

Abstract
T cell-specific adapter protein (TSAd) and adapter protein in lymphocytes of unknown function (ALX) are two related src-homology-2 (SH2) domain-containing signaling adapter molecules that have both been shown to regulate TCR signal transduction in T cells. TSAd is required for normal TCR-induced synthesis of IL-2 and other cytokines in T cells and acts at least in part by promoting activation of the LCK protein tyrosine kinase at the outset of the TCR signaling cascade. By contrast, ALX functions as a negative-regulator of TCR-induced IL-2 synthesis through as yet undetermined mechanisms. Herein, we report a novel T cell-expressed adapter protein named SH2D4A that contains an SH2 domain that is highly homologous to the TSAd and ALX SH2 domains and which shares other structural features with these adapters. To examine the function of SH2D4A in T cells we produced SH2D4A-deficient mice by homologous recombination in embryonic stem cells. T cell development, homeostasis, proliferation and function were all found to be normal in these mice. Furthermore, knockdown of SH2D4A expression in human T cells did not impact upon their function. We conclude that in contrast to TSAd and ALX, SH2D4A is dispensable for TCR signal transduction in T cells.
Keywords: T cell, signal transduction, transgenic/knockout mice
Adapter proteins are central to the process of cellular signal transduction in eukaryotes (1, 2). By definition, adapter proteins lack catalytic activity. Nonetheless, they participate in signal transduction through formation of complexes with catalytically-active signaling molecules. Complex formation results in modulation of the enzymatic activity of catalytically-active molecules, directly or indirectly, and/or forces their juxtaposition to substrates. Physical interaction of adapters with other signaling molecules is mediated by modular binding domains and/or conserved peptide motifs within the adapter molecule.
Adapter proteins are known to play important roles in signal transduction through the TCR (3). Well studied adapter proteins in the canonical TCR signaling pathway include LAT, Grb-2, GADS, SLP-76, FYB and SKAP-55. In co-operation with protein kinases and phosphatases, phospholipases, guanine nucleotide exchange factors and other catalytically-active molecules, these adapters couple the TCR to the activation of transcription factors such as NFκB, NFAT, AP-1, T-bet and GATA-3. In turn, these transcription factors initiate programs of gene expression that drive T cell cytokine synthesis, proliferation and differentiation into effector or memory cells.
Another, relatively less well-studied, adapter in T cells is the T cell-specific adapter (TSAd)3 protein, also known as SH2D2A. TSAd is an intracellular signaling molecule that contains an NH2-terminal region of unknown function, a centrally-located Src-homology-2 (SH2) domain and a COOH region with conserved tyrosine residues in consensus phosphorylation motifs and a proline-rich region (4, 5). TSAd participates in TCR signal transduction at different levels. First, at the outset of TCR signal transduction, TSAd facilitates TCR-mediated activation of the LCK protein tyrosine kinase (PTK). In response to phosphorylation by LCK, TSAd physically associates with the SH2 and SH3 domains of LCK, thereby preventing the kinase from closing into an inactive conformation (6). Second, based upon the finding that TSAd is actively transported to the cell nucleus, TSAd likely also functions at a more distal point in the TCR signaling pathway, possibly functioning directly in the process of gene transcription (7, 8).
Evidence for the importance of TSAd in T cell activation comes from the finding that TSAd-deficient mice are susceptible to spontaneous and experimentally-induced lupus-like autoimmune disease (9). The cause of autoimmunity in TSAd-deficient mice is uncertain. Despite that TSAd is well expressed in thymocytes, T cell development is normal and no defects in positive or negative selection are apparent (9–11). In addition, no deficiencies in the generation or function of T regulatory cells (Treg), which normally protect against autoimmune disease in the periphery, are evident (unpublished observations). Thus far, the only defect that has been identified in TSAd-deficient mice that could account for autoimmunity is a relative resistance of T cells to antigen-induced death in vivo (9). As determined in gene profiling experiments, TSAd-deficient T cells synthesize substantially reduced amounts of mRNA for the T cell growth factor, IL-2 (9). Moreover, impaired IL-2 secretion by TSAd-deficient T cells has been demonstrated at the protein level in vitro and in vivo (9, 10). Potentially, impaired IL-2 secretion could account for resistance to cell death, although this has yet to be proven (12).
Related to TSAd is the adapter protein in lymphocytes of unknown function (ALX), also known as HSH2D (13, 14). Like TSAd, ALX contains an SH2 domain, tyrosine residues in consensus phosphorylation motifs and proline-rich regions. At the amino acid sequence level, the SH2 domains of TSAd and ALX are 65% homologous in mice. Upon over-expression, ALX has been shown to inhibit TCR and CD28 costimulatory receptor induction of IL-2 synthesis in the human Jurkat T leukemic cell line suggesting function as a negative-regulator of T cell signaling (14). In addition, over-expression of ALX in the WEHI-231 murine B lymphoma cell line was shown to protect cells from BCR-induced apoptosis suggesting a role for ALX as an anti-apoptotic molecule in this cell type (15, 16). However, the recent report of ALX-deficient mice indicates that ALX may be a more important regulator of T cell function than B cell function (17). Thus, T cells from ALX-deficient mice were shown to synthesize increased amounts of IL-2 in response to TCR and CD28 engagement which is consistent with a function for ALX as a negative-regulator of signal transduction in T cells. In contrast, serum antibody concentrations and B cell antibody responses to a nominal antigen were unaffected in ALX-deficient mice as was B cell expansion in response to a number of different stimuli. The mechanism by which ALX controls IL-2 synthesis in T cells is largely unknown but may be linked to an ability of ALX to regulate activation of the p38 mitogen-activated protein kinase.
During the course of our examination of the function of TSAd, we identified another SH2 domain-containing adapter protein with significant homology to TSAd and ALX. This ubiquitously-expressed protein, named SH2D4A, has not previously been studied in T cells or in any other cell type. To examine the function of this protein, we generated SH2D4A-deficient mice by homologous recombination in embryonic stem (ES) cells. In addition, we examined the effect of knockdown of SH2D4A expression in human T cells using the technique of RNA interference (RNAi). The effect of gene deletion or knockdown upon T cell responses was then examined.
Materials and Methods
Mice
All mice used in these studies were bred in-house. For experiments, unless otherwise noted, all mice were 2–3 mo of age at the time of sacrifice. All experiments were performed in compliance with University of Michigan guidelines and were approved by the University Committee on the Use and Care of Animals.
SH2D4A antiserum
Human SH2D4A cDNA was ligated into the BamHI and XhoI sites of pSMT3 (Invitrogen) to generate a construct encoding SH2D4A with an NH2-terminal SMT3 tag. The construct was transformed into BL21 cells (Stratagene) which were used to produce recombinant SH2D4A- SMT3 protein following induction with IPTG. Recombinant protein was purified from cell lysates with the use of Ni-NTA agarose beads (Qiagen) and was injected into rabbits with CFA to generate an SH2D4A antiserum (Pocono Rabbit Farm and Laboratory).
SH2D4A expression
Expression of SH2D4A in C57BL/6J mouse cells and human PBMC was determined by Western blotting. Mouse CD4+ T cells, CD8+ T cells and B cells were prepared by negative selection from whole splenocytes using Miltenyi kits. Bone marrow-derived macrophages and DC were prepared by growth of bone marrow cells in L-cell and GMCSF-conditioned medium respectively. Mouse T cells were stimulated or not with plate-bound (1 µg/ml) CD3 mAb (145-2C11; eBioscience) and 0.5 µg/ml of soluble CD28 mAb (37.5.1; BD Biosciences) in mouse complete medium (RPMI 1640 supplemented with 10% FCS, 2-ME and antibiotics). Murine B cells and bone marrow macrophages and DC were stimulated or not with LPS (1 µg/ml) in mouse complete medium. Human PBMC were stimulated or not with CD3 (OKT3) and CD28 (CD28.2) mAb (both BD Biosciences) (1 µg/ml each) in human complete medium (RPMI 1640 supplemented with 10% FCS and antibiotics). Following stimulation, cells were lysed in 1% NP-40 lysis buffer and aliquots of lysates were subject to Western analysis using an SH2D4A antiserum. NH2-terminal hemagglutinin (HA)-tagged murine or human SH2D4A were run as controls in experiments. For this purpose, SH2D4A cDNAs were cloned into the BamHI and XhoI sites of HA-pcDNA3.1 (Invitrogen) and constructs were transfected into Jurkat T leukemia cells (ATCC) by electroporation in a Gene Pulser (BioRad). Cell lysates, prepared 24 h after transfection, were run in Western blot experiments.
Fluorescence microscopy
Human SH2D4A cDNA was ligated into the BamHI and XhoI sites of pEGFP-N1 (Clontech) to generate a construct encoding SH2D4A with a COOH-terminal GFP tag. The construct was transfected in to 293T cells by electroporation using a Gene Pulser or into PBMC by nucleofection using an Amaxa nucleoporation device. After overnight culture, cells were spun onto glass slides, fixed and permeabilized in PBS, 3.75% formaldehyde, 0.1% Triton X-100 and stained with 1 µg/ml Hoechst 33258 (Molecular Probes). GFP and Hoechst fluorescence was observed by fluorescence microscopy on an Olympus IX70 microscope.
Gene targeting
The targeting vector for the generation of sh2d4a gene-targeted mice was constructed by inserting DNA fragments of a sh2d4a genomic BAC clone into p-loxP-2FRT-PGKneo (18). In this construct, exon 8 of sh2d4a was flanked by loxP sites and a FRT site-flanked neomycin-resistance (NeoR) selection cassette was inserted into intron 8 (see Fig. 3, further details are available upon request). Linearized vector was electroporated into the Bruce4 C57BL/6 ES cell line (19). Correctly targeted ES cell clones were identified by Southern blotting using indicated 5’ and 3’ probes and were subsequently injected into B6(Cg)-Tyrc-2J/ J blastocysts. Resultant chimeras were bred to B6(Cg)-Tyrc-2J/J mice to achieve germline transmission of the targeted sh2d4a allele. Heterozygotes were bred with congenic C57BL/6J transgenic mice expressing the Flp recombinase under the control of an actin promoter to delete the NeoR cassette (20). Resulting heterozygote NeoR-deleted mice were then bred with C57BL/6J mice expressing the Cre recombinase under the control of a CMV promoter, to delete sh2d4a exon 8 of the NeoR-deleted targeted allele in all tissues (21). Heterozygote NeoR-deleted sh2d4a exon 8-deleted progeny were then intercrossed to generate wild-type, heterozygote and homozygote NeoR-deleted sh2d4a exon 8-deleted offspring on an inbred C57BL/6J background. Genotype was assessed by PCR using primer combinations shown in Fig. 3.
FIGURE 3.

Generation of sh2d4a gene-targeted mice. A, Shown is the organization of the endogenous sh2d4a locus, targeting vector and targeted sh2d4a allele before and after Cre and Flp-mediated recombination. Locations of HindIII restriction sites and 5’ and 3’ probes used in Southern blotting experiments and positions of PCR primers used in genotyping experiments are indicated. B, Shown is a Southern blot of HindIII digested genomic DNA from two ES cell clones electroporated with the sh2d4a targeting vector. The blot was probed with 5’ and 3’ probes simultaneously. Positions of bands from wild type and targeted sh2d4a alleles are shown. Clone B is correctly targeted. C, Mice that carried a germline targeted sh2d4a allele were crossed with actin-Flp transgenic mice to delete the NeoR cassette. Progeny were subsequently crossed with CMV-Cre transgenic mice to delete exon 8 of the sh2d4a gene in all tissues. Heterozygote NeoR-deleted sh2d4a exon 8-deleted mice were then intercrossed to generate wild type (+/+), heterozygote (+/−) and homozygote (−/−) NeoR-deleted sh2d4a exon 8-deleted progeny. Progeny were genotyped by PCR of tail DNA using the indicated primer combinations to detect wild-type and NeoR-deleted sh2d4a exon 8-deleted alleles. D, Expression of SH2D4A protein in whole splenocytes of littermate wild-type, heterozygote and homozygote NeoR-deleted sh2d4a exon 8-deleted mice was determined by Western blotting using an anti-SH2D4A antiserum. NS, non-specific. Note the absence of the 48 kDa SH2D4A band in the homozygous mutant animals. The membrane was re-probed with a GAPDH antibody to demonstrate equivalent protein loading.
Flow cytometry
Single-cell suspensions from mouse thymi and spleen were stained with the following conjugated mAb (BD BioSciences): H57-597-CyChrome (TCRβ chain), RA3-6B2-PE (CD45R/B220), IM7-FITC (CD44), GK1.5-PE and H129.19-CyChrome (CD4), 53-6.7-CyChrome (CD8), PC61-PE (CD25), and M1/70-PE (CD11b). Cell staining was analyzed by flow cytometry using a FACScan (BD BioSciences).
Mouse T cell cytokine production and proliferation
Splenocytes (2 × 105/well) were stimulated with varying amounts of soluble CD3 mAb and 0.5 µg/ml of a soluble CD28 mAb in 96-well round-bottom plates in mouse complete medium. Concentrations of cytokines in the supernatants were determined by ELISA after 24 or 48 h of culture (R&D Systems). For proliferation assays, T cells were purified from spleen by negative selection and were labeled with 1 µM CFSE (Molecular Probes) before stimulation in complete medium with plate-bound (1 µg/ml) CD3 mAb and soluble CD28 mAb (1 µg/ml). CFSE fluorescence intensity was then analyzed by flow cytometry after 72 h. To assess cytokine secretion upon re-stimulation of T cells, splenocytes (2 × 106/well) were first activated with soluble CD3 and CD28 mAb (0.5 µg/ml each) in complete medium in 6-well plates. After 48 h, recombinant IL-2 (10 U/ml; R&D Systems) was added to cultures and cells were incubated for an additional 48 h. Cytokine production was then assayed as above using different concentrations of plate-bound CD3 mAb and soluble CD28 mAb (0.5 µg/ml).
Apoptosis assays
Purified T cells (2 × 106/well) were stimulated with CD3 and CD28 mAb (0.5 µg/ml each) plus 10 U/ml IL-2 in complete medium in 6-well plates. After 48 h, cells were washed and re-plated in complete medium or RPMI 1640 alone and cultured for a further 24 h. Cells were then stained with Annexin-V-PE and 7AAD (BD Biosciences) and analyzed by flow cytometry. The percentage of Annexin-V positive cells among 7AAD-negative cells was determined and used an indicator of the extent of T cell apoptosis.
Listeria infection
Listeria monocytogenes were grown to 6 × 108 CFU/ml in Brain-Heart Infusion medium. 5 × 105 CFU in 250 µl were injected i.p in to male mice. At 48 h and 96 h post-infection, mice were sacrificed and the liver and spleen were harvested. Organs were homogenized in 10ml of PBS, 0.2% NP-40 and different dilutions of the homogenate were plated on LB agar. Colonies were counted after 14 h incubation at 37°C. The statistical significance of differences in colony counts was determined using the Student’s two sample t-test.
siRNA knockdown experiments
Human PBMCs were mock transfected or were transfected with 2.5µg SH2D4A siRNA (Qiagen) by nucleofection and were then incubated in complete medium for 4 h. Cells were then stimulated or not with soluble CD3 and CD28 mAb (1 µg/ml each) in complete medium for 24 h or 48 h. Expression of SH2D4A protein was determined by Western blotting of lysates using an SH2D4A antiserum. IL-2 and IFN-γ concentrations in culture supernatants were determined by ELISA. For proliferation assays, transfected cells were labeled with CFSE and stimulated with CD3 and CD28 mAb in complete medium for 96 h. The extent of cell division was then determined by flow cytometry. For protein tyrosine phosphorylation assays, transfected cells were first cultured in complete medium with IL-2 (10 U/ml) for 48 h. Cells were then washed and stimulated with 0.5 µg/ml each of CD3 and CD28 mAb for varying times before lysis. Protein tyrosine phosphorylation was determined by Western blotting of lysates with a phosphotyrosine antibody (PY99; Santa Cruz Biotechnology).
Results
Identification of SH2D4A
The TSAd and ALX SH2 domains are 73% homologous in mice. To identify any other adapter proteins with related SH2 domains, we performed standard blast program searches with the murine TSAd SH2 domain. One such adapter that was identified was SH2D4A that has not been characterized previously with the exception of a recent report documenting expression in kidney glomeruli (22). SH2D4A possesses a single SH2 domain located at the COOH end of the protein (Fig. 1). In mice, the SH2 domain of SH2D4A is 66% homologous to the TSAd SH2 domain and 71% homologous to the ALX SH2 domain (Fig. 1B). All three SH2 domains are classified as type I SH2 domains and show conservation of several residues predicted to form contacts with side chains of amino acids carboxyl to phosphorylated tyrosine in protein targets (23). Outside of the SH2 domain, the SH2D4A amino acid sequence does not show any significant homology to TSAd or ALX in the same way that TSAd and ALX also do not show any significant similarity to one another in these regions. However, like TSAd and ALX, SH2D4A does possess conserved tyrosines in consensus phosphorylation motifs (NetPhos program) and a proline-rich region that have the potential to be recognized by the SH2 and SH3 domains respectively of other signaling molecules (Fig. 1A and C).
FIGURE 1.

The SH2D4A adapter protein. A, Protein sequence alignment of human and murine SH2D4A. B, Protein sequence alignment of the SH2 domains of murine SH2D4A, TSAd, and ALX. C, Domain organization of murine SH2D4A, TSAd and ALX proteins. In A and B, residues that are identical or homologous between sequences are boxed. In A and C, the locations of SH2 domains, proline-rich regions (PRR) and conserved tyrosine residues in consensus phosphorylation motifs are indicated.
Analysis of expressed sequence tag databases at the NCBI and of public microarray databases (e.g. GNF SymAtlas; http://symatlas.gnf.org/SymAtlas) indicate that SH2D4A is expressed ubiquitously at low levels in a variety of tissues. To confirm that SH2D4A protein was expressed in T cells, we generated an SH2D4A polyclonal antibody using full-length recombinant human SH2D4A as an immunogen. The antibody detected a band of approximately 52 kDa in Western blots of human PBMC which is in agreement with the predicted molecular mass of 52.7 kDa (Fig. 2A). Proof that this band is indeed SH2D4A was shown in RNAi experiments (see later). In whole PBMC (approximately 70% T cells), expression of SH2D4A was low. Notably, however, stimulation of PBMC with CD3 mAb (directed to the TCR complex) and CD28 mAb resulted in a substantial upregulation of SH2D4A expression in T cells. Increased expression was apparent as soon as 4 h, peaked at 48 h and persisted until at least 72 h after simulation (Fig. 2A and data not shown).
FIGURE 2.

SH2D4A expression. A, Anti-SH2D4A Western blot of human PBMC stimulated with CD3 and CD28 mAb for the indicated times in hours (0 represents 16 h mock-stimulated cells). NS is a non-specific reactive band that serves as an equal loading control (recombinant HA-tagged human SH2D4A was run in the leftmost track). B, Expression of SH2D4A in murine thymocytes, purified splenic CD4+ and CD8+ T cells, purified splenic B cells and bone marrow-derived macrophages (Mac) and DC was determined by Western blotting using an anti-SH2D4A antiserum. Cells were stimulated with CD3/CD28 mAb (T cells) or LPS (all other cells) or were mock-stimulated for different times as indicated. Blots were reprobed with a GAPDH antibody (below). C, Intracellular localization of SH2D4A. 293T cells or human peripheral blood T cells (PBT) were transfected with SH2D4A-GFP or GFP control. Intracellular localization of transfected proteins was determined by fluorescent microscopy. The position of nuclei was determined by Hoechst staining.
The same polyclonal antiserum was also used to monitor expression of SH2D4A in mouse T cells (Fig. 2B). In Western blots of mouse cells, the antiserum detected a protein band at approximately 48 kDa (predicted molecular mass = 48.5 kDa). Proof that this band is SH2D4A was shown in gene-targeting experiments (see later). In the T cell lineage, SH2D4A was expressed constitutively in each of thymocytes and purified CD4+ and CD8+ T cells. In contrast to human T cells, CD3 and CD28 mAb stimulation of murine T cells did not increase SH2D4A expression significantly. Some induction was noted in CD4+ T cells. However, this increase largely paralleled an increase in expression of control GAPDH in CD4+ T cells in these experiments. Aside from T cells, we confirmed that SH2D4A was expressed in other immune cell types including B cells, macrophages and dendritic cells (Fig. 2B). In each of these non-T cell populations, SH2D4A expression was not significantly increased in response to stimulation with LPS.
TSAd and ALX have been shown to reside in the cell cytoplasm and nucleus (7, 24). To determine if SH2D4A also occupied both of these cellular compartments, we transfected 293T cells and primary human T cells with SH2D4A-GFP and examined protein localization by fluorescence microscopy (Fig. 2C). Unlike TSAd and ALX, SH2D4A was found to reside almost exclusively within the cytoplasm of transfected cells. Furthermore, SH2D4A was not mobilized to the nucleus in 293T cells in response to treatment with a variety of stimuli or to the nucleus of T cells in response to stimulation with CD3 and CD28 mAb (data not shown).
Generation of SH2D4A-deficient mice
In order to address if SH2D4A was essential for TCR signal transduction in primary T cells, we produced SH2D4A-deficient mice (Fig. 3). By homologous recombination in ES cells, a targeted sh2d4a allele was generated in which exon 8, which encodes for the entire SH2 domain of the SH2D4A protein, was flanked by loxP recognition sites and in which a NeoR cassette flanked by FRT recognition sites was inserted into intron 8. Following germline transmission of the targeted allele, heterozygote mice were crossed with actin promoter-driven Flp recombinase transgenic mice to delete the NeoR cassette in all cells and generate a conditional sh2d4a allele. First, we were interested to examine the influence of disruption of the sh2d4a gene in all tissues. Therefore, targeted NeoR-deleted progeny were crossed with CMV promoter-driven Cre transgenic mice to delete exon 8 of the NeoR-deleted targeted sh2d4a allele in all cell types. Heterozygote NeoR-deleted sh2d4a exon 8-deleted mice were then intercrossed to generate wild-type, heterozygote and homozygote NeoR-deleted sh2d4a exon 8-deleted progeny.
Wild-type, heterozygote and homozygote sh2d4a exon 8-deleted mice were born in normal Mendelian ratios and no gross abnormalities in growth or development were observed up until 6 mo of age. At the most, transcripts generated from the sh2d4a exon 8-deleted allele would direct the synthesis of an SH2D4A protein that completely lacked the SH2 domain which is likely an important functional component of the protein. As confirmed in Western blots of whole splenocytes using our SH2D4A polyclonal antibody, full-length murine SH2D4A protein was not detected in homozygote mice (Fig. 3D). Furthermore, no truncated forms of the SH2D4A protein were detected in homozygote mice in these experiments, even after long exposure of blots (data not shown). Most likely, in transcripts generated from this mutant allele, exon 7 would remain unspliced or would be spliced to exons downstream of exon 8, none of which are in frame with exon 7. Thus, transcripts would likely be degraded by the process of non-sense-mediated RNA decay resulting in complete loss of SH2D4A protein (25).
T cell development and function in SH2D4A-deficient mice
We examined T cell development and homeostasis in homozygote NeoR-deleted sh2d4a exon 8-deleted mice hereafter referred to as SH2D4A-deficient mice (Fig. 4). In thymus, no abnormalities in the number or ratio of CD4-CD8- double-negative (DN), CD4+CD8+ double-positive and CD4+CD8− or CD4−CD8+ single-positive thymocytes were apparent. In addition, within the DN population, numbers and ratios of CD44+CD25− (DN1), CD44+CD25+ (DN2), CD44−CD25+ (DN3) and CD44−CD25− (DN4) thymocytes were normal. In spleen, normal numbers and ratios of CD4+ and CD8+ T cells, B220+ B cells and CD11b+ macrophages were observed and there was no evidence for any increased activation of CD4+ or CD8+ T cells as judged by increased expression of CD69 or reduced expression of CD62L (Fig. 4 and data not shown). Memory cell development in CD4 and CD8 compartments was also normal as determined by CD44 expression (Fig. 4 and data not shown). The same findings were observed in lymph nodes (data not shown). We also examined the frequency of other populations in lymphoid organs including Treg, γδ T cells, NK cells and NK T cells. No alteration in the frequency of these populations was noted.
FIGURE 4.
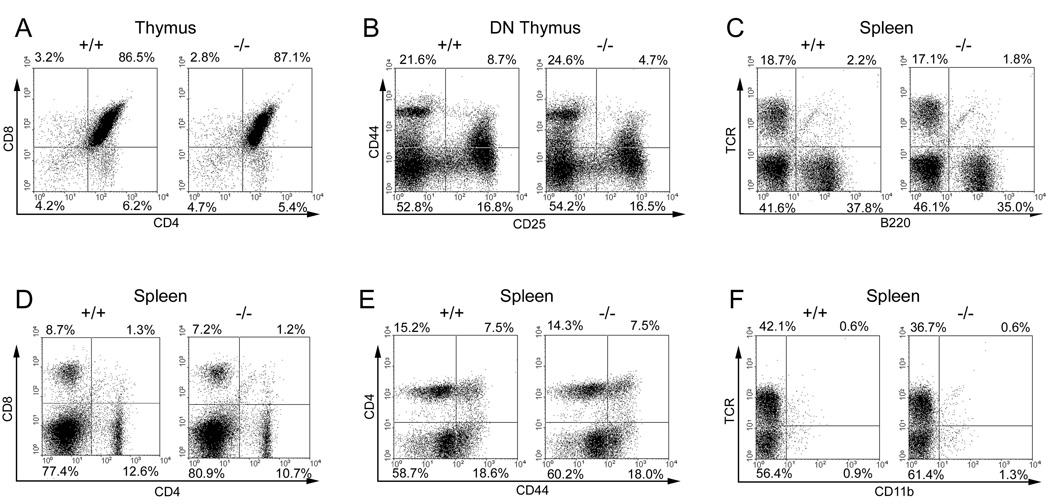
Flow cytometric analysis of SH2D4A-deficient mice. Depicted are flow cytometric dot plots of thymocytes and splenocytes from littermate wild-type and SH2D4A-deficient mice showing expression of the indicated markers on live populations. DN, CD4-CD8- double-negative thymocytes. Percentages of cells in quadrants are indicated.
Next, we examined T cell cytokine synthesis and proliferation in SH2D4A-deficient mice. Splenocytes were stimulated in vitro with CD3 and CD28 mAb and supernatant concentrations of IL-2, the Th1 cytokine, IFN-γ, and the Th2 cytokine, IL-4, were determined by ELISA (Fig. 5A). As shown, SH2D4A-deficient T cells and control T cells synthesized similar quantities of all three cytokines over a range of tested CD3 mAb concentrations. We also examined cytokine responses of recently-stimulated SH2D4A-deficient T cells. Splenocytes from SH2D4A-deficient and control mice were thus stimulated with CD3 and CD28 mAb for 48 h and then grown in IL-2 for a further 48 h before washing and restimulation with CD3 and CD28 mAb (Fig. 5B). Similar to unstimulated SH2D4A-deficient T cells, recently-stimulated SH2D4A-deficient T cells synthesized normal quantities of IL-2, IFN-γ and IL-4 (the apparent reduced amount of IL-4 synthesized by SH2D4A-deficient T cells in response to suboptimal concentrations of CD3 mAb is not a reproducible finding) (Fig. 5B). Consistent with the finding that CD3 and CD28 mAb-induced IL-2 secretion is unaffected, purified T cells from SH2D4A-deficient mice were found to proliferate normally in response to CD3 and CD28 mAb stimulation in vitro (Fig. 5C).
FIGURE 5.
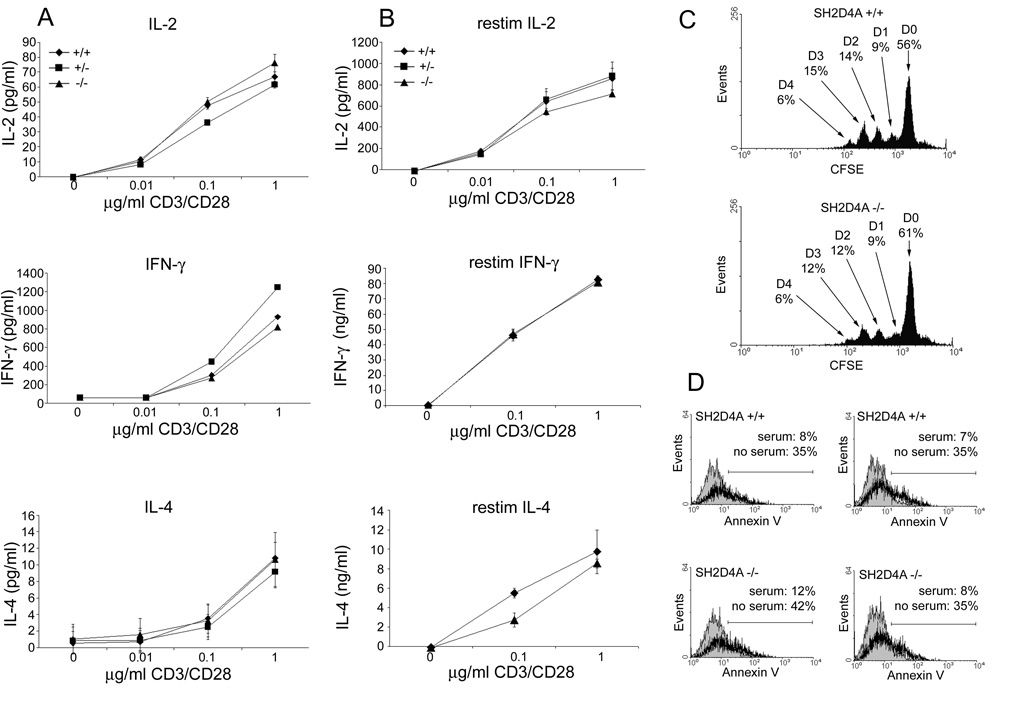
T cell function in SH2D4A-deficient mice. A, Splenocytes from littermate wild-type, heterozygote or homozygote SH2D4A-deficient mice were stimulated with the indicated concentrations of a CD3 mAb and 0.5µg/ml of a CD28 mAb. Concentrations of IL-2 (at 24 h), IFN-γ and IL-4 (at 48 h) in culture supernatants were determined by ELISA. Data are represented as means +/− 1 SD of triplicate determinations. B, Splenocytes from littermate mice of the indicated genotypes were stimulated with CD3 and CD28 mAb for 48 h and then grown in IL-2 for a further 48 h. Cytokine synthesis in response to restimulation with CD3 and CD28 mAb was then determined as in A. C, Purified splenic T cells from littermate wild type and SH2D4A-deficient mice were stained with CFSE and stimulated with plate-bound CD3 mAb and 0.5µg/ml of soluble CD28 mAb. After 72 h, CFSE fluorescence was analyzed by flow cytometry. Indicated is the percentage of cells at successive cell division numbers, D0 through D4. D, Purified splenic T cells from littermate wild type and SH2D4A-deficient mice were stimulated with CD3 and CD28 mAb plus IL-2 for 48 h and then cultured in serum-containing (filled histogram) or serum-free (bold histogram) medium for a further 24 h. The percentage of Annexin-V-positive cells in cultures was then determined by flow cytometry. Shown are results from two different wild-type and SH2D4A-deficient mice.
We also examined the ability of SH2D4A-deficient T cells to undergo activation-induced apoptotic cell death (AICD) initiated by CD3/CD28 mAb stimulation in vitro (Fig. 5D). In these experiments, purified splenic T cells from SH2D4A-deficient and control mice were stimulated with CD3 and CD28 mAb for 48 h in the presence of IL-2. Cells were then washed and cultured in either complete medium or serum free medium for 24 h after which time the extent of cell death was determined by annexin-V staining and flow cytometry. As shown, upon serum withdrawal, CD3/CD28 mAb-stimulated SH2D4A-deficient T cells underwent AICD to a similar extent as that observed with CD3/CD28 mAb-stimulated wild type T cells.
Listeria infection of SH2D4A-deficient mice
Since SH2D4A-deficient T cells responded normally to CD3 and CD28 mAb stimulation in vitro, we next asked if loss of SH2D4A expression would impact upon an ability of T cells to function in an immune response in vivo. For this purpose, we used the well characterized Listeria monocytogenes infection model in which T cells as well as other immune cell types have been shown to play an important role (26). Groups of wild-type and SH2D4A-deficient mice were inoculated with Listeria intraperitoneally. At different time points thereafter, mice were sacrificed, liver and spleen were harvested and the number of Listeria CFU in each organ were determined (Fig. 6). Two days after inoculation, there were no significant differences in the number of colonies in spleen or liver between wild-type and SH2D4A-deficient mice. This finding indicates that the innate immune response to Listeria is intact in SH2D4A-deficient mice since it is the innate immune response that is primarily responsible for control of infection at this time point. Likewise, 4 d after inoculation, the number of Listeria in spleen and liver was similar between wild-type and SH2D4A-deficient mice. By 7 d, 4/4 wild type and 3/3 SH2D4A-deficient mice had completely cleared Listeria organisms from the spleen (data not shown). At these later time points, T cells are considered to be the principal effector cells involved in the control of infection. Therefore, these findings show that SH2D4A is not necessary for the normal functioning of T cells in the Listeria immune response.
FIGURE 6.
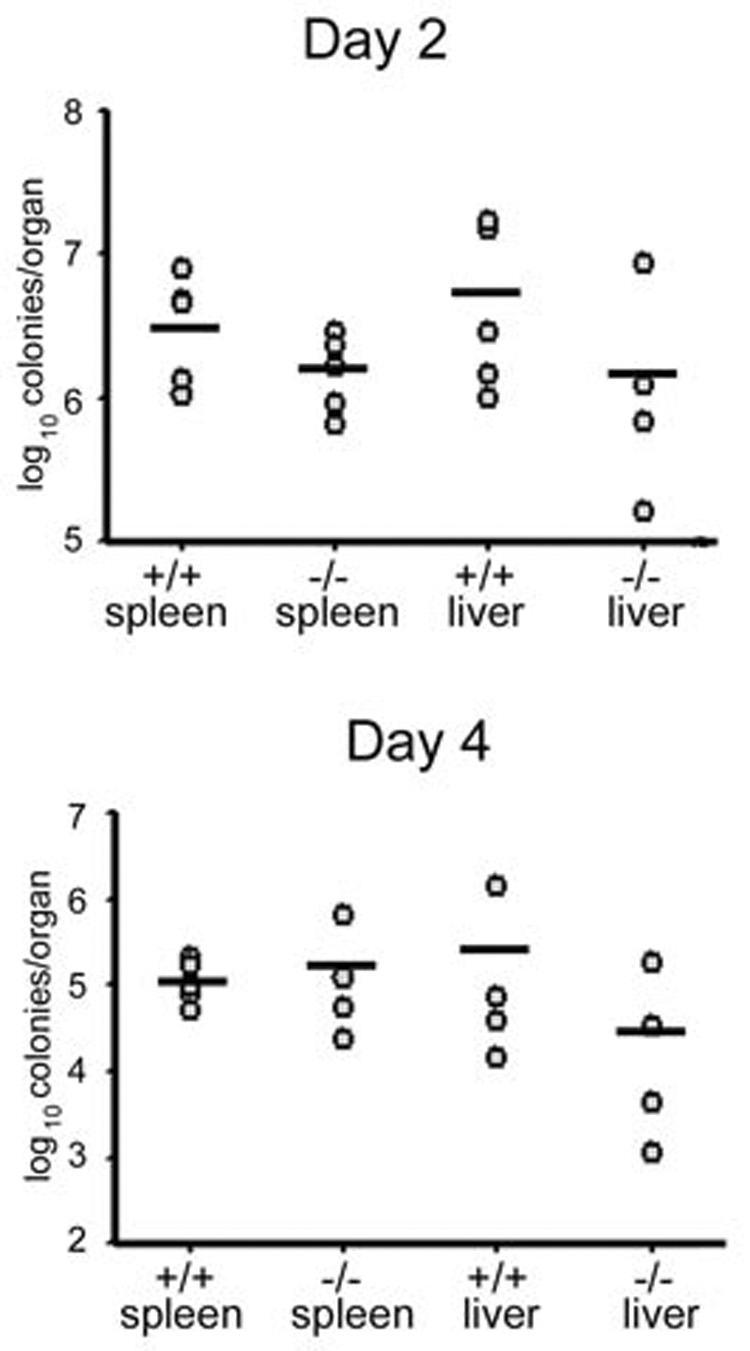
Listeria infection of SH2D4A-deficient mice. Wild type or SH2D4A-deficient mice were challenged i.p. with 5 × 105 CFU of Listeria monocytogenes. At the indicated times post-infection, the number of CFU in spleen and liver was determined. Points represent data from individual mice. Means are indicated with a bar. Differences in CFU between mice within an organ are not statistically significant at either time point.
Knockdown of SH2D4A expression in human T cells
To confirm that the apparent lack of a requirement of SH2D4A in TCR signal transduction was not a peculiarity of mouse T cells, we also examined the influence of knockdown expression of SH2D4A in human T cells (Fig. 7). Human PBMC were transfected with an SH2D4A siRNA before stimulation with CD3 and CD28 mAb. After 24 h and 48 h mAb stimulation, expression of SH2D4A was then examined by Western blotting (Fig. 7A). As shown, the SH2D4A siRNA was effective at reducing expression levels of SH2D4A in human T cells at both time points. However, despite this reduction in expression, CD3 and CD28 mAb-induced synthesis of IL-2 and IFN-γ was unaffected (Fig. 7B). Similarly, CD3/CD28 mAb-induced proliferation of T cells was unaffected by knockdown of SH2D4A (Fig. 7C).
FIGURE 7.
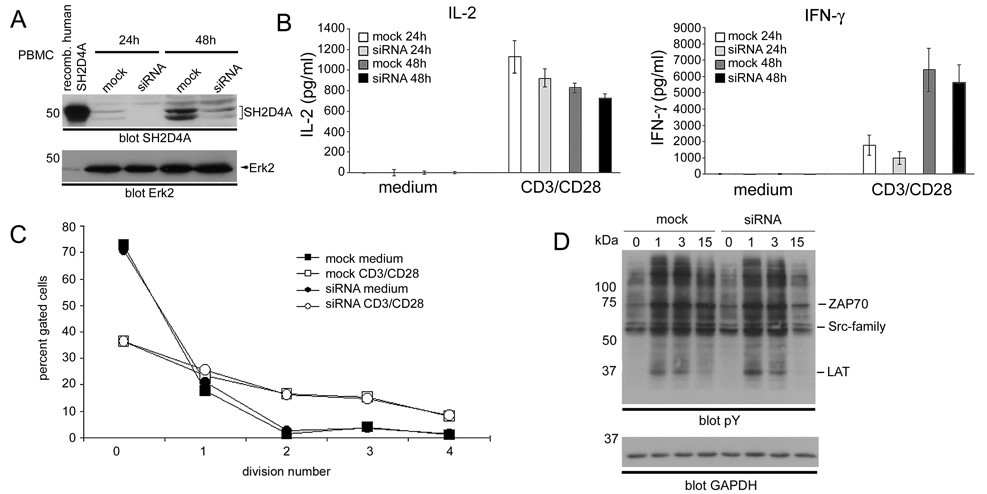
Effect of siRNA knockdown of SH2D4A expression in human T cells. A, Human PBMC were transfected with SH2D4A siRNA or mock transfected by nucleofection. Cells were then stimulated with CD3 and CD28 mAb for 24 h or 48 h and lysed. SH2D4A expression was determined by Western blotting using an anti-SH2D4A antiserum. The blot was reprobed with an ERK antibody to demonstrate equal loading. B, SH2D4A siRNA-transfected and mock-transfected PBMC were stimulated or not with CD3 and CD28 mAb for 24 or 48 h. Concentrations of IL-2 and IFN-γ in culture supernatants were determined by ELISA. Data are represented as means +/− 1 SE of triplicate determinations. C, SH2D4A siRNA-transfected and mock-transfected PBMC were labeled with CFSE and stimulated or not with CD3 and CD28 mAb. After 96 h, the extent of cell division was determined by flow cytometry. For each cell type and condition of stimulation is shown is the percentage of cells that have undergone the indicated number of divisions. D, SH2D4A siRNA-transfected and mock-transfected PBMC were cultured in IL-2 for 48 h, washed and stimulated with CD3 and CD28 mAb for the indicated times. Protein tyrosine phosphorylation was then determined by Western blotting of whole cell lysates. Phosphotyrosine bands corresponding to the expected positions of the LAT phosphoprotein, Src-family and ZAP-70 PTK are indicated. Blots were reprobed with a GAPDH antibody to demonstrate equal protein loading.
Lastly, we also examined the impact of SH2D4A knockdown upon CD3/CD28 mAb-induced protein tyrosine phosphorylation in T cells. In these experiments, following transfection with SH2D4A siRNA, PBMC were cultured in the presence of IL-2 for 48 h (to maintain cell viability). Cells were then stimulated with CD3/CD28 mAb for different times and protein phosphotyrosine responses were assessed by Western blotting (Fig. 7D). As shown, CD3/CD28 mAb-induced phosphotyrosine responses were comparable in magnitude and kinetics between SH2D4A-knockdown and mock-transfected T cells. These findings are consistent with the lack of an influence of SH2D4A-knockdown upon cytokine production and proliferation.
Discussion
We report here a novel adapter protein, SH2D4A, which is structurally related to the TSAd and ALX adapter proteins that have been shown to regulate TCR signal transduction in T cells. SH2D4A possesses tyrosine residues with potential for phosphorylation by PTK, a proline-rich region and an SH2 domain that shows strong homology to the TSAd and ALX SH2 domains. Despite that the location of the SH2 domain within the linear sequence differs, all three adapter proteins clearly belong to the same family. However, there are differences in the expression pattern of these adapters and in their subcellular localization. TSAd is expressed predominantly in T lineage cells and in human T cells expression levels are increased upon T cell activation (4, 5, 10, 27). ALX, by contrast, is expressed in other hematopoietic cells as well (13–15). Indeed, one report has indicated that within the lymphocytic compartment, ALX is expressed predominantly in B cells. Also, it has been shown that in B cells, the expression level of ALX can be up- or down-regulated in response to anti-apoptotic or pro-apoptotic stimuli respectively (15, 16). However, that ALX is in fact also expressed in T cells, even if at low levels, and is functional in that cell type is shown by the phenotype of ALX-deficient mice (17). SH2D4A is the most ubiquitously expressed of the three adapters in that it is found both in hematopoeitic and non-hematopoeitic cell types. Within the hematopoeitic compartment, we have observed expression in T cells, B cells, macrophages and dendritic cells. In human T cells, expression levels are increased in response to T cell activation. By contrast, in mouse T cells, expression of SH2D4A is not increased upon activation. With regards to subcellular localization, TSAd and ALX are found in cytoplamic and nuclear compartments and are transported between compartments through active mechanisms (7, 24). By contrast, SH2D4A resides almost exclusively in the cell cytoplasm.
To address definitively if SH2D4A is necessary for T cell development and function, we produced SH2D4A-deficient mice. Using Cre-LoxP recombination methodology, we produced a mouse in which exon 8 of the sh2d4a gene which encodes the SH2 domain was deleted in all tissues. Western blot analysis of tissues from these mice confirmed the loss of full-length protein containing the SH2 domain. In fact, no unique SH2D4A antiserum-reactive bands were detected in these mice indicating complete loss of expression of the SH2D4A protein. Most likely, the complete loss of expression is a result of nonsense-mediated RNA decay. In addition, truncated forms of SH2D4A may be unstable and rapidly degraded.
No defects in T cell immunity were apparent in SH2D4A-deficient mice. T cell development was normal and there was no alteration in the number or ratio of different T cell subsets in peripheral lymphoid organs. As determined in vitro, SH2D4A-deficient T cells synthesized normal quantities of cytokines and proliferated normally in response to CD3 plus CD28 mAb stimulation. The same results were obtained when the superantigen, staphylococcal Enterotoxin B, was used to stimulate T cells showing that normal responses are not peculiar to the use of mAb to stimulate T cells. These results differ from those obtained with TSAd-deficient and ALX-deficient T cells which show impaired and exaggerated proliferative and cytokine responses respectively (9, 10, 17). In addition, SH2D4A-deficient T cells were found to undergo normal AICD triggered by CD3/CD28 mAb. By contrast, TSAd-deficient T cells show impaired T cell death responses (9). We also demonstrated that SH2D4A-deficient T cells are fully competent as orchestrators of an adaptive immune response against Listeria monocytogenes in vivo. Furthermore, knockdown of SH2D4A expression in human peripheral blood T cells had no influence upon their ability to secrete cytokines or proliferate in response to CD3 and CD28 mAb stimulation. Neither did SH2D4A knockdown affect CD3/CD28 mAb-induced protein tyrosine phosphorylation in T cells. We conclude that, in contrast to TSAd and ALX, SH2D4A is dispensable for the normal functioning of T cells in mice and humans.
Given its ubiquitous expression, we have also examined if other cell types show functional defects in SH2D4A-deficient mice. However, no defects have thus far been found (data not shown). Concerning B cells, antibody responses and antibody class-switching in response to antigen immunization are normal. Also, we have not observed any functional defects in other hematopoeitic cell types. In non-lymphoid tissues, extensive histological analysis of multiple organ systems has revealed no significant lesions. In addition, SH2D4A-deficient mice perform normally in various tests of neurological and neuromuscular function. It is conceivable that SH2D4A could perform a non-redundant functional role in cellular processes that we have not yet examined. Also, it is possible that functional influences of the loss of SH2D4A expression might only become apparent once the homologous adapters, TSAd and ALX, have also been deleted from mice. This last possibility is currently being examined in the laboratory.
Acknowledgements
Laura Bauler and Mary O’Riordan are thanked for assistance and advice with Listeria infection experiments.
Footnotes
This work was supported by American Heart Association Grants 0615514Z and 08501702 and National Institute of Health grant AI050699
Abbreviations used in this paper: TSAd, T cell-specific adapter protein; PTK, protein tyrosine kinase; Treg, T regulatory cell; ES cell, embryonic stem cell; RNAi, RNA interference; HA, hemagglutinin; DN, double-negative
Disclosures The authors have no financial conflict of interest.
References
- 1.Leo A, Wienands J, Baier G, Horejsi V, Schraven B. Adapters in lymphocyte signaling. J Clin Invest. 2002;109:301–309. doi: 10.1172/JCI14942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simeoni L, Kliche S, Lindquist J, Schraven B. Adaptors and linkers in T and B cells. Curr Opin Immunol. 2004;16:304–313. doi: 10.1016/j.coi.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 3.Samelson LE. Signal transduction mediated by the T cell antigen receptor: the role of adapter proteins. Annu Rev Immunol. 2002;20:371–394. doi: 10.1146/annurev.immunol.20.092601.111357. [DOI] [PubMed] [Google Scholar]
- 4.Spurkland A, Brinchmann JE, Markussen G, Pedeutour F, Munthe E, Lea T, Vartdal F, Aasheim HC. Molecular cloning of a T cell-specific adapter protein (TSAd) containing an Src homology (SH) 2 domain and putative SH3 and phosphotyrosine binding sites. J Biol Chem. 1998;273:4539–4546. doi: 10.1074/jbc.273.8.4539. [DOI] [PubMed] [Google Scholar]
- 5.Choi YB, Kim CK, Yun Y. Lad, an adapter protein interacting with the SH2 domain of p56lck, is required for T cell activation. J Immunol. 1999;163:5242–5249. [PubMed] [Google Scholar]
- 6.Marti F, Garcia GG, Lapinski PE, MacGregor JN, King PD. Essential role of the T cell-specific adapter protein in the activation of LCK in peripheral T cells. J Exp Med. 2006;203:281–287. doi: 10.1084/jem.20051637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marti F, Post NH, Chan E, King PD. A transcription function for the T cell-specific adapter (TSAd) protein in T cells: critical role of the TSAd Src homology 2 domain. J Exp Med. 2001;193:1425–1430. doi: 10.1084/jem.193.12.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marti F, King PD. The p95-100 kDa ligand of the T cell-specific adaptor (TSAd) protein Src-homology-2 (SH2) domain implicated in TSAd nuclear import is p97b Valosin-containing protein (VCP) Immunol Lett. 2005;97:235–243. doi: 10.1016/j.imlet.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 9.Drappa J, Kamen LA, Chan E, Georgiev M, Ashany D, Marti F, King PD. Impaired T cell death and lupus-like autoimmunity in T cell-specific adapter protein-deficient mice. J Exp Med. 2003;198:809–821. doi: 10.1084/jem.20021358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rajagopal K, Sommers CL, Decker DC, Mitchell EO, Korthauer U, Sperling AI, Kozak CA, Love PE, Bluestone JA. RIBP, a novel Rlk/Txk- and itk-binding adaptor protein that regulates T cell activation. J Exp Med. 1999;190:1657–1668. doi: 10.1084/jem.190.11.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lapinski PE, MacGregor JN, Marti F, King PD. The T cell-specific adapter protein functions as regulator of peripheral but not central immunological tolerance. Adv Exp Med Biol. 2006;584:73–88. doi: 10.1007/0-387-34132-3_6. [DOI] [PubMed] [Google Scholar]
- 12.Marti F, Lapinski PE, King PD. The emerging role of the T cell-specific adaptor (TSAd) protein as an autoimmune disease-regulator in mouse and man. Immunol Lett. 2005;97:165–170. doi: 10.1016/j.imlet.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 13.Oda T, Muramatsu MA, Isogai T, Masuho Y, Asano S, Yamashita T. HSH2: a novel SH2 domain-containing adapter protein involved in tyrosine kinase signaling in hematopoietic cells. Biochem Biophys Res Commun. 2001;288:1078–1086. doi: 10.1006/bbrc.2001.5890. [DOI] [PubMed] [Google Scholar]
- 14.Greene TA, Powell P, Nzerem C, Shapiro MJ, Shapiro VS. Cloning and characterization of ALX, an adaptor downstream of CD28. J Biol Chem. 2003;278:45128–45134. doi: 10.1074/jbc.M306283200. [DOI] [PubMed] [Google Scholar]
- 15.Herrin BR, Groeger AL, Justement LB. The adaptor protein HSH2 attenuates apoptosis in response to ligation of the B cell antigen receptor complex on the B lymphoma cell line, WEHI-231. J Biol Chem. 2005;280:3507–3515. doi: 10.1074/jbc.M407690200. [DOI] [PubMed] [Google Scholar]
- 16.Herrin BR, Justement LB. Expression of the adaptor protein hematopoietic Src homology 2 is up-regulated in response to stimuli that promote survival and differentiation of B cells. J Immunol. 2006;176:4163–4172. doi: 10.4049/jimmunol.176.7.4163. [DOI] [PubMed] [Google Scholar]
- 17.Perchonock CE, Fernando MC, Quinn WJ, 3rd, Nguyen CT, Sun J, Shapiro MJ, Shapiro VS. Negative regulation of interleukin-2 and p38 mitogen-activated protein kinase during T-cell activation by the adaptor ALX. Mol Cell Biol. 2006;26:6005–6015. doi: 10.1128/MCB.02067-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Charles MA, Saunders TL, Wood WM, Owens K, Parlow AF, Camper SA, Ridgway EC, Gordon DF. Pituitary-specific Gata2 knockout: effects on gonadotrope and thyrotrope function. Mol Endocrinol. 2006;20:1366–1377. doi: 10.1210/me.2005-0378. [DOI] [PubMed] [Google Scholar]
- 19.Kontgen F, Suss G, Stewart C, Steinmetz M, Bluethmann H. Targeted disruption of the MHC class II Aa gene in C57BL/6 mice. Int Immunol. 1993;5:957–964. doi: 10.1093/intimm/5.8.957. [DOI] [PubMed] [Google Scholar]
- 20.Rodriguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R, Stewart JF, Dymecki SM. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet. 2000;25:139–140. doi: 10.1038/75973. [DOI] [PubMed] [Google Scholar]
- 21.Dupe V, Davenne M, Brocard J, Dolle P, Mark M, Dierich A, Chambon P, Rijli FM. In vivo functional analysis of the Hoxa-1 3′ retinoic acid response element (3′RARE) Development. 1997;124:399–410. doi: 10.1242/dev.124.2.399. [DOI] [PubMed] [Google Scholar]
- 22.Patrakka J, Xiao Z, Nukui M, Takemoto M, He L, Oddsson A, Perisic L, Kaukinen A, Szigyarto CA, Uhlen M, Jalanko H, Betsholtz C, Tryggvason K. Expression and subcellular distribution of novel glomerulus-associated proteins dendrin, ehd3, sh2d4a, plekhh2, and 2310066E14Rik. J Am Soc Nephrol. 2007;18:689–697. doi: 10.1681/ASN.2006060675. [DOI] [PubMed] [Google Scholar]
- 23.Kuriyan J, Cowburn D. Modular peptide recognition domains in eukaryotic signaling. Annu Rev Biophys Biomol Struct. 1997;26:259–288. doi: 10.1146/annurev.biophys.26.1.259. [DOI] [PubMed] [Google Scholar]
- 24.Shapiro MJ, Chen YY, Shapiro VS. The carboxyl-terminal segment of the adaptor protein ALX directs its nuclear export during T cell activation. J Biol Chem. 2005;280:38242–38246. doi: 10.1074/jbc.M507441200. [DOI] [PubMed] [Google Scholar]
- 25.Conti E, Izaurralde E. Nonsense-mediated mRNA decay: molecular insights and mechanistic variations across species. Curr Opin Cell Biol. 2005;17:316–325. doi: 10.1016/j.ceb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 26.Pamer EG. Immune responses to Listeria monocytogenes. Nat Rev Immunol. 2004;4:812–823. doi: 10.1038/nri1461. [DOI] [PubMed] [Google Scholar]
- 27.Dai KZ, Johansen FE, Kolltveit KM, Aasheim HC, Dembic Z, Vartdal F, Spurkland A. Transcriptional activation of the SH2D2A gene is dependent on a cyclic adenosine 5′-monophosphate-responsive element in the proximal SH2D2A promoter. J Immunol. 2004;172:6144–6151. doi: 10.4049/jimmunol.172.10.6144. [DOI] [PubMed] [Google Scholar]
- 28.Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]