist-enhancing actions leading to receptor desensitization or nerve terminal inactivation.3,17,21 The biological activity of this novel class of enhancers represents a new type of pharmacological activity for TRPV1 function and reinforces the idea of dynamic pore modulation.28 The TRPV1 receptor structure is not known, and therefore the structure activity relationship (SAR) described in this study is more descriptive than mechanistic.
Results
Chemical Synthesis
The synthesis of the DHP derivatives (7 – 32,Table 1) was carried out by two variations of standard method for a three-component Hantzsch condensation (Scheme 1).29 Both 3-esters 7 – 11 and 3-thioesters 12 – 32 were prepared. For unsubstituted 6-phenyl DHPs, the precursor was an ethyl 3- aminocinnamate 33. The β-enamino esters 38 or thioesters 39 required for the route to synthesize unsubstituted 6-phenyl DHPs (Scheme 1B) were prepared from a dioxinone derivative 41 or Meldrum’s acid 43 as shown in Scheme 2. Many of these DHPs and related analogues were prepared for use in previous studies of antagonists of adenosine and P2 receptors.30–36
Table 1.
Activities of the 1,4-dihydropyridine derivatives as enhancers at the TRPV1 receptor ectopically expressed in NIH3T3 cells.
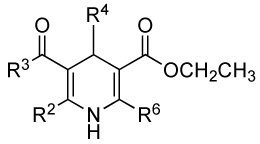 | ||||||||
|---|---|---|---|---|---|---|---|---|
| No | R2 | R3 | R4 | R6 | EC50(µM) ± SEM |
Hill Slope ± SEM |
Emax(%)a ± SEM |
nb |
| 3-Esters | ||||||||
| 7d | CH3 | CH3CH2O | CH3CH2 | Ph | — c | — | 383 ± 39 | 3 |
| 8d | CH3 | CH3CH2O | (CH3O) 2CH | Ph | — | — | 114 ± 24 | 2 |
| 9 | CH3 | CH3CH2O | CH3CH2 | 3-Cl-Ph | 17.1 ± 1.2 | 1.45 ± 0.28 | 347 ± 24 | 3 |
| 10 | CH3 | CH3CH2O | CH3CH2 | 3-F-Ph | 26.7 ± 1.1 | 1.98 ± 0.30 | 403 ± 21 | 3 |
| 11 | CH3 | CH3CH2O | CH3CH2 | 4-F-Ph | 29.2 ± 1.2 | 1.86 ± 0.42 | 372 ± 33 | 3 |
| 3-Thioesters | ||||||||
| 12d | CH3 | CH3CH2S | CH3CH2 | Ph | 28.1 ± 1.0 | 1.55 ± 0.17 | 435 ± 11 | 3 |
| 13 | CH3 | CH3CH2S | CH3CH2 | 2-F-Ph | — | — | 198 ± 2 | 2 |
| 14e | CH3 | CH3CH2S | CH3CH2 | 3-F-Ph | 21.8 ± 1.2 | 1.59 ± 0.33 | 446 ± 39 | 3 |
| 15 | CH3 | CH3CH2S | CH3CH2 | 4-F-Ph | 28.4 ± 1.2 | 1.82 ± 0.33 | 409 ± 32 | 3 |
| 16 | CH3 | CH3CH2S | CH3CH2 | 4-CN-Ph | — | — | 129 ± 2 | 2 |
| 17 | CH3 | CH3CH2S | CH3CH2 | cyclohexyl | — | — | 87 ± 8 | 2 |
| 18d | CH3 | CH3CH2S | CH3CH2CH2 | Ph | 22.4 ± 1.2 | 1.38 ± 0.23 | 364 ± 16 | 3 |
| 19 | CH3 | CH3CH2S | CH3CH2CH2 | 3-Cl-Ph | — | — | 177 ± 19 | 2 |
| 20 | CH3 | CH3CH2S | cPr | 4-F-Ph | — | — | 175 ± 4 | 2 |
| 21d | CH3CH2 | CH3CH2S | CH3CH2 | Ph | — | — | 257 ± 17 | 3 |
| 22d | CH3CH2CH2 | CH3CH2S | CH3CH2 | Ph | — | — | 148 ± 3 | 2 |
| 23d,e | CH3 | CH3OCH2CH2S | CH3CH2 | Ph | 21.3 ± 1.1 | 1.79 ± 0.17 | 626 ± 25 | 3 |
| 24 | CH3 | (CH3)2CHS | CH3CH2 | 2-F-Ph | — | — | 124 ± 4 | 2 |
| 25 | CH3 | PhCH2S | CH3CH2 | 2-F-Ph | — | — | 123 ± 1 | 2 |
| 26 | CH3 | PhCH2CH2S | CH3CH2CH2 | Ph | 27.6 ± 1.5 | 1.14 ± 0.32 | 360 ± 35 | 3 |
| 27 | CH3 | PhCH2CH2S | CH3CH2 | 2-F-Ph | — | — | 117 ± 4 | 2 |
| 28 | CH3 | PhCH2CH2S | CH3CH2 | 3-Cl-Ph | — | — | 194 ± 29 | 2 |
| 29 | CH3 | PhCH2CH2S | CH3CH2 | 4-F-Ph | 51.6 ± 1.6 | 1.18 ± 0.25 | 232 ± 7 | 3 |
| 30 | CH3 | PhCH2CH2S | CH3CH2 | 4-NO2-Ph | — | — | 101 ± 3 | 2 |
| 31 | CH3 | PhCH2S | CH3CH2 | 4-NO2-Ph | — | — | 98 ± 3 | 2 |
| 32 | CH3 | CH3CH2S | CH3CH2 | 2-naphthyl | — | — | 116 ± 19 | 2 |
Emax = TRPV1 activation at [Enhancer]max (100 µM), expressed as % of control (2 µM capsaicin).
“n” represents the number of separate assay plate runs, with compounds in duplicate wells per assay.
For compounds where EC50 and Hill Slope could not be calculated (i.e., the data could not be curve fitted), only the Emax is shown.
Ki value in binding to the human A3 adenosine receptor, in µM (corresponding compound number in ref. 27): 7, 2.27 (9); 8, 15.3 (14); 12, 2.01 (10); 18, 2.17 (12); 21, 0.907 (21); 22, 2.09 (22); 23, 4.58 (11).
Compound 23 is MRS1477; 14 is MRS3625.
Scheme 1.

Synthesis of 1,4-DHP derivatives. Reagents and conditions: i) EtOH, 24 – 48 h, 90 – 95 °C.
Scheme 2.

Synthesis of β-ketoester 34, β-ketothioester 35, and β-amino-α,β -unsaturated ester intermediates 38 and 39. Reagents and conditions: i) RSH, toluene, 100 – 110 °C; ii) ammonium acetate, EtOH, 95 – 100 °C; iii) DMAP, CH2Cl2, 0 – 25 °C, 2 – 4 h; iv) pyridine, CH2Cl2, 0 – 25 °C, 2 – 4 h; v) EtOH, toluene, reflux, 18 h; vi) ammonium acetate, EtOH, 95 °C, sealed tube.
Biological Activity
The effects of compounds in a small, targeted library of DHP derivatives on capsaicin-induced 45Ca2+-uptake were studied in a stably transfected TRPV1ε-NIH3T3 mouse fibroblast cell line and in primary cultures of dorsal root ganglion (DRG) neurons that express the TRPV1 channel endogenously. The enhancing effect of the DHP derivatives was demonstrable in both systems: the TRPV1ε-NIH3T3 cell line (Figure 1) and the DRG primary cells (Figure 2). For the various DHP derivatives with enhancing activity, the concentration response curves in the presence and absence of capsaicin clearly demonstrate that in this assay the enhancers have no observable activity when added alone (data not shown). Rather, they augment the maximal activity obtained with the vanilloid agonist.
Figure 1.

Representative dose-response kinetics of DHPs in the presence of a vanilloid evaluated in NIH3T3 cells ectopically expressing TRPV1. In the presence of 2 µM capsaicin, strong, dosedependent effects of some DHPs, e.g., compounds 14, 18, or 23, were observed. Other DHPs were slightly active in the dose range evaluated (e.g., 29), while some were neutral even in the presence of capsaicin (e.g., 31). The y-axis is normalized to the effect of capsaicin alone.
Figure 2.

Dose-response kinetics of DHPs evaluated in primary DRG cells. Active DHPs retained similar kinetics in primary DRGs (e.g., 18), while inactive compounds were also inactive in DRGs (e.g., 31). Due to DMSO solvent sensitivity, the maximum DHP concentration evaluated in DRGs was 50 µM. The y-axis is normalized to the effect of capsaicin alone.
The SAR was probed through a comparison of analogues in which the 3- and 5-ester groups and the substitution at the 1, 2, 4, and 6 positions, including aryl substitutions of the 6-phenyl group, were varied. Some of the members of the DHP library proved to be enhancers of the action of the TRPV1 agonist capsaicin, which led to further synthesis based on chemical leads. DHP derivatives that were substituted at the 4-position with phenyl rings and at the 6-position with small alkyl groups typically did not show enhancing activity (data not shown), and some of the DHP derivatives that were substituted at the 6-position with phenyl rings and with small alkyl groups at the 2- and 4-positions (7 – 32, Table 1) enhanced the effects of capsaicin.
The potency (EC50) and fold of augmentation of Emax elicited by 2 µM (ED200) capsaicin in the TRPV1ε-NIH3T3 cell line are shown in Table 1. The EC50 values of the enhancers were typically in the range of 15 to 30 µM. The most potent enhancer, using the stably expressing TRPV1ε-NIH3T3 cell line as the basis for the assay, was the 6-(3-chlorophenyl) derivative 9 with an EC50 of 17.1±1.2 µM. This compound did not achieve as high an efficacy (augmentation of Emax) as compound 23, which contained 3-methyloxyethylthio ester and 6-phenyl groups. Thus, in the TRPV1ε-NIH3T3 cells, the most efficacious compound was 23, which had an EC50 of 21.3±1.1 µM (Figure 1), and produced a 5.3- fold increase in calcium influx.
In both the 3-ester and 3-thioester series of DHP derivatives, an ethyl ester was always present at the 5 position. In the ester series, an unsubstituted 6-phenyl group, as in 18, was associated with relatively high efficacy as an enhancer. Branching at the 4-position as in acetal 8 was not tolerated. Halo substitution of the 6-phenyl ring in 9 – 11 enhanced potency as enhancers, with the pattern 3-Cl > 3-F or 4-F. Substitution of the 6-phenyl ring with fluoro 13 – 15 or cyano 16 did not enhance potency over that of unsubstituted 6-phenyl 12. 2-Fluoro and 4-cyano substitution of the 6-phenyl moiety greatly reduced the enhancing activity. The aromatic character of the 6-aryl group was essential for enhancer activity, since the 6-cyclohexyl analogue 17 was inactive.
Homologation of the 4-ethyl group in 18 did not favor increased potency or efficacy, in comparison to 12. Compound 19, the 6-(3-chlorophenyl) analogue of 18, was greatly reduced in percent activation, while the 4-cyclopropyl-6-(3-chlorophenyl) analogue 20 was less attenuated in activity. Homologation of the 2-substitution from methyl appeared to decrease maximal enhancement (ethyl 21 and propyl 22 analogues in comparison to methyl 12).
At the 3-thioester position, extension of the ester moiety was well tolerated at TRPV1, as in the highly potent and efficacious 2-methoxyethyl analogue 23. 3-Phenylalkylthio substitution, such as a 3- (2-phenylethylthio) ester 26, was tolerated. However, the 3-benzylthio substituted analogue 25 was greatly reduced in maximal enhancement. Substitution of the 6-phenyl ring in the 3-(2-phenylethylthio) series in compounds 27 – 30 produced either a reduction of potency in the 4-fluoro analogue 29 or a marked reduction of efficacy in 27 and 30.
In the primary DRG cells (Table 2), a 3-ethylthio-4-propyl-6-phenyl derivative 18 was highly efficacious and showed comparatively greater potency than in the TRPV1ε-NIH3T3 cell line, with an EC50 of 10.5±1.1 µM (Figure 2). The enhancers increased the maximal effect on 45Ca2+-uptake in response to vanilloid agonists in the DRG cells by typically 2- to 3-fold, and compound 18 produced a 3.2-fold increase in the calcium influx.
Table 2.
Activities of DHPs evaluated in primary dorsal root ganglion cells.a
| Compound | EC50 ± SEM, µM | Hill Slope ± SEM | Emax ± SEM, % of control |
|---|---|---|---|
| 9 | 17.0 ± 1.2 | 2.24 ± 0.53 | 259 ± 6 |
| 10 | 20.5 ± 1.1 | 2.64 ± 0.59 | 206 ± 7 |
| 11 | 15.7 ± 1.3 | 2.01 ± 0.77 | 175 ± 9 |
| 14 | — | — | 198 ± 16 |
| 18 | 10.5 ± 1.1 | 1.47 ± 0.16 | 318 ± 8 |
| 26 | — | — | 231 ± 13 |
| 29 | — | — | 148 ± 10 |
| 30 | — | — | 117 ± 5 |
Results from a single assay run in quadruplicate wells. The DMSO concentration during the assay did not exceed 0.2%.
Various other DHP derivatives tested did not enhance the action of capsaicin, and their pharmacological properties will be described later. Thus, the L-type calcium channel blocker nicardipine and the L-type calcium channel activator S(–)-Bay K 8644 were not enhancers in either cell model described above.
In order to confirm the enhancer effect using a more physiologically informative method, we examined 23 using live cell imaging of primary DRG neurons, a subpopulation of which expresses TRPV1. Because repetitive exposures to capsaicin can produce acute desensitization,2 the comparison was made between cultures exposed to capsaicin only and separate cultures exposed to a combination of capsaicin and 23. This experiment was designed using a low concentration (200 nM) of capsaicin in order to observe enhancement, yet avoid calcium cytotoxicity. Figure 3A shows the typical response to capsaicin using the dye Fura-4F AM and ratiometric imaging, the left panel at baseline, the right panel after capsaicin alone. Progressively higher intracellular calcium [Ca2+]i is depicted as a transition from blue to green to red. Comparison of the action of capsaicin to capsaicin + enhancer is depicted graphically in Figure 3B. The traces are the average of 3 separate experiments comprising recordings from a total of 167 neurons. Co-administration of 23 with capsaicin significantly increased the peak Ca2+ influx by 13% compared to capsaicin alone. The peak ratio values were 1.518±0.001 for capsaicin alone and 1.701±0.001 in the presence of 10 µM 23. While this number is lower than the augmentation of Emax obtained with the 45Ca2+-uptake screen, several variables differ between the two methods, notably the dose of capsaicin is lower (200 nM versus 500 nM) and the duration of exposure is much shorter (30 sec vs 10 min). The culture contains capsaicin-insensitive neurons as well and these non- TRPV1-expressing neurons do not respond to either capsaicin or 23 when morphology or changes in [Ca2+]i are used as the endpoints.
Figure 3.

Intracellular calcium measurements of primary embryonic DRG neurons in mixed cultures. A. False color representation of calcium measurements using MetaFluor software. Cells marked with “#” are capsaicin-insensitive (i.e., TRPV1-negative) DRG neurons. “*” and “**” mark cells that are, respectively, sensitive or highly sensitive to capsaicin. B. Cells in a closed perfusion system were exposed to either 200 nM capsaicin or 200 nM capsaicin in the presence of µM 23. Baseline was recorded for 2 min, and ratios were normalized to the baseline. Each trace is the mean of activated (i.e., capsaicin sensitive) DRG cells from 3 individual experiments (98 DRGs total for 200 nM capsaicin; 69 DRGs for 200 nM capsaicin + 10 µM 23). Drugs were added at 2 min for 30 sec (indicated by horizontal bar). In cells concomitantly exposed to capsaicin and 23, intracellular calcium influx increases compared to capsaicin alone.
DHP derivatives have been adapted through chemical modification to bind to various receptor sites, in addition to the classical L-type calcium channels. Among the receptors to which other DHP derivatives have been shown to bind are the adenosine receptors, and certain DHP derivatives have been engineered to bind with nanomolar affinity to the A3 adenosine receptor. Among the derivatives investigated here, several were shown to bind to the human (but not rat) A3 adenosine receptor with a high micromolar affinity. For example, the L-type calcium channel blocker nicardipine, which was not an enhancer at TRPV1, displayed a Ki value at the A3 receptor of 3.25 µM.30 Thus, the TRPV1- enhancing activity does not correlate with A3 adenosine receptor antagonism. Furthermore, the potent A3 receptor antagonist 1,4-dihydro-2-methyl-6-phenyl-4-(phenylethynyl)-3,5-pyridinedicarboxylic acid, 3-ethyl 5-(phenylmethyl) ester33 (MRS1191) had no enhancing effect. Therefore, potency as an A3 adenosine receptor antagonist or as an L-type calcium channel antagonist did not correlate with TRPV1 enhancement.
Discussion
The present set of observations delineates a new agonist-enhancing activity present in TRPV1 that can be elicited by DHP compounds. We refer to these agents as enhancers since, at least in the in vitro assays used (the ectopically expressing TRPV1ε-NIH3T3 cell line and primary DRG cultures), no intrinsic activity of the enhancers alone was detected. The enhancing compounds acted only in the presence of a vanilloid agonist. As such, these compounds present a novel approach to selective manipulation of TRPV1 function. The enhancers operate in an activity-dependent fashion and, thereby, are hypothesized to only target nerve endings that are actively engaged in nociceptive transduction. The agonist-dependent actions of the enhancers is consistent with recent observations showing modulation of the TRPV1 pore selectivity filter by vanilloid agonist activation.28 It is possible that the DHP enhancers increase the elasticity of the pore even further when the pore has been opened initially by vanilloid agonists and allow not only cations, but other large molecules to enter the nociceptive ending.37,38
The elucidation of the biological mechanism of the enhancers will be the topic of future studies. The present work explores the initial SAR pattern following the observation of this novel action, and allows the identification of one or more enhancer molecules that could be used as pharmacological tools. The structural requirements for TRPV1 enhancement by DHPs are distinct from the structural requirements of DHPs to interact with several other protein targets,30 and also delineated from the structure of the DHP derivative nifedipine (a 4-aryl-6-methyl-DHP), which is reported to inhibit TRPV1.11 Compound 23 was the most efficacious enhancer in the transfected cells, and this enhancement was confirmed using calcium imaging of primary DRG neuronal cultures.
We have recently shown that selective analgesia with vanilloid agonists such as resiniferatoxin (RTX) can be achieved through anatomically localized administration, either by peripheral injection,21 perineural application,20,39 direct intraganglionic injection,17,52 or injection into the spinal CSF17,18 for treatment of pain unilaterally or at individual or multiple spinal segments. All of these applications depend on excessive opening of TRPV1 to produce a local calcium overload.19 Potential therapeutic actions of an enhancer may operate through the same calcium overload mechanism. Moreover, the enhancer would be efficacious only at nerve endings where TRPV1 is actively occupied by an endovanilloid, rather than at all susceptible nociceptive nerve endings within the volume of distribution a traditional agonist might reach. Thus, selective, localized actions may be achieved, but without the need for local administration and without blockade or inhibition of the important protective aspects of pain elsewhere in the body,40 as might be encountered with an antagonist. Consideration of the potential pharmacological actions of the enhancers raises several interesting questions. One is pharmacodynamic: would administration of an enhancer block pain, or would it enhance pain before it is blocked? We currently are investigating these issues using inflammatory hyperalgesia41 and operant methods to assess pain sensations behaviorally.42 A second question is whether there is an endogenous ligand at this site. TRPV1 is known to be sensitive to a variety of modulators such as pH, endovanilloids, lipid derivatives and a variety of natural products and venoms (as reviewed27), and it is possible that some endogenous compound acts as a TRPV1 enhancer. In such a case, the action of an enhancer of the endogenous activator, such as the present DHP derivatives, might be more event-specific and tissue-specific than a directly acting ligand. Thus, in theory the novel enhancers might be useful clinically either alone or in conjunction with exogenously administered vanilloid activators. Lastly, there is the question of where on TRPV1 the enhancers exert their action; in this regard it is also prudent to consider the possibility that the enhancers might be acting on some other, occult cellular process that produces positive feedback on TRPV1 once it is in the vanilloid agonist-activated state.
Recently, an amino acid sequence required for vanilloid binding was tentatively defined between transmembrane domains (TD) 3 and 4,1 using both domain swapping and mutagenesis. Additional studies emphasize the importance of both the N- and C-terminal regions of TRPV1 as determinants of receptor function.14,43 The detailed three-dimensional structure of TRPV1 is currently unknown although portions of the structure and the functional roles that these regions play (e.g., to sensitize or promote or inhibit desensitization) are being elucidated.14,43
A variety of sites exist that can positively modulate function and potentially interact with DHP enhancers. A brief consideration of the quaternary structure and post-translational modifications is also important. Immunoprecipitation experiments suggest that TRPV1 exists as a homotetramer and, in this regard, exhibits similarities to a wide variety of other cation channels with four individual monomeric subunits or four linked repeating domains, with each monomer or domain having six transmembrane regions with a pore loop and an ion-conducting pore in the center, faced by transmembrane domain 6.44 Theoretically, pharmacological enhancement could be exerted through modulation of the quaternary structure. In addition, multiple phosphorylation sites exist on TRPV1 for protein kinase A, protein kinase C and the proline directed kinase CDK5.45–47 Post-translational phosphorylation of all of these sites can produce sensitization of the receptor and an enhanced sensitivity to noxious chemical and inflammatory agents creating a very broad combinatorial palate from which to produce, maintain, and modulate nociceptive transmission.
Distinct structural features of the DHP pharmacophore are required to produce enhancer activity. In future studies, the effects in three-dimensional space of alterations of charge distribution, hydrophobicity, rotational constraints, π electron clouds, aromaticity, planarity, etc. can be modeled in order to optimize biological activity. Also, it would be desirable to reduce the hydrophobicity of this DHP series. For example, the cLog p values of 9, 14, and 23 are 5.75, 5.55 and 4.71, respectively. Solubility could be a potentially major factor that limits activity.
The idea of selective suppression of pain is an attractive concept. NSAIDs fulfill this role to some degree, but have the drawback of inhibiting the cyclooxygenases everywhere in addition to sites of tissue injury, and the consequences can produce serious, even fatal side effects in the case of cycloxygenase II inhibitors.48 The potential for selectivity based on TRPV1 activity status was recently examined by co-administration of capsaicin, to open the TRPV1 channel, with the quaternary local anesthetic QX-314 (N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide), which enters the cell via the open TRPV1 channel.38 While selectivity for pain-sensing fiber types may be achieved, a major drawback to this approach is the delay in establishing inhibition (~5 min), during which time the algesic actions of capsaicin were behaviorally evident. These can be quite intense: human behavioral and functional brain imaging studies3,49,50 following intradermal capsaicin administration demonstrate that it potently activates the brain pain network within this time frame and triggers central sensitization. However, in active pain states, an enhancer may obviate the use of a vanilloid agonist.
In conclusion, the finding that a well-described compound class, i.e. the DHPs, can modulate TRPV1 channels in a positive fashion represents a useful lead for future pain therapeutics. This study has focused on the SAR of the novel DHP enhancers, which differs considerably from their known action at other target sites, such as L-type calcium channels and adenosine receptors. In future studies it will be desirable to further separate the activity of the TRPV1 enhancers from activity at the purine receptors. The present study revealed that 6-phenyl-, 3-O/S-ester-, and small alkyl-substitutions at the 2 and 4 positions of the DHP pharmacophore are required for the maximal enhancing effect. The development of analogues that are more potent and selective in their action at the TRPV1 channel and the validation of this approach in pain models, including in vivo studies, will be required.
Experimental Procedures
Chemical Synthesis
Materials and Instrumentation
Ethyl-3-aminocrotonate, aldehydes, ethylacetoacetate, 2,2,6-trimethyl-4H-1,3-dioxin-4-one, all acid chlorides, 2,2-dimethyl-1,3-dioxane-4,6-dione, ethanethiol, propanethiol, and other general reagents were purchased from Aldrich (Milwaukee, WI), and synthetic intermediates were of analytical grade. Compounds 7, 8, 12, 18, and 21 – 23 were prepared as reported by Li, et al.34 All other materials were obtained from commercial sources. Proton nuclear magnetic resonance spectroscopy was performed on a Varian GEMINI-300 spectrometer, and all spectra were obtained in CDCl3. Chemical shifts (δ) relative to TMS are given. Chemical ionization (CI) mass spectrometry was performed with a Finnigan 4600 mass spectrometer, and electron-impact (EI) mass spectrometry with a VG7070F mass spectrometer at 6kV. High-resolution FAB (fast atom bombardment) mass spectra were taken with a JEOL SX102 spectrometer using nitrobenzoic acid as matrix. cLog p values were calculated using ChemDraw Ultra, version 11.0.
General Procedure for preparation of substituted 1,4-DHP derivatives
The synthesis of DHP derivatives was carried out by known methods.29,34 The methods shown in Scheme 1 and Scheme 2 were used depending on the substitution on C-6 position of DHP. Equimolar amounts (0.5–1.0 mmol) of the appropriate β-enaminoester (33, 38, or 58), aldehyde (36 or 40), and β-ketoester (34, 35, or 37) were dissolved in 2–5 mL of absolute ethanol. The mixture was sealed in a Pyrex tube and heated, with stirring, to 80–90°C for 18–36 h. After the mixture was cooled to room temperature, the solvent was evaporated, and the residue was purified using preparative TLC (silica 60, Aldrich; petroleum ether-ethyl acetate (5:1-10:1)). The products were shown to be homogeneous by analytical TLC and were stored at −20°C.
3,5-Diethyl-2-methyl-4-ethyl-6-(3-chlorophenyl)-1,4-(±)-dihydropyridine-3,5-dicarboxylate, 9
C20H24ClNO4 1H NMR (CDCl3) 7.17-7.41 (m, 4H), 5.57 (s, 1H), 4.15-4.24 (m, 2H), 4.03 (t, J = 5.7 Hz, 1H), 3.94 (q, J = 7.1 Hz, 2H), 2.32 (s, 3H), 1.47- 1.54 (m, 2H), 1.31 (t, J = 7.2 Hz, 3H), 0.86-0.97 (m, 6H) 348.2 (M-ethyl, base). Anal. (C20H24ClNO4) C, H, N: calcd 63.57, 6.40, 3.71; found 63.29, 6.59, 3.53.
3,5-Diethyl-2-methyl-4-ethyl-6-(3-fluorophenyl)-1,4-(±)-dihydropyridine-3,5-dicarboxylate, 10
C20H24FNO4 1H NMR (CDCl3) 6.95-7.41 (m, 4H), 5.58 (s, 1H), 4.18-4.24 (m, 2H), 4.03 (t, J = 5.7 Hz, 1H), 3.93 (q, J = 7.1 Hz, 2H), 2.32 (s, 3H), 1.47-1.54 (m, 2H), 1.32 (t, J = 7.2 Hz, 3H), 0.86-0.97 (m, 6H) MS : 332.2 (M-ethyl, base). Anal. (C20H24FNO4) C, H, N: calcd 66.47, 6.69, 3.88; found 66.39, 6.69, 3.91.
3,5-Diethyl-2-methyl-4-ethyl-6-(4-fluorophenyl)-1,4-(±)-dihydropyridine-3,5-dicarboxylate, 11
C20H24FNO4 1H NMR (CDCl3) 7.26-7.31 (m, 2H), 7.05-7.11 (m, 2H), 5.57 (s, 1H), 4.18-4.24 (m, 2H), 4.02 (t, J = 5.6 Hz, 1H), 3.90-3.97 (m, 2H), 2.32 (s, 3H), 1.48-1.54 (m, 2H), 1.31 (t, J = 7.2 Hz, 3H), 0.96 (t, J = 7.2 Hz, 3H), 0.88 (t, J =7.5 Hz, 3H) MS : 362.2 (M+H), 332.2 (M-ethyl, base) Anal. (C20H24FNO4) C, H, N: calcd 66.47, 6.69, 3.88; found 66.41, 6.93, 3.76.
3-Ethylthio-5-ethyl-2-methyl-4-ethyl-6-(2-fluorophenyl)-1,4-(±)-dihydropyridine-3,5-dicarboxylate, 13
C20H24FNO3S 1H NMR (CDCl3) 7.31-7.34 (m, 1H), 7.01-7.19 (m, 3H), 5.62 (s, 1H), 4.16 (t, J = 5.4 Hz, 1H), 3.87-3.94 (m, 2H), 2.87 (q, J = 7.5 Hz, 2H), 2.25 (s, 3H), 1.47-1.57 (m, 2H), 1.22 (t, J = 7.4 Hz, 3H), 0.82-0.92 (m, 6H) MS : 348.2 (M-ethyl, base). Anal. (C20H24FNO3S) C, H, N: calcd 63.64, 6.41, 3.71; found 63.47, 6.39, 3.64.
3-Ethylthio-5-ethyl-2-methyl-4-ethyl-6-(3-fluorophenyl)-1,4-(±)-dihydropyridine-3,5-dicarboxylate, 14
C20H24FNO3S 1H NMR (CDCl3) 7.34-7.38 (m, 1H), 7.01-7.12 (m, 3H), 5.72 (s, 1H), 4.18 (t, J = 5.7 Hz, 1H), 3.93-4.01 (m, 2H), 2.94 (q, J = 7.2 Hz, 2H), 2.33 (s, 3H), 1.54-1.60 (m, 2H), 1.26-1.32 (m, 3H), 0.89-0.99 (m, 6H). HRMS calcd for C20H25FNO3S 378.1539, found 378.1525.
3-Thioethyl-5-ethyl-2-methyl-4-ethyl-6-(4-fluorophenyl)-1,4-(±)-dihydropyridine-3,5-dicarboxylate, 15
C20H24FNO3S 1H NMR (CDCl3) 7.27-7.32 (m, 2H), 7.06-7.12 (m, 2H), 5.69 (s, 1H), 4.18 (t, J = 6.0 Hz, 1H), 3.93-4.00 (m, 2H), 2.94 (q, J = 7.4 Hz, 2H), 2.33 (s, 3H), 1.53-1.60 (m, 2H), 1.29 (t, J = 7.2 Hz, 3H), 0.89-1.02 (m, 6H) MS : 348.2 (M-Et) base, 316.2 (M-Et-S). Anal. (C20H24FNO3S) C, H, N: calcd 63.64, 6.41, 3.71; found 63.49, 6.43, 3.58.
3-Ethylthio-5-ethyl-2-methyl-4-ethyl-6-(4-cyanophenyl)-1,4-(±)-dihydropyridine-3,5-dicarboxylate, 16
C21H24N2O3S 1H NMR (CDCl3) 7.69 (d, J = 8.1 Hz, 2H), 7.43 (d, J = 8.1 Hz, 2H), 5.71 (s, 1H), 4.19 (t, J = 5.9 Hz, 1H), 3.94-3.99 (m, 2H), 2.95 (q, J = 7.5 Hz, 2H), 2.32 (s, 3H), 1.56-1.61 (m, 2H), 1.29 (t, J = 7.4 Hz, 3H), 0.89-1.02 (m, 6H) MS : 355.2 (M-Et), 323.2 (M-EtS). HRMS calcd for C21H24N2O3NaS 407.1403, found 407.1385.
3-Ethylthio-5-ethyl-2-methyl-4-ethyl-6-cyclohexyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate, 17
C20H31NO3S 1H NMR (CDCl3) 5.81 (s, 1H), 4.08-4.27 (m, 3H), 3.72-3.86 (m, 1H), 2.90 (q, J = 7.2 Hz, 2H), 2.30 (s, 3H), 1.78-1.81 (m, 4H), 1.15-1.43 (m, 14H), 0.77 (t, J = 7.7 Hz, 3H) 336.3 (M-ethyl, base). HRMS calcd for C20H31NO3SNa 388.1922, found 388.1914.
3-Ethylthio-5-ethyl-2-methyl-4-propyl-6-(3-chlorophenyl)-1,4-(±)-dihydropyridine-3,5-dicarboxylate, 18
C21H26ClNO3S 1H NMR (CDCl3) 7.17-7.41 (m, 4H), 5.74 (s, 1H), 4.19 (t, J = 5.9 Hz, 1H), 3.98 (q, J = 7.2 Hz, 2H), 2.92 (q, J = 7.5 Hz, 2H), 2.32 (s, 3H), 1.26-1.55 (m, 7H), 0.89-1.17 (m, 6H) MS : 364.2 (M-Propyl), 346.2 (M-EtS). Anal. (C21H26ClNO3S) C, H, N: calcd 61.83, 6.42, 3.43; found 61.56, 6.47, 3.33.
3-Ethylthio-5-ethyl-2-methyl-4-cyclopropyl-6-(4-fluorophenyl)-1,4-(±)-dihydropyridine-3,5-dicarboxylate, 19
C21H24FNO3S 1H NMR (CDCl3) 7.28-7.33 (m, 2H), 7.07 - 7.13 (m, 2H), 5.74 (s, 1H), 3.94-4.03 (m, 3H), 2.95 (q, J = 7.2 Hz, 2H), 2.35 (s, 3H), 1.29 (t, J = 7.2 Hz, 3H), 1.01 (t, J = 7.2 Hz, 3H), 0.25-0.45 (m, 5H) 348.2 (M-cyclopropyl), 328.2 (M-EtS). Anal. (C21H24FNO3S) C, H, N: calcd 64.76, 6.21, 3.60; found 64.85, 6.22, 3.54.
3,5-Diethyl-2-methyl-4-ethyl-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate, 23
C21H27NO4S 1H NMR (CDCl3) 7.37-7.41 (m, 1H), 7.07-7.26 (m, 3H), 5.87 (s, 1H), 4.20 (t, J =5.4 Hz, 1H), 3.91-3.95 (m, 2H), 3.55 (m, 2H), 3.14 (m, 2H), 2.32 (s, 3H), 1.59 (m, 2H), 1.31-1.38 (m, 6H), 0.90-0.95 (m, 6H). m.p. 118-119°C. Anal. (C21H27NO4S) C, H, N: calcd C, 64.75; H, 6.99; N, 3.60; found C, 64.81; H, 7.05; N, 3.43.
3-Isopropylthio-5-ethyl-2-methyl-4-ethyl-6-(2-fluorophenyl)-1,4-(±)-dihydropyridine-3,5-dicarboxylate, 24
C21H26FNO3S 1H NMR (CDCl3) 7.37-7.41 (m, 1H), 7.07-7.26 (m, 3H), 5.71 (s, 1H), 4.19 (t, J =5.4 Hz, 1H), 3.94-4.00 (m, 2H), 3.65-3.70 (m, 1H), 2.31 (s, 3H), 1.56-1.61 (m, 2H), 1.31-1.38 (m, 6H), 0.89- 0.99 (m, 6H). HRMS calcd for C21H26FNO3SNa 414.1515, found 414.1516.
3-Thiobenzyl-5-ethyl-2-methyl-4-ethyl-6-(2-fluorophenyl)-1,4-(±)-dihydropyridine-3,5-dicarboxylate, 25
C25H26FNO3S 1H NMR (CDCl3) 7.07-7.41 (m, 9H), 5.74 (s, 1H), 4.17-4.23 (m, 3H), 3.92-4.00 (m, 2H), 2.34 (s, 3H), 1.20-1.22 (m, 2H), 0.87-0.98 (m, 6H). MS : 440.1 (M+H), 462.1 (M+Na) HRMS calcd for C25H26FNO3SNa 462.1515, found 462.1514.
3-(2-Phenylethylthio)-5-ethyl-2-methyl-4-propyl-6-phenyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate, 26
C27H31NO3S 1H NMR (CDCl3) 7.21-7.41 (m, 10H), 5.84 (s, 1H), 4.17-4.22 (m, 1H), 3.89-3.97 (m, 2H), 3.13-3.19 (m, 2H), 2.88-2.93 (t, J = 7.2 Hz, 2H), 2.33 (s, 3H), 1.31-1.58 (s, 4H), 0.88-0.97 (m, 6H). HRMS calcd for C27H31NO3NaS 472.1922, found 472.1932.
3-(2-Phenylethylthio)-5-ethyl-2-methyl-4-ethyl-6-(2-fluorophenyl)-1,4-(±)-dihydropyridine-3,5-dicarboxylate, 27
C26H28FNO3S 1H NMR (CDCl3) 8.27 (d, J = 8.7 Hz, 2H), 7.47-7.50 (m, 2H), 7.23-7.29 (m, 5H), 5.64 (s, 1H), 4.21 (t, J = 5.7 Hz, 1H), 3.93-4.14 (m, 2H), 3.17-3.22 (m, 2H), 2.83-2.94 (m, 2H), 2.34 (s, 3H), 1.53-1.60 (m, 2H), 0.90-1.03 (m, 6H) HRMS calcd for C26H28FNO3SNa 476.1672, found 476.1685.
3-(2-Phenylethylthio)-5-ethyl-2-methyl-4-ethyl-6-(3-chlorophenyl)-1,4-(±)-dihydropyridine-3,5-dicarboxylate, 28
C26H28ClNO3S 1H NMR (CDCl3) 7.17-7.41 (m, 9H), 5.75 (s, 1H), 4.18 (t, J = 5.4 Hz, 1H), 3.96 (q, J = 7.2 Hz, 2H), 3.13-3.19 (m, 2H), 2.87-2.93 (m, 2H), 2.33 (s, 3H), 1.55-1.59 (m, 2H), 0.88-0.98 (m, 6H). HRMS calcd for C26H29ClNO3S 470.1557, found 470.1573.
3-(2-Phenylethylthio)-5-ethyl-2-methyl-4-ethyl-6-(4-fluorophenyl)-1,4-(±)-dihydropyridine-3,5-dicarboxylate, 29
C26H28FNO3S 1H NMR (CDCl3) 7.06-7.33 (m, 9H), 5.71 (s, 1H), 4.18 (t, J = 5.7 Hz, 1H), 3.93-4.18 (m, 2H), 3.14-3.19 (m, 2H), 2.90 (t, J = 7.8 Hz, 2H), 2.33 (s, 3H), 1.53-1.60 (m, 2H), 0.99 (t, J = 7.1 Hz, 3H), 0.91(t, J = 7.4 Hz, 3H) HRMS calcd for C26H28FNO3SNa 476.1672, found 476.1671.
3-(2-Phenylethylthio)-5-ethyl-2-methyl-4-ethyl-6-(4-nitrophenyl)-1,4-(±)-dihydropyridine-3,5-dicarboxylate, 30
C26H28N2O5S 1H NMR (CDCl3) 8.27 (d, J = 8.7 Hz, 2H), 7.49 (d, J = 8.7Hz, 2H), 7.23-7.32 (m, 5H), 5.67 (s, 1H), 4.21 (t, J = 5.7 Hz, 1H), 3.96-4.01 (m, 2H), 3.15-3.22 (m, 2H), 2.89-2.94 (m, 2H), 2.23 (s, 3H), 1.56-1.62 (m, 2H), 1.01 (t, J = 7.2 Hz, 3H), 0.93 (t, J = 7.5 Hz, 3H). HRMS calcd for C26H29N2O5S 481.1797, found 481.1789.
3-(2-Benzylthio)-5-ethyl-2-methyl-4-ethyl-6-(4-nitrophenyl)-1,4-(±)-dihydropyridine-3,5-dicarboxylate, 31
C25H26N2O5S 1H NMR (CDCl3) 8.26 (d, J = 9.0 Hz, 2H), 7.49 (d, J = 9.0 Hz, 2H), 7.22-7.37 (m, 5H), 5.69 (s, 1H), 4.15-4.21 (m, 3H), 3.94-3.99 (m, 2H), 2.35 (s, 3H), 1.28-1.62 (m, 2H), 0.88-1.02 (m, 6H). HRMS calcd for C26H29N2O5S 489.1460, found 489.1461.
3-Ethylthio-5-ethyl-2-methyl-4-ethyl-6-(2-naphthyl)-1,4-(±)-dihydropyridine-3,5-dicarboxylate, 32
C24H27NO3S 1H NMR (CDCl3) 7.77-7.87 (m, 4H), 7.51-7.53 (m, 2H), 7.38-7.42 (dd, J = 1.5, 8.4 Hz, 1H), 5.86 (s, 1H), 4.23 (t, J = 5.4 Hz, 1H), 3.94(m, 2H), 2.96(q, J = 7.5 Hz, 2H), 2.36 (s, 3H), 1.58- 1.66 (m, 2H), 1.22-1.30 (m, 3H), 0.83-0.99 (m, 6H). HRMS calcd for C24H27NO3SNa 432.1609, found 489.1596.
General Procedure for preparation of β-amino-α,β -unsaturated esters
A β-ketoester (3 mmol) and ammonium acetate (4.5 mmol) were mixed in 5 mL of absolute ethanol and refluxed at 80°C for 24 h. The solvent was removed and the residue was chromatographed to give the desired compounds in moderate yields.
Synthesis of β-ketoesters and β-ketothioesters
β-Ketoesters 34 and β-ketothioesters 35 were prepared by the reaction of 2,2,6-trimethyl-4H-1,3- dioxin-4-one 41 and an alcohol or a thiol. Equimolar amounts of 2,2,6-trimethyl-4H-1,3-dioxin-4-one and an alcohol or a thiol were heated with a small volume of toluene at 100°C in a sealed tube overnight. After the mixture was cooled to room temperature, the solvent was removed under reduced pressure and the residue was purified by chromatography in satisfactory yields.
Synthesis of β-ketoesters via Meldrum’s acid
The preparation of ethyl–2-fluorobenzoylacetate 34 (R = 2-F-Ph) is provided as an example. 2,2- Dimethyl-1,3-dioxane-4,6-dione (Meldrum’s acid, 42) and 2-fluorobenzoyl chloride were dissolved in dry CH2Cl2. At 0°C, 4-(dimethylamino)pyridine was added. The reaction mixture was stirred at 0°C for 1 h and then at room temperature for an additional hour. The reaction mixture was washed with 1 N HCl and water and then dried with anhydrous Na2SO4. Removal of the solvent left the desired product, which was directly used for the next reaction without purification. This acylated Meldrum’s acid and ethanol were mixed in toluene. This mixture was heated at 90°C in a flask with an effective reflux condenser for 24 h. The solvent and the excess ethanol were removed and the residue was purified by column chromatography to give the desired product.
Pharmacological Methods
Cell Culture
A TRPV1ε-NIH3T3 cell line, ectopically expressing the C-terminally ε-epitope tagged vanilloid receptor 1, was cultured as described previously.13,19 Cells were seeded in DMEM + 10% FBS one day before use and cultured at 35°C in a humidified incubator. Mixed primary cultures from the DRG of rat embryos were prepared and characterized similar to the procedures employed in earlier Ca2+-transport and imaging studies.13,19,22 Briefly, cells were harvested from E15 rat embryos, enzymatically dissociated, and then plated onto 96-well microtiter plates pre-coated with poly-D-lysine and mouse laminin. DRGs were maintained in culture at 35°C for 5–6 days before assaying.
Vanilloid-induced 45Ca-Uptake
Assays were performed using an automated high-throughput liquid handling workstation (Biomek FX, Beckman Coulter) equipped with a 96-well pipeting head. Compounds were initially screened for calcium transport activity using the NIH3T3 cells stably expressing TRPV1ε. The ED100 for capsaicin activation of TRPV1 in TRPV1ε-NIH3T3 cells was determined to be ~2 µM at pH 7.5. Comparisons to the parental NIH3T3 cell line established the specificity of the capsaicin activation.13,22 Assays with DRG cells were similar, but due to their increased sensitivity, the optimal capsaicin concentration for DRG activation was determined to be ~0.5 µM.
Immediately before assay, cells were adapted to room temperature (20°C) for 5 min in 25 mM Tris-HCl (pH 7.5) buffered Hank’s balanced salt solution, supplemented with 1 mM sodium ascorbate, 10 µM CaCl2 and 0.8 mM MgCl2 (HCM). 45Ca2+-uptake was performed for 10 min at 20°C in HCM using 0.5 µCi/well 45Ca2+ (MP Biomedicals) as radioactive tracer in 100 µL/well final volume. To stop 45Ca2+-uptake, the assay mix was removed, and the cells were rapidly changed back into HCM, washed 5 additional times with 100 µL/well HCM, and then lysed in 100 µL/well RIPA buffer (50 mM Tris- HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.5% deoxycholate, 0.1% SDS, 5 mM EDTA) for 30 min. Aliquots (80 µL) of the solubilized cell extracts were transferred to an OptiPlate-96 (PerkinElmer, Inc.) containing 120 µL/well Microscint-40 (PerkinElmer, Inc.), sealed, mixed on a plate shaker for 15 min, and then counted in a microplate liquid scintillation counter (TopCount NXT, PerkinElmer, Inc.).
DMSO Solvent Sensitivity
In primary DRG cultures, we found a noticeable concentration dependent increase of capsaicin-induced calcium influx from the DMSO solvent (Fig. S1, Supporting Information). During our initial screening in the TRPV1ε-NIH3T3 cells, DHP stock solutions were formulated to a concentration of 5 mM, and the DMSO concentration in the assay buffer was kept constant at 2% (since the highest DHP concentration evaluated was 100 µM). This DMSO concentration did not affect results with the TRPV1ε-NIH3T3 cells, but it was found to greatly increase the baseline capsaicin activation in DRGs. Therefore, for the DRG assays, we reformulated more concentrated stock solutions (25 mM) from the remaining DHP powder. Due to limited quantities, we were unable to re-assay all DHPs in the primary DRG cells. The maximum DHP concentration evaluated in DRGs was 50 µM, with a corresponding DMSO concentration of 0.2% in the assay buffer.
Dose Response Curves
Data was analyzed using GraphPad Prism software. Raw counts from each assay were normalized as percentages to in-plate controls that contained only capsaicin (CPM [X]DHP/CPM capsaicin-only × 100). Capsaicin-only activation (Emax) was designated as 100% (MEAN of 4 wells). Data was plotted as MEAN ± SEM (%) vs. Log[Enhancer] (M), and then fit using a standard non-linear dose-response algorithm supplied by the GraphPad software (“Bottom to Top, variable slope”; Y = Bottom + (Top – Bottom)/1 + 10((logEC50 – X) × Hill Slope)). The EC50, Hill Slope and ΔEmax were extracted from data that could be curve fitted, typically with r2 ≥ 0.90, in the concentration range evaluated. For compounds that showed activity at only the higher concentrations, and therefore were not closely sigmoidal in this range, only the ΔEmax at the highest concentration ([Enhancer]max) is shown (see Fig. S2, Supporting Information, for plots of all assay runs in TRPV1ε-NIH3T3 cells).
Intracellular Ca2+ Measurements
Adapted from published methods with a few changes.51 Primary DRG neurons (prepared as described above) were plated onto poly-ornithine/laminin (Sigma) coated 25-mm diameter coverslips and cultured 4 to 6 days before measurement. Cells were loaded with 2.5 µM of the high-Kd calcium indicator Fura-4F AM ester (Molecular Probes) in the presence of 0.0025% pluronic in normal perfusion medium (in mM: 2.5 CaCl2, 10 glucose, 10 Hepes, 3 KCl, 0.6 MgCl2, 130 NaCl, pH adjusted with 1 M Tris base to 7.4, osmolality adjusted with sucrose to 325 mosmol/L) for 20 min at room temperature, followed by a wash and further incubation in perfusion medium for an additional 20 min. The coverslip was inverted and mounted onto a closed flow-through perfusion chamber (Warner Instruments) with a nominal bath volume of 358 µL, then perfused continuously at a rate of 0.5 mL/min using a peristaltic pump (Minipuls 3, Gilson). All experiments were performed at room temperature.
Fluorescence data were acquired with MetaFluor software (Molecular Devices) via a CCD camera (ORCA-ER, Hamamatsu) connected to an upright microscope (BX60, Olympus) using a 20× water-immersion objective. The ratio of fluorescence emission (510 nm) in response to 340/380 nm excitation, controlled by a filter changer (Lambda 10-2, Sutter Instruments), was acquired continuously at 0.5 Hz. Drugs were administered via an in-line manual sample injector (Rheodyne) equipped with a 500 µL sample loop. During experiments, the perfusion medium was supplemented with 1 mM ascorbate. Capsaicin (1 mM EtOH stock) was diluted to 200 nM, and 23 (25 mM DMSO stock) was diluted to 10 µM. Capsaicin-only experiments were supplemented with an equivalent amount of DMSO (0.04%). Cells were recorded for 17 min total: baseline for 2 min, followed by a 15-min recovery. Drugs were administered for 30 sec beginning at 2 min (designated as a horizontal bar in Fig. 3B).
Activated cells were individually identified and their corresponding 340/380 ratios measured using the MetaFluor analysis software. Data from activated cells on six separate coverslips (3 with capsaicin only; 3 with capsaicin plus enhancer), from a total of 98 cells analyzed in capsaicin only conditions and 69 cells analyzed when capsaicin plus enhancer were administered, were normalized to baseline and plotted as a function of time. The traces were normalized to baseline by dividing by the mean of the first 2 min. Mean calculations and statistical analysis were performed using Excel and SigmaPlot software, and plots were generated using GraphPad Prism software. Peak ratio was determined from the mean of 10 data points located at the peak response, determined qualitatively.
Supplementary Material
Figure S1. Capsaicin activation of primary dorsal root ganglion cells is enhanced with DMSO alone. For this reason, we formulated higher concentration DHP stocks to minimize the influence of the solvent during assays with DRG cells.
{kind=link}
Figure S2. Plots of individual assay runs for DHPs evaluated for enhancer activity on TRPV1ε-NIH/3T3 cells. Each plot shows normalized 45Ca2+ influx vs. log [DHP]. Calcium influx was normalized to in-plate controls of activation with capsaicin alone, designated as 100%. Assays contained duplicate wells at each point in the DHP concentration range. Additional Information on Chemical Synthesis of DHP Analogues.
{kind=link}
Acknowledgments
This research was supported in part by the Intramural Research Programs of the NIDDK and NIDCR, National Institutes of Health. We thank Dr. William Trenkle for advice on the chemical synthesis of compound 23 and Dr. Herman Yeh for the assistance with the NMR characterization.
Abbreviations
- DHP
1,4-dihydropyridine
- DMEM
Dulbecco modified Eagle’s medium’
- DRG
dorsal root ganglion
- ER
endoplasmic reticulum
- FAB
fast atom bombardment
- FBS
fetal bovine serum
- NSAID
nonsteroidal antiinflammatory drug
- RTX
resiniferatoxin
- SAR
structure activity relationship
- TLC
thin layer chromatography
- TMS
tetramethylsilane
- TRP
transient receptor potential
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information Available: Effect of DMSO on capsaicin activation of primary DRG cells, plots of individual assay runs for enhancers TRPV1ε-NIH/3T3 cells.
References
- 1.Jordt SE, Julius D. Cell. 2002;108:421. doi: 10.1016/s0092-8674(02)00637-2. [DOI] [PubMed] [Google Scholar]
- 2.Szallasi A, Cortright DN, Blum CA, Eid SR. Nat. Rev. Drug Discov. 2007;6:357. doi: 10.1038/nrd2280. [DOI] [PubMed] [Google Scholar]
- 3.Iadarola MJ, Max MB, Berman KF, Byas-Smith MG, Coghill RC, Gracely RH, Bennett GJ. Pain. 1995;63:55. doi: 10.1016/0304-3959(95)00015-K. [DOI] [PubMed] [Google Scholar]
- 4.Gunthorpe MJ, Rami HK, Jerman JC, Smart D, Gill CH, Soffin EM, Luis Hannan S, Lappin SC, Egerton J, Smith GD, Worby A, Howett L, Owen D, Nasir S, Davies CH, Thompson M, Wyman PA, Randall AD, Davis J. Neuropharmacology. 2004;46:133. doi: 10.1016/s0028-3908(03)00305-8. [DOI] [PubMed] [Google Scholar]
- 5.Sauerberg P, Olesen PH, Sheardown MJ, Rimvall K, Thogersen H, Shannon HE, Sawyer BD, Ward JS, Bymaster FP, DeLapp NW, Calligaro DO, Swedberg MD. J. Med. Chem. 1998;41:109. doi: 10.1021/jm9705216. [DOI] [PubMed] [Google Scholar]
- 6.Wang Y, Szabo T, Welter JD, Toth A, Tran R, Lee J, Kang SU, Suh YG, Blumberg PM. Mol. Pharmacol. 2002;62:947. doi: 10.1124/mol.62.4.947. [DOI] [PubMed] [Google Scholar]
- 7.Szallasi A, Appendino G. J. Med. Chem. 2004;47:2717. doi: 10.1021/jm030560j. [DOI] [PubMed] [Google Scholar]
- 8.Krause JE, Chenard BL, Cortright DN. Curr. Opin. Investig. Drugs. 2005;6:48. [PubMed] [Google Scholar]
- 9.Levine JD, Alessandri-Haber N. Biochim. Biophys. Acta - Molecular Basis of Disease. 2007;1772:989. doi: 10.1016/j.bbadis.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 10.Hucho T, Levine JD. Neuron. 2007;55:365. doi: 10.1016/j.neuron.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 11.Abernethy DR, Schwartz JB. Clin Pharmacokinet. 1988;15:1. doi: 10.2165/00003088-198815010-00001. [DOI] [PubMed] [Google Scholar]
- 12.Huang SM, Bisogno T, Trevisani M, Al-Hayani A, De Petrocellis L, Fezza F, Tognetto M, Petros TJ, Krey JF, Chu CJ, Miller JD, Davies SN, Geppetti P, Walker JM, Di Marzo V. Proc. Natl. Acad. Sci. U. S. A. 2002;99:8400. doi: 10.1073/pnas.122196999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karai L, Russell JT, Iadarola MJ, Olah Z. J. Biol. Chem. 2004;279:16377. doi: 10.1074/jbc.M310891200. [DOI] [PubMed] [Google Scholar]
- 14.Lishko PV, Procko E, Jin X, Phelps CB, Gaudet R. Neuron. 2007;54:905. doi: 10.1016/j.neuron.2007.05.027. [DOI] [PubMed] [Google Scholar]
- 15.Grycova L, Lansky Z, Friedlova E, Vlachova V, Kubala M, Obsilova V, Obsil T, Teisinger J. Arch. Biochem. Biophys. 2007;465:389–398. doi: 10.1016/j.abb.2007.06.035. [DOI] [PubMed] [Google Scholar]
- 16.Malmberg AB, Mizisin AP, Calcutt NA, von Stein T, Robbins WR, Bley KR. Pain. 2004;111:360. doi: 10.1016/j.pain.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 17.Karai L, Brown DC, Mannes AJ, Connelly ST, Brown J, Gandal M, Wellisch OM, Neubert JK, Olah Z, Iadarola MJ. J. Clin. Invest. 2004;113:1344. doi: 10.1172/JCI20449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown DC, Iadarola MJ, Perkowski SZ, Hardem E, Shofer F, Karai L, Olah Z, Mannes AJ. Anesthesiology. 2005;103:1052. doi: 10.1097/00000542-200511000-00020. [DOI] [PubMed] [Google Scholar]
- 19.Olah Z, Szabo T, Karai L, Hough C, Fields RD, Caudle RM, Blumberg PM, Iadarola MJ. J. Biol. Chem. 2001;276:11021. doi: 10.1074/jbc.M008392200. [DOI] [PubMed] [Google Scholar]
- 20.Caudle RM, Karai L, Mena N, Cooper BY, Mannes AJ, Perez FM, Iadarola MJ, Olah Z. Neurotoxicology. 2003;24:895. doi: 10.1016/S0161-813X(03)00146-3. [DOI] [PubMed] [Google Scholar]
- 21.Neubert JK, Kim H-S, Jun JH, Karai L, Iadarola MJ. Pain. 2003;104:219. doi: 10.1016/s0304-3959(03)00009-5. [DOI] [PubMed] [Google Scholar]
- 22.Olah Z, Karai L, Iadarola MJ. J. Biol. Chem. 2001;276:31163. doi: 10.1074/jbc.M101607200. [DOI] [PubMed] [Google Scholar]
- 23.Han P, McDonald HA, Bianchi BR, El Kouhen R, Vos MH, Jarvis MF, Faltynek CR, Moreland RB. Biochem. Pharmacol. 2007;73:1635. doi: 10.1016/j.bcp.2006.12.035. [DOI] [PubMed] [Google Scholar]
- 24.Turner H, Fleig A, Stokes A, Kinet JP, Penner R. Biochem. J. 2003;371:341. doi: 10.1042/BJ20021381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu M, Liu MC, Magoulas C, Priestley JV, Willmott NJ. J. Biol. Chem. 2003;278:5462. doi: 10.1074/jbc.M209111200. [DOI] [PubMed] [Google Scholar]
- 26.Wisnoskey BJ, Sinkins WG, Schilling WP. Biochem. J. 2003;372:517. doi: 10.1042/BJ20021574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cortright DN, Krause JE, Broom DC. Biochim. Biophys. Acta. 2007;1772:978. doi: 10.1016/j.bbadis.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 28.Chung MK, Güler AD, Caterina MJ. Nat. Neurosci. 2008;11:555. doi: 10.1038/nn.2102. [DOI] [PubMed] [Google Scholar]
- 29.Stout DM, Myers AI. Chem. Rev. 1982;82:223. [Google Scholar]
- 30.van Rhee AM, Jiang J-l, Melman N, Olah ME, Stiles GL, Jacobson KA. J. Med. Chem. 1996;39:2980. doi: 10.1021/jm9600205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang J-l, van Rhee AM, Melman N, Ji Xd, Jacobson KA. J. Med. Chem. 1996;39:4667–4675. doi: 10.1021/jm960457c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang J-l, van Rhee AM, Chang L, Patchornik A, Evans P, Melman N, Jacobson KA. J. Med. Chem. 1997;40:2596–2608. doi: 10.1021/jm970091j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li A-H, Moro S, Forsyth N, Melman N, Ji X-d, Jacobson KA. J. Med. Chem. 1999;42:706. doi: 10.1021/jm980550w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li A-H, Moro S, Melman N, Ji X-d, Jacobson KA. J. Med. Chem. 1998;41:3186. doi: 10.1021/jm980093j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jacobson KA, Kim YC, King BF. J. Auton. Nerv. Syst. 2000;81:152. doi: 10.1016/s0165-1838(00)00128-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harper JL, Camerini-Otero CS, Li AH, Kim SA, Jacobson KA, Daly JW. Biochem. Pharmacol. 2003;65:329. doi: 10.1016/s0006-2952(02)01488-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leffler A, Fischer MJ, Rehner D, Kienel S, Kistner K, Sauer SK, Gavva NR, Reeh PW, Nau C. J. Clin. Invest. 2008;118:763. doi: 10.1172/JCI32751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Binshtok AM, Bean BP, Woolf CJ. Nature. 2007;449:607. doi: 10.1038/nature06191. [DOI] [PubMed] [Google Scholar]
- 39.Neubert JK, Mannes AJ, Karai L, Jenkins AC, Zawatski L, Abu-Asab M, Iadarola MJ. Mol. Pain. 2008;4:3. doi: 10.1186/1744-8069-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mannes AJ, Iadarola MJ. Nat. Clin. Pract. Neurol. 2007;3:648. doi: 10.1038/ncpneuro0660. [DOI] [PubMed] [Google Scholar]
- 41.Iadarola MJ, Brady LS, Draisci G, Dubner R. Pain. 1988;35:313. doi: 10.1016/0304-3959(88)90141-8. [DOI] [PubMed] [Google Scholar]
- 42.Neubert JK, Rossi HL, Malphurs W, Vierck CJ, Jr, Caudle RM. Behav. Brain. Res. 2006;170:308. doi: 10.1016/j.bbr.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 43.Vlachova V, Teisinger J, Suankova K, Lyfenko A, Ettrich R, Vyklicky L. J. Neurosci. 2003;23:1340. doi: 10.1523/JNEUROSCI.23-04-01340.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kedei N, Szabo T, Lile JD, Treanor JJ, Olah Z, Iadarola MJ, Bumberg PM. J. Biol. Chem. 2001;276:28613. doi: 10.1074/jbc.M103272200. [DOI] [PubMed] [Google Scholar]
- 45.Numazaki M, Tominaga T, Toyooka H, Tominaga M. J. Biol. Chem. 2002;277:13375. doi: 10.1074/jbc.C200104200. [DOI] [PubMed] [Google Scholar]
- 46.Bhave G, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW., 4th Neuron. 2002;35:721. doi: 10.1016/s0896-6273(02)00802-4. [DOI] [PubMed] [Google Scholar]
- 47.Pareek TK, Keller J, Kesavapany S, Agarwal N, Kuner R, Pant HC, Iadarola MJ, Brady RO, Kulkarni AB. Proc. Natl. Acad. Sci. U.S.A. 2007;104:660. doi: 10.1073/pnas.0609916104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ray WA, Stein CM, Daugherty JR, Hall K, Arbogast PG, Griffin MR. Lancet. 2002;360:1071. doi: 10.1016/S0140-6736(02)11131-7. [DOI] [PubMed] [Google Scholar]
- 49.LaMotte RH, Lundberg LE, Torebjork HE. J. Physiol. 1992;448:749. doi: 10.1113/jphysiol.1992.sp019068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sang CN, Gracely RH, Max MB, Bennett GJ. Anesthesiology. 1996;85:491. doi: 10.1097/00000542-199609000-00007. [DOI] [PubMed] [Google Scholar]
- 51.Lu S, Zhang X, Gold MS. J. Physiol. 2006;577:169. doi: 10.1113/jphysiol.2006.116418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tender GC, Walbridge S, Olah Z, Karai L, Iadarola M, Oldfield EH, Lonser RR. J. Neurosurg. 2005;102:522. doi: 10.3171/jns.2005.102.3.0522. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Capsaicin activation of primary dorsal root ganglion cells is enhanced with DMSO alone. For this reason, we formulated higher concentration DHP stocks to minimize the influence of the solvent during assays with DRG cells.
Figure S2. Plots of individual assay runs for DHPs evaluated for enhancer activity on TRPV1ε-NIH/3T3 cells. Each plot shows normalized 45Ca2+ influx vs. log [DHP]. Calcium influx was normalized to in-plate controls of activation with capsaicin alone, designated as 100%. Assays contained duplicate wells at each point in the DHP concentration range. Additional Information on Chemical Synthesis of DHP Analogues.