

Abstract
Most patients with hormone-responsive breast cancer eventually develop resistance to traditional antiestrogens such as tamoxifen, and this has become a major obstacle in their treatment. We prepared and characterized the activity of a series of 16 guanylhydrazone small molecules that are designed to block estrogen receptor (ER) activity through a non-traditional mechanism, by directly interfering with coactivator binding to agonist-liganded ER. The inhibitory activity of these compounds was determined in cell-based transcription assays using ER-responsive reporter gene and mammalian two hybrid assays. Several of the compounds gave IC50 values in the low micromolar range. Two secondary assays were used to confirm that these compounds were acting through the proposed non-traditional mode of estrogen inhibitory action and not as conventional antagonists at the ligand binding site.
Keywords: Estrogen Receptor, Coactivator Binding, Tamoxifen Resistance, Protein-protein Interaction
Introduction
The estrogen receptor (ER), a member of the nuclear receptor superfamily of ligand-modulated transcription factors, mediates the activity of estrogens in both reproductive and non-reproductive tissues and appears to drive cell proliferation in about half of human breast cancers.1 Traditional endocrine therapies for breast cancer rely on ER-antagonists such as tamoxifen or raloxifene, which block ER transcription and thus prevent tumor growth. Resistance to tamoxifen and related drugs, however, has become a major obstacle in the treatment of breast cancer, with a majority of patients showing resistance at some point during treatment;2 so, the development of alternative therapies is essential. Herein we report the exploration of a series of guanylhydrazone small molecules, designed to be coactivator binding inhibitors (CBIs), which block ER transcription through a different mechanism than traditional ER antagonists.
In basic terms, ER can be considered a switch that turns gene transcription on or off depending on the nature of the molecule bound in its ligand binding pocket: When the endogenous steroidal ligand, estradiol (E2), or another ER agonist ligand binds to ER, it induces a conformational change in the receptor, forming a hydrophobic groove on the surface where coactivator proteins bind and initiate transcription. Conversely, an ER antagonist (such as tamoxifen or raloxifene) displaces estradiol from the binding pocket, altering the ER conformation: The bulky and basic side chain of the antagonist ligand interacts sterically with helix 12 of ER, forcing it over the coactivator binding groove, thus occluding coactivator binding and preventing transcription.3, 4 A CBI bypasses this ER conformational switch because it does not displace estradiol, but instead interferes with the interaction between ER and coactivator by direct competition with the coactivator. Various assays can be used to distinguish CBIs from conventional ligand antagonists.5, 6
Previously reported ER CBIs were based on peptidomimetics, small molecules that mimic the key LXXLL binding sequence of the coactivator protein.7, 8, 9 Such CBIs were found to block ER interaction with coactivators with Ki values in the micromolar range, but the poor cellular uptake of peptides and many peptidomimetics constrain their utility.10
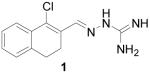
We were intrigued by a report that guanylhydrazone 1 (Scheme 1) showed CBI activity against ER, with an IC50 of 5.5 μM determined by a mammalian two-hybrid (M2H) assay, and that it inhibited ER transactivation in human breast cancer MCF-7 cells.11 In further studies, the investigators confirmed that 1 was not a conventional antagonist. Because this compound appeared to possess the pharmacological footprint of a CBI, we chose to prepare 1 and a series of related derivatives and analogs for further investigation.
Scheme 1.

Results and Discussion
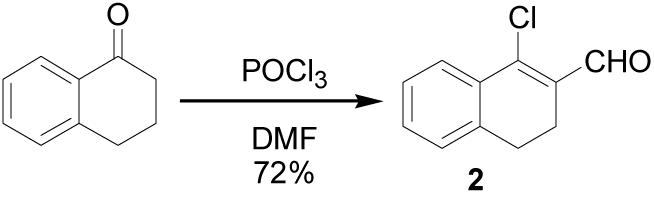
Although the synthesis of 1 was not reported, it can be prepared easily by hydrazone formation from the corresponding β-chlorovinyl aldehyde (2), which is accessible from α-tetralone via a Vilsmeier-Haack reaction (Scheme 1). To prepare chloro-enal 2, the Vilsmeier-Haack reagent was generated by refluxing POCl3 in anhydrous DMF for 30 to 60 minutes.12 α-Tetralone was then added to the mixture and reflux continued for another 4-6 hours until the reaction was shown to be complete by TLC. An aqueous workup hydrolyzed the iminium species to aldehyde 2, which was then purified by either bulb-to-bulb (Kugelrohr) distillation or column chromatography.
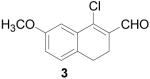
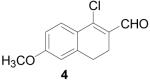
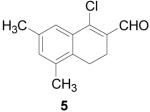
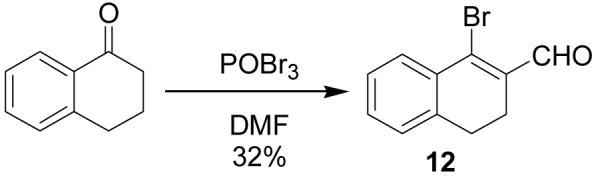
To further explore the structure-activity relationships (SAR) of this class of compounds, several analogs of chloro-enal 2 were prepared from various aryl ketones (Table 1) using similar conditions. The α-tetralone (3, 4, 5, 8) and benzosuberone (7) derivatives were obtained in good to excellent yield. The aldehyde product from the Vilsmeier reaction with propiophenone (10) was obtained as a 6:1 mixture of E/Z isomers, as determined by 1H NMR. The Vilsmeier products from indanone (6), acetophenone (9) and acetonaphthone (11) required much milder conditions; these reactions were conducted at 0 °C (instead of reflux), and products were obtained in lower yields.13 Products 9 and 11 were isolated as single isomers. The bromo-analog of 2 (12) was obtained by using POBr3 in place of POCl3 in the Vilsmeier reaction (Scheme 2) in lower yield.
Table 1.
Chloro-enal products from the Vilsmeier reaction of various aryl ketones.
 | |||||
|---|---|---|---|---|---|
| Product | Yield | Product | Yield | Product | Yield |
 |
93% | 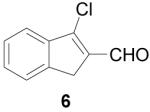 |
25% | 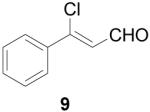 |
49% |
 |
93% | 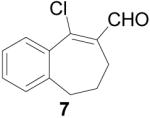 |
66% | 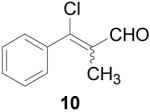 |
67% |
 |
93% | 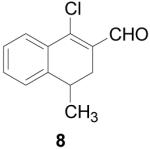 |
98% | 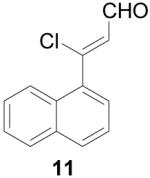 |
16% |
Scheme 2.
It is known that β-chlorovinyl aldehydes are susceptible to nucleophilic substitution at the β position by an addition-elimination mechanism (Scheme 3).14 This reactivity was exploited to prepare derivatives from halo-enals 2 and 12 with different substitutions of the halogen. By stirring 2 at room temperature for three hours in the presence of sodium methoxide in methanol, the methoxy-enal 13 (Scheme 4) was obtained in good yield. Similarly, refluxing 2 with phenol and potassium carbonate in DMF for 24 hours gave the phenoxide derivative 14.15
Scheme 3.

Scheme 4.

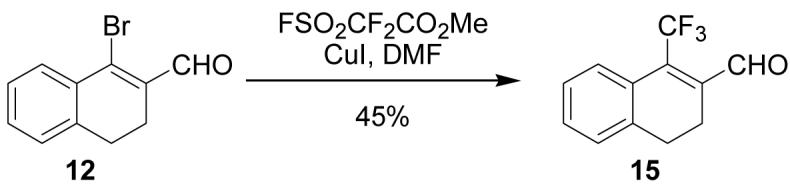
Several attempts were made to prepare the fluoro analog of 2 through nucleophilic fluorination of the chloro- or bromo-enals 2 and 12. Numerous reagents (TBAF, KF with 18-crown-6 or 2.2.2.Kryptofix, AgF), temperatures and solvents (DMF, CH3CN) were employed, but none furnished the fluoro product. The trifluoromethyl derivative (15, Scheme 5) was obtained from bromo-enal 12 in moderate yield by stirring copper iodide and methyl difluoro(fluorosulfonyl)acetate in DMF at 80 °C for 28 hours. 16, 17 Efforts to prepare 15 from the chloro analog (2) were unsuccessful.
Scheme 5.
In an attempt to prepare a β-methyl derivative of 2, methyl lithium was added to a solution of 2 in ether at −10 °C; however, the desired product was not obtained. Instead, the methyl anion attacked the aldehyde carbon, giving secondary alcohol 16 in high purity and excellent yield. It is known that copper(I) can catalyze the 1,4-addition of alkyl nucleophiles to unsaturated ketones;18,19 however, all attempts to effect conjugate addition of various methyl carbanions with 2 resulted in formation of allylic alcohol 16. Although not the desired product, alcohol 16 was still useful: oxidation using Dess-Martin periodinane gave ketone 17 in good yield,20 in effect, replacing the aldehyde with a methyl ketone.
Hydrazone formation occurred in good yield by refluxing the β-chlorovinyl aldehyde (or ketone) in a 2:1 mixture of methanol/acetic acid with aminoguanidine hydrochloride (Table 2).21 Once deemed complete by TLC, the reaction was quenched with water. The basic guanylhydrazone partitioned into the acidic water layer, and organic impurities (including any unreacted starting material) were extracted with chloroform. The aqueous layer was then neutralized, and the product was extracted with ethyl acetate. Addition of HCl in ether to a concentrated solution of the guanylhydrazone in ethyl acetate promoted precipitation of the hydrochloride salt, which was collected by filtration, washed with ether and dried thoroughly. The compounds thus obtained appeared to be pure based on NMR, TLC and elemental analysis, and further purification was unnecessary.
Table 2.
Guanylhydrazones prepared from the aldehydes and ketones.
| Guanylhydrazone | Yield | Guanylhydrazone | Yield | Guanylhydrazone | Yield |
|---|---|---|---|---|---|
 |
78% | 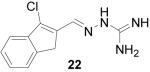 |
- |  |
81% |
 |
63% | 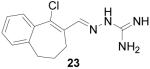 |
59% | 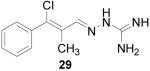 |
67% |
 |
33% | 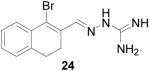 |
52% | 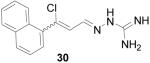 |
63% |
 |
56% | 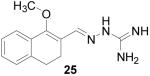 |
- |  |
97% |
 |
- |  |
66% | 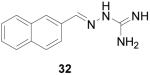 |
87% |
 |
81% | 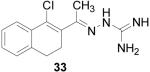 |
14% |
Although aldehyde 10 was a 6:1 mixture of isomers, guanylhydrazone 29 was isolated as a single isomer, likely due to isomerization of the alkene under highly acidic conditions. Because entries 31 and 32 utilized 1- and 2-naphthaldehyde as the respective starting materials, the Vilsmeier-Haack reaction was not necessary. These compounds were prepared because they are similar in structure to 1. Guanylhydrazones 21, 22 and 25 did not form precipitates upon HCl addition; these products were tested in their free-base form.
The ability of these compounds to act as inhibitors of estrogen receptor/coactivator interaction was assessed using an in vitro fluorescence polarization assay that we have previously described.7 Briefly, the assay uses recombinant estrogen receptor ligand binding domain, an 8-mer fluorescein-labeled SRC peptide containing the LXXLL interaction sequence (ILRKLLQE), and the agonist ligand estradiol. In the presence of an agonist, the coactivator peptide is recruited to ER, giving it large polarization values. However, CBI binding to the coactivator binding groove of the receptor displaces the coactivator peptide, causing a dose-dependent decrease in polarization values. A high concentration (1 μM) of estradiol was used in the assay to ensure full coactivator recruitment and also to prevent any potential binding of tested compounds in the ligand binding pocket of the receptor. The results of this assay with selected guanylhydrazones (1, 23, 29) and the control peptide (SRC) are shown in Figure 2; the clear inhibition curves establish that compounds in this class do, in fact, act by blocking estrogen receptor/coactivator binding.
Figure 2.
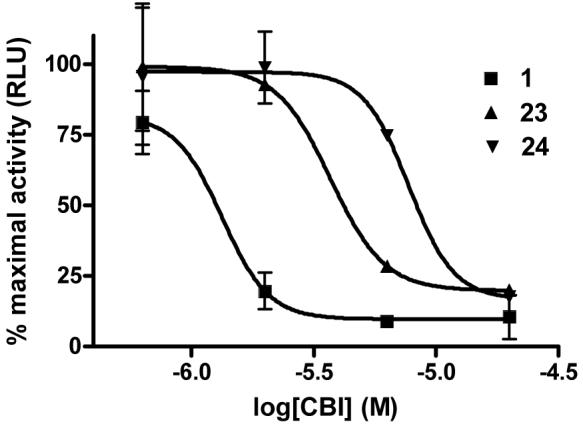
Representative inhibition curves for 1 (IC50 = 1.3 μM), 23 (IC50 = 4.1 μM) and 24 (IC50 = 7.7 μM) in the luciferase reporter gene assay in the presence of 1 nM estradiol.
The Ki values we report for these compounds are higher than those we determine using cell-based assay methods (see below). The fluorescence polarization assay format, however, necessitates use of a high concentration of estrogen receptor, and this results in a less-sensitive assay. Unfortunately, the highly sensitive assay we currently implement to assess the CBI activity of compounds from different functional classes, namely, a time-resolved fluorescence resonance energy transfer assay (TR-FRET), cannot be used with this compound set, because the guanylhydrazones have a fluorescence pattern that interferes with the TR-FRET assay.
A more complete analysis of the coactivator binding inhibitory (CBI) activity of all of the guanylhydrazones in Table 2 was made by assaying their inhibition of transcription in human endometrial cancer (HEC-1) cells transfected with plasmids for expression of estrogen receptor α (ERα) and an estrogen-responsive luciferase reporter gene.22 The assay monitored the progressive decrease in reporter gene activity, stimulated by 1 nM estradiol, as increasing concentrations of the candidate CBI were added. The results from the reporter gene assay are reported in Table 3, and representative inhibition curves are shown in Figure 2.
Table 3.
Potency of guanylhydrazone CBI inhibition in cell-based assays of reporter gene activity.
| CBI | IC50 (μM) | CBI | IC50 (μM) | CBI | IC50 (μM) |
|---|---|---|---|---|---|
| 1 | 1.4 ± 0.1 | 23 | 3.3 ± 0.8 | 29 | 1.4 ± 0.4 |
| 18 | 3.9 ± 0.3 | 24 | 7.8 ± 0.1 | 30 | ≥ 50 |
| 19 | 2.0 ± 0.1 | 25 | 3.1 ± 0.8 | 31 | 7.5 ± 0.0 |
| 20 | 1.2 ± 0.1 | 26 | 3.5 ± 1.1 | 32 | 7.5 ± 0.5 |
| 21 | 1.5 | 27 | 12 | 33 | ≥ 50 |
| 22 | 1.3 ± 0.1 | 28 | 0.9 ± 0.4 |
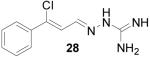
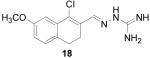
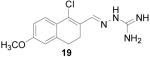
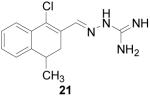
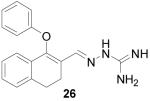
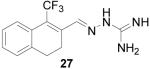
It is evident that derivatives of 1 with 5,7-dimethyl (20) or 6-methoxy (19) substitution on the aromatic ring or methyl at the 4-position (21) show potency similar to that of 1. The 7-methoxy derivative (18) has three-fold lower potency (three-fold higher IC50) than 1. Changing the ring size from 6-membered (1) to 5-membered (22) has little effect on the activity, while enlarging to the 7-membered derivative (23) decreases the potency. In all cases, replacement of the chlorine with other substituents (24, 25, 26, 27) decreases CBI potency, with the bromo (24) and trifluoromethyl (27) derivatives showing the largest increase in IC50. The naphthaldehyde derivatives (31, 32) showed lower potency, indicating that a saturated ring system and/or chlorine are essential for CBI action. Larger substituents on the hydrazone carbon are not tolerated, because the methyl ketone derivative (33) showed the lowest activity of the entire series. Acyclic derivatives (28 and 29) performed well, with the simplest analog, 28, having the greatest potency of the series. This work thoroughly explored the SAR for the CBI activity of the α-tetralone guanylhydrazone scaffold, which is summarized in Figure 3.
Figure 3.

Graphical depiction of the SAR for guanylhydrazone CBIs based on reporter gene assay results
The CBIs were also tested in a mammalian two hybrid (M2H) assay using HEC-1 cells transfected with pFR luciferase, pCMV β-galactosidase, Gal4-SRC-1nrd, and ERαdef-VP16.23 After transfection, the cells were treated with 1 nM estradiol and titrated with increasing concentrations of CBI. The results from the M2H assay are presented in Table 4. The observed potencies of the compounds are, in most cases, consistent between the assays, both qualitatively and quantitatively, with the M2H IC50 values being generally somewhat higher than those from the reporter gene assays. The slight differences in IC50 values between the two assays could arise from the use of fusion proteins with a truncated ER (domains DEF) in the M2H assay versus full-length ER in the reporter gene assay. The variation might also reflect the difference in ability of the compounds to inhibit ER/coactivator interaction depending upon the predominant coactivator protein; the M2H assay uses an SRC1-Gal4 fusion protein, whereas SRC3 comprises the majority of the endogenous coactivator population used in the reporter gene assay. For these reasons, we chose to analyze the structure-activity relationships (Figure 3) based on data from the reporter gene assay (Table 3).
Table 4.
Potency of guanylhydrazone CBI inhibition in the M2H assay. (NA = not assayed)
| CBI | IC50 (μM) | CBI | IC50 (μM) | CBI | IC50 (μM) |
|---|---|---|---|---|---|
| 1 | 3.6 ± 0.6 | 23 | 4.0 ± 2.1 | 29 | 2.6 |
| 18 | 3.8 ± 0.4 | 24 | 4.9 ± 1.5 | 30 | ≥ 50 |
| 19 | 2.1 ± 0.2 | 25 | ≥ 50 | 31 | 11.5 ± 0.7 |
| 20 | 2.5 ± 1.1 | 26 | 5.9 ± 3.4 | 32 | 11.3 ± 3.8 |
| 21 | 9.4 ± 3.6 | 27 | NA | 33 | NA |
| 22 | 6.8 | 28 | 4.6 ± 0.5 |
In both of these cell-based assays, an internal control, beta-galactosidase, was expressed and evaluated as a monitor of cell-toxicity (data not shown). After treatment for a 24-hour period, these compounds produced a decrease in the beta-galactosidase values (50%) only at the highest concentration tested, 20 μM. Because these compounds inhibit the activity of ER at concentrations approximately an order of magnitude lower than this observed toxicity, we conclude that the decrease in luciferase values for both the reporter gene assay and the M2H assay are due to specific inhibition of ER/coactivator interaction, not to generalized cell toxicity.
It is important to differentiate between compounds that function as CBIs from those that function as conventional antagonist ligands. A conventional antagonist will compete with estradiol for the ligand binding pocket and, upon binding, induce a conformational change that prevents coactivator from interacting with receptor. Thus, a traditional antagonist would give a false positive reading in the CBI assay. By contrast, a CBI blocks coactivator binding directly, without displacing estradiol. Therefore, to distinguish antagonists from CBIs, we measured the affinity of the guanylhydrazones for the ligand binding pocket using a radiometric competitive binding assay with tritiated estradiol as the tracer. The binding affinity of each compound is expressed as a percentage relative to the binding of the endogenous ligand, estradiol, which is set at 100%. None of the guanylhydrazones showed relative binding affinities (RBAs) high enough to suspect that their inhibitory activity was caused by traditional antagonist activity. For example, one of the highest potency compounds in the ligand-binding assay, compound 1, has an RBA of 0.004%; direct competition in the CBI assay against estradiol (1 μM) would produce an inhibition curve with an IC50 of 25,000 μM (1 μM/(0.004% / 100)). Because we see IC50 values significantly lower than 25 mM in the CBI assay, we conclude that these compounds are not acting as antagonists by binding to the ligand binding pocket and displacing estradiol, but rather that their inhibitory activity most likely derives from a CBI mechanism.
As additional support that guanylhydrazones act as CBIs and not traditional antagonists, a member of the guanylhydrazone class of compounds (23) was subjected to further testing in the reporter gene assay (Figure 3). In this experiment, increasing concentrations of hydrazone 23 were added to HEC-1 cells as before, but in the presence of two different concentrations of estradiol (1 nM and 100 nM). With a traditional ER antagonist such as hydroxytamoxifen (TOT), one would expect a corresponding 100-fold increase in the IC50 of the compound when assayed with a 100-fold higher estradiol concentration, due to the direct competition of both compounds for the same site in the ligand binding pocket; this is, in fact, observed, as shown in Figure 4b. In contrast, the IC50 of a true CBI, which binds in the coactivator groove and not in the ligand binding pocket, should not be significantly changed by altering the estradiol concentration, as is seen with compound 23, shown in Figure 4a. In this experiment, the IC50 changed only slightly, from 2.5 μM to 3.6 μM, with the 100-fold increase in estradiol; this increase is small enough to be attributed to experimental variability. Thus, the inhibitory activity of the guanylhydrazones presented in these confirmatory assays is insurmountable by excess estradiol, providing further evidence that the activity of these compounds is due to a CBI mechanism.
Figure 4b.
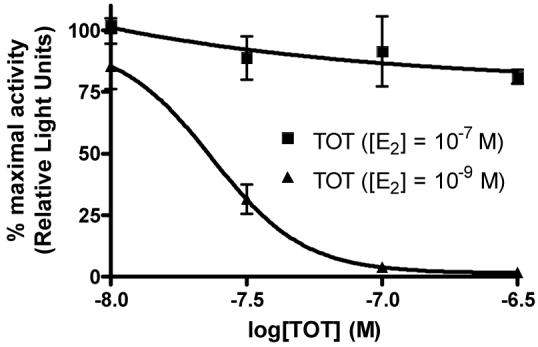
Inhibition curves for hydroxytamoxifen (TOT) with different estradiol (E2) concentrations in the reporter gene assay.
Figure 4a.
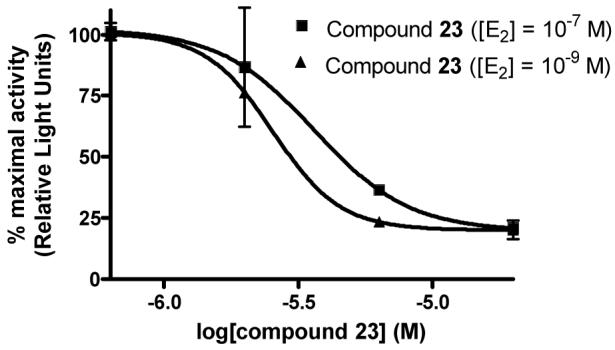
Inhibition curves for hydrazone 23 with different estradiol concentrations in the reporter gene assay. (E2 = estradiol)
Because the β-chlorovinyl imine functionality in many of the most potent guanylhydrazones is potentially electrophilic, it is possible that these compounds are undergoing a covalent reaction with nucleophilic residues in ERα. In fact, a small molecule Mannich base, discovered as an inhibitor of coactivator binding to the thyroid receptor, was found to undergo an elimination reaction during the assay to form an unsaturated ketone that covalently alkylated the receptor through a Michael addition reaction.24 To examine the possibility that our β-chlorovinyl imines were reacting covalently with ERα, we incubated guanylhydrazone 1 or vehicle with ERα for 30 min at 4 °C in phosphate-buffered saline, dialyzed into water overnight, and then determined the molecular weight of the ER by electrospray ionization mass spectrometry (low-res ESI). A change in the molecular weight of the ER protein MS would have been indicative of covalent attachment, but no change in molecular weight was observed (data not shown). Therefore, we consider it unlikely that the guanylhydrazones we have studied are interacting with ER covalently.
Conclusions
In conclusion, we have prepared a series of sixteen guanylhydrazone coactivator binding inhibitors (CBIs) for ER. A direct in vitro assay of estrogen receptor-coactivator interaction confirmed that these compounds were functioning, as expected, by blocking this interaction. Derivatives with substitutions at several positions were tested in cell-based assays for the inhibition of transcriptional activity of the estrogen receptor. In this assay, the best compound in the series, the simple phenylchloro-enal (30), had higher potency than the parent guanylhydrazone (1). The structure-activity relationships for the α-tetralone-based compounds are summarized in Figure 3. Because they had very low ligand binding affinities relative to that of estradiol and their inhibition of transcription was insurmountable by a 100-fold increase in estradiol concentration, it is apparent that these compounds are acting as true CBIs, and not conventional antagonists. These CBIs are currently being tested in further cell-based assays to probe their ability to inhibit estrogen action. Additional work related to the activity and receptor specificity of these and other CBIs will be the subject of a future publication.
Experimental
All reagents were purchased from commercial suppliers and were used without further purification. Anhydrous solvents (with the exception of DMF) were obtained from an anhydrous solvent dispensing system, and anhydrous DMF was obtained by distillation over molecular sieves. For all reactions employing anhydrous solvents, glassware was oven-dried overnight and cooled under vacuum, then purged with argon; all of these reactions were conducted under argon.
1H NMR spectra were recorded on either a 400 or 500 MHz Varian Oxford instrument and 13C NMR were recorded at either 100 or 125 MHz on the same instruments. NMR spectra are reported in ppm and were referenced to the solvent peak and processed using ACD Labs 5.0 software. EI mass spectra were recorded at 70 eV using the 70-VSE mass spectrometer, which was purchased in part with a grant from the Division of Research Resources, National Institutes of Health (RR 04648); ESI mass spectra were recorded using the Quattro mass spectrometer, which was purchased in part with a grant from the Division of Research Resources, National Institutes of Health (RR 07141). Melting points are uncorrected and were obtained using a Thomas Hoover Uni-Melt capillary melting point apparatus.
1-Chloro-3,4-dihydronaphthalene-2-carboxaldehyde (2)
POCl3 (5.49 mL, 60 mmol) was added slowly to anhydrous DMF (10 mL) at 0 °C, and the solution was warmed to 70 °C and stirred for 30 min. α-Tetralone (1.33 mL, 10 mmol) was added and stirring at 70 °C continued for 5 h until complete by TLC (4:1 hexanes/EtOAc). After cooling to 0 °C, 30% w/v NaOAc (10 mL) was added very slowly, followed by water (20 mL), and the product was extracted with EtOAc (3 × 50 mL). The organic layer was washed with saturated LiCl (2 × 30 mL) and brine (30 mL), dried over Na2SO4, filtered and concentrated. Purification by silica gel column (4% EtOAc/hexanes) gave the product as a yellow oil (1.38 g, 72%). 1H NMR (500 MHz, chloroform-d) δ: 10.39 (s, 1H), 7.87 (d, J = 9.0 Hz, 1H), 7.36 (m, 2H), 7.22 (d, J = 6.2 Hz, 1H), 2.85 (dd, J = 8.5, 7.5 Hz, 2H), 2.64 (dd, J = 8.5, 7.5 Hz; 13C NMR (125 MHz, chloroform-d) δ: 190.9, 146.1, 139.2, 132.2, 132.1, 131.6, 127.9, 127.3, 126.5, 27.2, 21.7. HRMS: Calc'd for C11H9OCl [M+]: 192.0342. Found: 192.0339.
1-Chloro-7-methoxy-3,4-dihydronaphthalene-2-carboxaldehyde (3)
POCl3 (4.6 mL, 30 mmol) was added to an oven-dried flask under argon and cooled to −10 °C. DMF (10 mL, 50 mmol) was slowly added and the mixture was then gradually heated to reflux and stirred for 1 h. 7-Methoxy-α-tetralone (881 mg, 5 mmol) was added and refluxed for an additional 2 h until complete by TLC (20% EtOAc/hexanes). The reaction mixture was allowed to cool and then carefully poured into 15% w/v NaOAc and left overnight. The product was extracted with CHCl3 (3 × 50 mL) and washed with water (50 mL) and brine (50 mL), dried over MgSO4, filtered and concentrated to give the product as a brown oil (1.03 g, 93%). 1H NMR (500 MHz, chloroform-d) δ: 2.57 - 2.64 (m, 2 H) 2.73 - 2.79 (m, 2 H) 3.84 (s, 3 H) 6.91 (dd, J=8.15, 2.57 Hz, 1 H) 7.12 (d, J=8.36 Hz, 1 H) 7.40 (d, J=2.57 Hz, 1 H) 10.37 (s, 1 H). 13C NMR (100 MHz, chloroform-d) δ: 21.93, 26.06, 55.48, 111.73, 116.84, 128.58, 131.07, 132.38, 132.90, 145.76, 158.61, 190.77. HRMS: Calc'd for C12H11ClO2 [M+]: 222.0448. Found: 222.0452.
1-Chloro-6-methoxy-3,4-dihydronaphthalene-2-carboxaldehyde (4)
POCl3 (4.6 mL, 30 mmol) was added to an oven-dried flask under argon and cooled to −10 °C. DMF (10 mL, 50 mmol) was slowly added, and then the mixture was gradually heated to reflux and stirred for 1 h. 6-Methoxy-α-tetralone (881 mg, 5 mmol) was added, and the reaction was refluxed for an additional 2 h until complete by TLC (20% EtOAc/hexanes). The reaction mixture was allowed to cool and then carefully poured into 15% w/v NaOAc and left overnight. The product was extracted with CHCl3 (3 × 50 mL) and washed with water (50 mL) and brine (50 mL), dried over MgSO4, filtered and concentrated to give the product as an orange oil (1.02 g, 93%). 1H NMR (500 MHz, chloroform-d) δ: 2.51 - 2.66 (m, 2 H) 2.74 - 2.82 (m, 2 H) 3.84 (s, 3 H) 6.73 (d, J=2.36 Hz, 1 H) 6.82 (dd, J=8.79, 2.57 Hz, 1 H) 7.78 (d, J=8.79 Hz, 1 H) 10.31 (s, 1 H). 13C NMR (100 MHz, chloroform-d) δ: 21.43, 27.48, 55.42, 112.03, 113.49, 124.81, 128.32, 129.73, 141.26, 146.08, 162.13, 190.44. HRMS: Calc'd for C12H12ClO2 [M]+: 222.0447. Found: 222.0441.
1-Chloro-5,7-dimethyl-3,4-dihydronaphthalene-2-carboxaldehyde (5)
POCl3 (4.6 mL, 30 mmol) was added to an oven-dried flask under argon and cooled to −10 °C. DMF (10 mL, 50 mmol) was slowly added, and then the mixture was gradually heated to reflux and stirred for 1 h. 5,7-Dimethyl-α-tetralone (871 mg, 5 mmol) was added, and the reaction was refluxed for an additional 2 h until complete by TLC (20% EtOAc/hexanes). The reaction mixture was allowed to cool and then carefully poured into 15% w/v NaOAc and left overnight. The product was extracted with CHCl3 (3 × 50 mL) and washed with water (50 mL) and brine (50 mL), dried over MgSO4, filtered and concentrated to give the product as a brown oil (1.03 g, 93%). 1H NMR (500 MHz, chloroform-d) δ: 2.27 (s, 3 H) 2.34 (s, 3 H) 2.55 - 2.62 (m, 2 H) 2.72 (t, J=7.93 Hz, 2 H) 7.08 (s, 1 H) 7.56 (s, 1 H) 10.36 (s, 1 H). 13C NMR (100 MHz, chloroform-d) δ: 19.39, 20.97, 21.26, 22.64, 124.89, 131.38, 131.69, 134.26, 134.42, 135.12, 135.82, 146.48, 190.82. HRMS: Calc'd for C13H13ClO [M]+: 220.0655. Found: 220.0655.
1-Chloro-3-dihydrobenzocyclopentene-2-carboxaldehyde (6)
POCl3 (1.22 mL, 13 mmol) and anhydrous DMF (5 mL) were added to an oven-dried flask under argon and cooled to 0 °C. A solution of indanone (1.32 g, 10 mmol) in anhydrous DMF (5 mL) was added and stirring continued at 0 °C for 4 h. The reaction was poured into ice water (200 mL) and allowed to sit overnight. The product was extracted with EtOAc (3 × 75 mL), washed with saturated LiCl (2 × 25 mL) and brine (25 mL), dried over MgSO4, filtered and concentrated. Purification by bulb-to-bulb distillation followed by silica gel column (15% EtOAc/hexanes) gave the product as an orange solid (450 mg, 25%). 1H NMR (400 MHz, chloroform-d) δ: 10.22 (s, 1H), 7.72 (m, 2H), 7.50 (m, 2H), 3.70 (s, 2H). HRMS: Calc'd for C10H7OCl [M]+: 178.0185. Found: 178.0185.
9-Chloro-6,7-dihydro-5-benzocycloheptene-8-carboxaldehyde (7)
POCl3 (5.49 mL, 60 mmol) was added slowly to anhydrous DMF (10 mL) at 0 °C, and the solution was warmed to 70 °C and stirred for 30 min 1-Benzosuberone (1.5 mL, 10 mmol) was added and stirring at 70 °C continued for 6 h until complete by TLC (4:1 hexanes/EtOAc). After cooling to 0 °C, 30% w/v NaOAc (10 mL) was added very slowly, followed by water (20 mL), and the product was extracted with EtOAc (3 × 50 mL). The organic layer was washed with saturated LiCl (2 × 30 mL) and brine (30 mL), dried over Na2SO4, filtered and concentrated. Purification by silica gel column (5% EtOAc/hexanes) gave the product as a yellow oil (1.37 g, 66%). 1H NMR (500 MHz, chloroform-d) δ: 10.39 (s, 1H), 7.67 (m, 1H), 7.37 (m, 2H), 7.26 (m, 1H), 2.61 (t, J = 7.1 Hz, 2H), 2.23 (dd, J = 7.07, 8.2 Hz, 2H), 2.15 (m, 2H). 13C NMR (125 MHz, chloroform-d) δ: 190.5, 170.4, 141.1, 138, 136.5, 130.8, 129.3, 128.6, 126.9, 34.0, 32.1, 22.7. HRMS: Calc'd for C12H11OCl [M]+: 206.0498. Found: 206.0497.
1-Chloro-3-dihydro-4-methylnaphthalene-2-carboxaldehyde (8)
POCl3 (4.6 mL, 30 mmol) was added to an oven-dried flask under argon and cooled to −10 °C. DMF (10 mL, 50 mmol) was slowly added then the mixture was gradually heated to reflux and stirred for 1 h. 4-Methyl-α-tetralone (0.743 mL, 5 mmol) was added and the reaction was refluxed for an additional 2 h until complete by TLC (20% EtOAc/hexanes). The reaction mixture was allowed to cool and then carefully poured into 15% w/v NaOAc and left overnight. The product was extracted with CHCl3 (3 × 50 mL) and washed with water (50 mL) and brine (50 mL), dried over MgSO4, filtered and concentrated to give the product as a brown oil (1.02 g, 98%). 1H NMR (500 MHz, chloroform-d) δ: 1.22 (d, J=6.86 Hz, 1 H) 2.52 (dd, J=16.62, 7.40 Hz, 1 H) 2.64 - 2.72 (m, 1 H) 2.95 - 3.05 (m, 1 H) 7.25 (d, J=7.50 Hz, 1 H) 7.30 - 7.36 (m, 1 H) 7.38 - 7.44 (m, 1 H) 7.88 (dd, J=7.72, 1.07 Hz, 1 H) 10.39 (s, 1 H). 13C NMR (100 MHz, chloroform-d) δ: 19.66, 28.90, 31.21, 126.36, 126.44, 126.91, 130.64, 130.99, 131.73, 143.90, 145.21, 190.95. HRMS: Calc'd for C12H11ClO [M]+: 206.0498. Found: 206.0492.
β-Chlorocinnamaldehyde (9)
POCl3 (1.22 mL, 1.33 mmol) was added to anhydrous DMF (10 mL), and the mixture was stirred for 30 min Acetophenone (1.17 mL, 10 mmol) was added and the mixture was stirred for 28 h at 0 °C. The reaction mixture was poured into ice water (80 mL) and allowed to sit overnight, then extracted with EtOAc (3 × 70 mL). The combined organic extracts were washed with sat. LiCl (2 × 40 mL) and brine (30 mL), dried over Na2SO4, filtered and concentrated. The mixture was purified by bulb-to-bulb distillation and 11 was isolated as a brown oil (807 mg, 49%). 1H NMR (400 MHz, chloroform-d) δ: 6.68 (d, J=7.08 Hz, 1 H) 7.42 - 7.56 (m, 4 H) 7.72 - 7.80 (m, 1 H) 10.23 (d, J=6.84 Hz, 1 H). 13C NMR (125 MHz, chloroform-d) δ: 191.5, 152.3, 135.5, 131.8, 128.9, 127.2, 124.4.
β-Chloro-α-methylcinnamaldehyde (10)
POCl3 (5.49 mL, 60 mmol) was added slowly to anhydrous DMF (10 mL) at 0 °C, and the solution was warmed to 70 °C and stirred for 30 min Propiophenone (1.3 mL, 10 mmol) was added and stirring at 70 °C continued for 4 h until complete by TLC (4:1 hexanes/EtOAc). After cooling to 0 °C, 30% w/v NaOAc (10 mL) was added very slowly, followed by water (20 mL), and the product was extracted with EtOAc (3 × 50 mL). The organic layer was washed with saturated LiCl (2 × 30 mL) and brine (30 mL), dried over Na2SO4, filtered and concentrated. The resulting oil was bulb-to-bulb distilled, giving the product as a 6:1 mixture of E to Z isomers as a yellow oil (1.2 g, 67%). 1H NMR (400 MHz, chloroform-d) E isomer δ: 9.48 (s, 1H), 7.42 (m, 5H), 2.09 (s, 3H); Z isomer δ: 10.41 (s, 1H), 7.42 (m, 5H), 1.85 (s, 3H). 13C NMR (125 MHz, chloroform-d) δ: 190.6, 170.4, 136.5, 135.9, 130.6, 130.3, 128.7, 13.5. HRMS: Calc'd for C10H9ClO [M]+: 179.0264. Found: 179.0273
3-Chloro-3-naphthalen-1-yl-propenal (11)
POCl3 (5.49 mL, 60 mmol) was added slowly to anhydrous DMF (10 mL) at 0 °C, and stirred for 30 min 1-Acetonaphthone (1.5 mL, 10 mmol) was added and stirring at 0 °C continued for 3 h until complete by TLC (4:1 hexanes/EtOAc). 30% w/v NaOAc (10 mL) was added very slowly, followed by water (20 mL), and the product was extracted with EtOAc (3 × 50 mL). The organic layer was washed with saturated LiCl (2 × 30 mL) and brine (30 mL), dried over Na2SO4, filtered and concentrated. Purification by silica gel column (15% EtOAc/hexanes) gave the product as a brown oil (340 mg, 16%). 1H NMR (500 MHz, chloroform-d) δ: 6.80 (d, J=6.65 Hz, 1 H) 7.51 - 7.63 (m, 2 H) 7.72 (dd, J=8.68, 2.04 Hz, 1 H) 7.82 - 7.88 (m, 2 H) 7.91 (d, J=7.07 Hz, 1 H) 8.32 (s, 1 H) 10.27 (d, J=6.86 Hz, 1 H). 13C NMR (125 MHz, chloroform-d) δ: 191.4, 152.1, 134.6, 132.6, 132.5, 129.1, 128.6, 128.5, 128.3, 127.2, 124.5, 123.0. HRMS: Calc'd for C13H9OCl [M]+: 216.0342. Found: 216.0336.
1-Bromo-3,4-dihydronaphthalene-2-carboxaldehyde (12)
POBr3 (10.4 g, 36 mmol) was added to an oven-dried flask under argon and cooled to 0 °C. DMF (18.6 mL, 240 mmol) was slowly added. Then the mixture was gradually heated to reflux and stirred for 1 h. α-Tetralone (0.743 mL, 5 mmol) was added and refluxed for an additional 2 h until complete by TLC (20% EtOAc/hexanes). The reaction mixture was allowed to cool and then carefully poured into ice water (100 mL) and left overnight. The product was extracted with EtOAc (2 × 200 mL) and washed with water (4 × 50 mL) and brine (2 × 50 mL), dried over MgSO4, filtered and concentrated. Purification by silica gel column gave the product as an orange solid (751 mg, 32%). 1H NMR (400 MHz, chloroform-d) δ: 2.58 - 2.70 (m, 2 H) 2.85 (t, J=7.93 Hz, 2 H) 7.16 - 7.25 (m, 1 H) 7.30 - 7.45 (m, 2 H) 7.84 - 7.96 (m, 1 H) 10.27 (s, 1 H). 13C NMR (100 MHz, chloroform-d) δ: 22.83, 27.14, 127.12, 127.58, 128.72, 131.35, 132.30, 132.96, 134.47, 139.03, 193.20. HRMS: Calc'd for C11H9BrO [M]+: 235.9843. Found: 235.9836.
1-Methoxy-3,4-dihydronaphthalene-2-carboxaldehyde (13)
1-Chloro-3,4-dihydronaphthalene-2-carboxaldehyde (2, 390 mg, 2 mmol) was added to an oven-dried flask under argon. Sodium methoxide in methanol (0.5 M, 6 mL, 3 mmol) was added and the reaction was stirred for 3 h at room temperature. The solvent was evaporated and the resulting residue redissolved in EtOAc (100 mL), washed with water (3 × 20 mL) and brine (20 mL), dried over MgSO4, filtered and concentrated to give the product as a clear oil (318 mg, 85%). 1H NMR (500 MHz, chloroform-d) δ: 2.47 - 2.55 (m, 2 H) 2.73 - 2.80 (m, 2 H) 3.87 - 3.91 (m, 3 H) 7.22 (dd, J=7.29, 0.86 Hz, 1 H) 7.26 - 7.36 (m, 2 H) 7.54 (dd, J=7.50, 1.07 Hz, 1 H) 10.26 (s, 1 H). 13C NMR (100 MHz, chloroform-d) δ: 18.79, 27.26, 62.75, 123.49, 124.03, 126.68, 128.16, 129.58, 130.77, 140.73, 167.53, 189.78. HRMS: Calc'd for C12H12O2 [M]+: 188.0837. Found: 188.0840.
1-Phenoxy-3,4-dihydronaphthalene-2-carboxaldehyde (14)
K2CO3 (3.22 g, 20.4 mmol) was suspended in DMF (20 mL) and phenol (960 mg, 10.2 mmol) was added. The mixture was refluxed for 1 h, then 1-chloro-3,4-dihydronaphthalene-2-carboxaldehyde (2, 490 mg, 2.54 mmol) was added and refluxed for another 24 h until complete by TLC (3:1 hexanes/EtOAc). Water (25 mL) was added and the product was extracted with CHCl3 (3 × 50 mL) and the combined organic layers were washed with 1 M KOH (50 mL) and brine (50 mL), dried over MgSO4, filtered and concentrated to give the product as a yellow solid (397 mg, 62%). 1H NMR (500 MHz, chloroform-d) δ: 2.63 - 2.75 (m, 2 H) 2.93 (t, J=7.93 Hz, 2 H) 6.94 - 7.06 (m, 3 H) 7.13 (t, J=7.50 Hz, 1 H) 7.20 - 7.33 (m, 4 H) 7.36 (d, J=7.72 Hz, 1 H) 10.18 (s, 1 H). 13C NMR (100 MHz, chloroform-d) δ: 19.08, 27.18, 115.51, 122.68, 124.86, 125.28, 126.73, 128.03, 129.34, 129.90, 130.88, 140.21, 158.27, 160.79, 189.86. HRMS: Calc'd for C17H14O2 [M]+: 250.994. Found: 250.995.
1-(1,1,1,-Trifluoromethyl)-3,4-dihydronaphthalene-2-carboxaldehyde (15)
1-Bromo-3,4-dihydronaphthalene-2-carboxaldehyde (12, 237 mg, 1 mmol) and CuI (10 mg, 0.05 mmol) were added to an oven-dried flask under argon and dissolved in anhydrous DMF (5 mL) and FSO2CF2CO2Me (0.161 mL, 1.1 mmol) was added. The reaction was heated to 80 °C for 4 h at which point starting material was still present by TLC (10% EtOAc/hexanes), so another portion of FSO2CF2CO2Me (0.161 mL, 1.1 mmol) was added and stirred for another 24 h at 80 °C. After cooling to room temperature, the reaction was diluted with EtOAc (100 mL) and washed with water (3 × 25 mL) and brine (2 × 25 mL), dried over MgSO4, filtered and concentrated. Purification by silica gel column (CH2Cl2) gave the product as a yellow oil (106 mg, 45%). 1H NMR (500 MHz, chloroform-d) δ: 2.59 - 2.67 (m, 2 H) 2.77 (t, J=7.83 Hz, 2 H) 7.24 - 7.29 (m, 1 H) 7.29 - 7.40 (m, 2 H) 7.54 - 7.61 (m, 1 H) 10.34 (q, J=2.36 Hz, 1 H). 13C NMR (100 MHz, chloroform-d) δ: 21.12, 26.89, 122.49, 124.70, 126.42 (q, J = 3.45 Hz), 126.94 (q, J = 0.92 Hz), 127.93, 128.73 (q, J = 1.61 Hz), 130.42, 138.22, 141.86 (q, J = 2.30 Hz), 189.81 (q, J = 5.52 Hz) HRMS: Calc'd for C12H9F3O [M]+: 226.0606. Found: 226.0606.
1-(1-Chloro-3,4-dihydro-naphthalen-2-yl)-ethanol (16)
1-Chloro-3,4-dihydronaphthalene-2-carboxaldehyde (2, 193 mg, 1 mmol) was added to an oven-dried flask under argon and dissolved in dry Et2O (10 mL) and cooled to −10 °C. CH3Li in Et2O (1.6 M, 3.12 mL, 5 mmol) was added and the mixture stirred for 1 ½ h at −10 °C until the reaction was complete by TLC (10% EtOAc/hexanes). The reaction was quenched with saturated NH4Cl (25 mL) and extracted with EtOAc (100 mL) and washed with water (2 × 25 mL) and brine (2 × 25 mL), dried over MgSO4, filtered and concentrated to give an off white solid (199 mg, 95%). 1H NMR (400 MHz, chloroform-d) δ: 1.36 (d, J=6.59 Hz, 3 H) 1.90 (s, 1 H) 2.36 - 2.49 (m, 1 H) 2.52 - 2.64 (m, 1 H) 2.76 - 2.88 (m, 2 H) 5.27 (q, J=6.35 Hz, 1 H) 7.09 - 7.18 (m, 1 H) 7.18 - 7.33 (m, 2 H) 7.63 (d, J=7.57 Hz, 1 H).
1-(1-Chloro-3,4-dihydro-naphthalen-2-yl)-ethanone (17)
1-(1-Chloro-3,4-dihydro-naphthalen-2-yl)-ethanol (16, 78 mg, 0.374 mmol) was dissolved in dry CH2Cl2 (5 mL). A solution of Dess-Martin reagent (206 mg, 0.486 mmol) in dry CH2Cl2 (5 mL) was added and the reaction was stirred for 25 min under open atmosphere until complete by TLC (2:1 hexanes/acetone). The mixture was diluted with EtOAc (100 mL) and washed with 1 M KOH (2 × 25 mL), water (2 × 25 mL) and brine (25 mL), dried over MgSO4, filtered and concentrated to give an orange oil (67 mg, 87%). 1H NMR (400 MHz, chloroform-d) δ: 2.60 (s, 3 H) 2.63 - 2.72 (m, 2 H) 2.80 - 2.93 (m, 2 H) 7.14 - 7.22 (m, 1 H) 7.28 - 7.37 (m, 2 H) 7.71 - 7.89 (m, 1 H). 13C NMR (100 MHz, chloroform-d) δ: 26.49, 27.51, 31.26, 126.02, 126.91, 127.21, 129.78, 132.29, 132.70, 135.41, 137.34, 201.23. HRMS: Calc'd for C12H11ClO [M]+: 206.0498. Found: 206.0497.
General procedure for hydrazone formation
The corresponding β-chlorovinylaldehyde (1.0 mmol) was dissolved in CH3OH (10 mL) and aminoguanidine HCl (122 mg, 1.1 mmol) was added, followed by acetic acid (5 mL). The mixture was refluxed until complete by TLC (3:1 hexanes/EtOAc), usually about 4 h. After cooling to room temperature, the solvent was evaporated under a stream of N2 and the resulting solid was suspended in water and was washed with EtOAc (3 × 50 mL). The aqueous layer was then neutralized with 1 M KOH and the product extracted with EtOAc (2 × 75 mL). The combined organic extracts were washed with brine (3 × 20 mL), dried over Na2SO4, filtered and concentrated. The product was redissolved in EtOAc (5 mL) and 1 M HCl in Et2O (5 mL) was added. The precipitated HCl salt was collected on a frit and washed with EtOAc and hexanes and dried thoroughly.
1-Chloro-3,4-dihydronaphthalene-2-carboxaldehyde aminoguanylhydrazone HCl (1)
Following the general procedure for hydrazone formation, 1 was obtained as a white powder (221 mg, 78%). mp 272-274 °C. 1H NMR (500 MHz, methanol-d4) δ: 8.42 (s, 1H), 7.59 (dd, J = 4.88, 4.15 Hz, 1H), 7.19 (dd, J = 4.88, 3.91 Hz, 2H), 7.12 (m, 1H), 2.73 (m, 4H). 13C NMR (125 MHz, methanol-d4) δ: 147.2, 139.2, 133.5, 131.2, 130.9, 128.5, 128, 126.7, 28.1, 24.3. HRMS: Calc'd for C12H14N4Cl [M+H]+: 249.0907. Found: 249.0898.
1-Chloro-7-methoxy-3,4-dihydronaphthalene-2-carboxaldehyde aminoguanylhydrazone HCl (18)
Following the general procedure for hydrazone formation, 18 was obtained as a white solid (200 mg, 63%). mp 238 °C. 1H NMR (500 MHz, methanol-d4) δ: 2.67 (s, 4 H) 3.69 (s, 3 H) 6.75 (d, J=6.22 Hz, 1 H) 6.96 - 7.16 (m, 2 H) 8.41 (s, 1 H). 13C NMR (125 MHz, methanol-d4) δ: 24.65, 27.18, 49.85, 55.88, 112.15, 115.88, 129.4, 131.16, 131.57, 134.45, 136.45, 147.28, 160.06. HRMS: Calc'd for C13H16N4OCl [M+H]+: 279.1013. Found: 279.1013.
1-Chloro-6-methoxy-3,4-dihydronaphthalene-2-carboxaldehyde aminoguanylhydrazone HCl (19)
Following the general procedure for hydrazone formation, 19 was obtained as a white solid (105 mg, 33%). mp 216 °C. 1H NMR (500 MHz, methanol-d4) δ: 2.61 - 2.80 (m, 4 H) 3.72 (s, 3 H) 6.65 - 6.78 (m, 2 H) 7.52 (d, J=8.58 Hz, 1 H) 8.39 (s, 1 H). 13C NMR (125 MHz, methanol-d4) δ: 24.17, 28.52, 55.89, 112.84, 114.44, 126.48, 128.12, 128.46, 136.75, 141.26, 147.54, 156.83, 162.50. HRMS: Calc'd for C13H16N4OCl [M+H]+: 279.1013. Found: 279.1009.
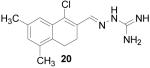
1-Chloro-5,7-dimethyl-3,4-dihydronaphthalene-2-carboxaldehyde aminoguanylhydrazone HCl (20)
Following the general procedure for hydrazone formation, 20 was obtained as a white solid (176 mg, 56%). mp decomposed at 242 °C. 1H NMR (500 MHz, methanol-d4) δ: 2.17 (d, J=13.72 Hz, 3 H) 2.19 (s, 3 H) 2.66 (s, 4 H) 6.91 (s, 1 H) 7.29 (s, 1 H) 8.41 (s, 1 H). 13C NMR (125 MHz, methanol-d4) δ: 19.45, 21.13, 23.79, 24.01, 49.85, 125.03, 130.42, 133.29, 133.61, 134.43, 136.07, 136.82, 147.51, 152.39. HRMS: Calc'd for C14H18N4Cl [M+H]+: 277.1220. Found: 277.1207.
1-Chloro-3-dihydro-4-methylnaphthalene-2-carboxaldehyde aminoguanylhydrazone HCl (21)
Following the general procedure for hydrazone formation, the product did not precipitate upon HCl addition. The crude free base was used for CBI testing, no yield recorded. 1H NMR (500 MHz, methanol-d4) δ: 1.19 (s, 3 H) 2.68 - 2.88 (m, 2 H) 2.87 - 3.00 (m, 1 H) 7.08 - 7.33 (m, 3 H) 7.57 - 7.73 (m, 1 H) 8.47 (s, 1 H).
1-Chloro-3-dihydrobenzocyclopentene-2-carboxaldehyde (22)
Following the general procedure for hydrazone formation, 22 was obtained as a white powder (96 mg, 63%). mp 206-210 °C. 1H NMR (500 MHz, methanol-d4) δ: 8.27 (s, 1H), 7.49 (m, 2H), 7.37 (m, 2H), 3.78 (m, 2H). ESI MS: C11H12ClN4 [M+H]+: 235.1.
9-Chloro-6,7-dihydro-5-benzocycloheptene-8-carboxaldehyde aminoguanylhydrazone HCl (23)
Following the general procedure for hydrazone formation, 23 was obtained as a white powder (176 mg, 59%). mp 214-216 °C. 1H NMR (500 MHz, methanol-d4) δ: 8.55 (s, 1H), 7.58 (m, 2H), 7.33 (m, 2H), 2.64 (t, J = 7.08 Hz, 2H), 2.46 (t, J = 6.96 Hz, 2H), 2.19 (t, 7.08 Hz, 2H). 13C NMR (125 MHz, methanol-d4) δ: 157.1, 147.1, 141.6, 136.5, 134.2, 131.1, 130.6, 129.7, 129.1, 127.5, 34.4, 25.0. ESI MS: [M+H]+ = 263.1.
1-Bromo-3,4-dihydronaphthalene-2-carboxaldehyde aminoguanylhydrazone HCl (24)
Following the general procedure for hydrazone formation, 24 was obtained as a white solid (170 mg, 52%). mp 230-232 °C. 1H NMR (500 MHz, methanol-d4) δ: 2.71 - 2.90 (m, 4 H) 7.12 - 7.21 (m, 1 H) 7.24 (dd, J=5.36, 3.43 Hz, 2 H) 7.62 - 7.76 (m, 1 H) 8.49 (s, 1 H). 13C NMR (125 MHz, methanol-d4) δ: 25.30, 28.18, 127.90, 127.93, 128.35, 128.76, 129.81, 130.79, 134.08, 134.59, 139.23, 149.93. HRMS: Calc'd for C12H14N4Br [M+H]+: 293.0402. Found: 293.0393.
1-Methoxy-3,4-dihydronaphthalene-2-carboxaldehyde aminoguanylhydrazone HCl (25)
Following the general procedure for hydrazone formation, the product did not precipitate upon HCl addition. The crude free base was used for CBI testing, no yield recorded. 1H NMR (500 MHz, methanol-d4) δ: 2.58 - 2.83 (m, 4 H) 3.67 (s, 3 H) 7.08 - 7.25 (m, 3 H) 7.31 - 7.40 (m, 1 H) 8.29 (s, 1 H).
1-Phenoxy-3,4-dihydronaphthalene-2-carboxaldehyde aminoguanylhydrazone HCl (26)
Following the general procedure for hydrazone formation, 26 was obtained as an orange solid (224 mg, 66%). mp 262-264 °C. 1H NMR (500 MHz, methanol-d4) δ: 2.75 (t, J=7.83 Hz, 2 H) 2.87 (t, J=7.93 Hz, 2 H) 6.80 - 6.92 (m, 3 H) 6.97 (t, J=7.50 Hz, 1 H) 7.05 - 7.22 (m, 5 H) 8.12 (s, 1 H). 13C NMR (125 MHz, methanol-d4) δ: 22.04, 28.26, 116.31, 123.34, 123.50, 124.61, 126.63, 127.60, 128.89, 130.51, 131.00, 131.21, 140.20, 145.83, 153.06, 159.54. HRMS: Calc'd for C18H19N4O [M+H]+: 307.1559. Found: 307.1563.
1-(1,1,1,-Trifluoromethyl)-3,4-dihydronaphthalene-2-carboxaldehyde aminoguanylhydrazone HCl (27)
Following the general procedure for hydrazone formation, 27 was obtained as a white solid (55 mg, 81%). mp decomposed at 230 °C. 1H NMR (400 MHz, methanol-d4) δ: 2.69 - 2.78 (m, 2 H) 2.78 - 2.87 (m, 2 H) 7.20 - 7.29 (m, 3 H) 7.36 - 7.45 (m, 1 H) 8.42 - 8.47 (m, 1 H). 13C NMR (125 MHz, methanol-d4) δ: 24.45, 28.35, 124.38, 126.51, 126.55, 127.12, 127.86, 127.90, 128.88, 130.34, 139.22, 145.95, 145.96. HRMS: Calc'd for C13H15N4F3 [M+H]+: 283.1171. Found: 283.1166.
β-Chlorocinnamaldehyde aminoguanylhydrazone HCl (28)
Following the general procedure for hydrazone formation, 28 was obtained as a white powder (210 mg, 81%). mp 182-184 °C. 1H NMR (500 MHz, methanol-d4) δ: 7.08 (d, J=8.79 Hz, 1 H) 7.45 (d, J=3.00 Hz, 3 H) 7.73 - 7.80 (m, 2 H) 8.32 (d, J=9.00 Hz, 1 H). 13C NMR (125 MHz, methanol-d4) δ: 147.2, 142.0, 137.6, 131.4, 129.8, 127.6, 122.5. ESI MS: [M+H]+ = 223.1.
β-Chloro-α-methylcinnamaldehyde aminoguanylhydrazone HCl (29)
Following the general procedure for hydrazone formation, 29 was obtained as a white solid (183 mg, 67%). Note: began with a 6:1 mixture of E to Z isomers, and only one isomer was obtained for the guanylhydrazone. mp 220-222 °C. 1H NMR (500 MHz, methanol-d4) δ: 2.25 (s, 3 H) 7.37 (d, J=7.72 Hz, 2 H) 7.41 - 7.50 (m, 3 H) 7.75 (s, 1 H). 13C NMR (125 MHz, methanol-d4) δ: 157.0, 148.6, 142.3, 138.6, 132.4, 131.0, 130.1, 15.2. ESI MS: [M+H]+: 237.1.
3-Chloro-3-naphthalen-1-yl-propenal aminoguanylhydrazone HCl (30)
Following the general procedure for hydrazone formation, 30 was obtained as a white powder (96 mg, 63%). mp 190-192 °C. 1H NMR (500 MHz, methanol-d4) δ: 6.85 (d, J=9.86 Hz, 1 H) 7.23 (d, J=9.00 Hz, 1 H) 7.51 - 7.67 (m, 2 H) 7.77 - 8.03 (m, 3 H) 8.30 (s, 1 H). ESI MS: C14H13N4Cl [M+H]+: 273.
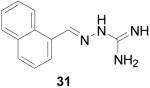
2-(1-Naphthalenylmethylene) hydrazinecarboximidamide HCl (31)
Following the general procedure for hydrazone formation, 31 was obtained as a white solid (241 mg, 97%). mp 162 °C. 1H NMR (500 MHz, methanol-d4) δ: 8.87 (s, 1H), 8.42 (d, J = 8.36 Hz, 1H), 8.09 (d, J = 7.08 Hz, 1H), 7.94 (d, J = 8.15 Hz, 1H), 7.89 (d, J = 8.15 Hz, 1H), 7.58 (m, 1H), 7.51 (t, J = 7.72 Hz, 2H). 13C NMR (125 MHz, methanol-d4) δ: 157.3, 148.4, 135.5, 132.7, 132.5, 130.2, 130.1, 128.6, 128.2, 127.5, 126.6, 124.3. ESI MS: [M+H]+: 213.2.
2-(2-Naphthalenylmethylene)hydrazinecarboximidamide HCl (32)
Following the general procedure for hydrazone formation, 32 was obtained as a white solid (216 mg, 87%). mp 260-262 °C. 1H NMR (500 MHz, methanol-d4) δ: 8.34 (s, 1H), 8.20 (m, 1H), 8.19 (2, 1H), 7.94 (m, 2H), 7.55 (m, 2H). 13C NMR (125 MHz, methanol-d4) δ: 155.5, 146.8, 133.9, 132.7, 131.3, 129.5, 128.4, 128.3, 127.8, 127.4, 126.8, 123.1. ESI MS: [M+H]+ = 213.2.
1-(1-Chloro-3,4-dihydro-naphthalen-2-yl)-ethanone aminoguanylhydrazone HCl (33)
Following the general procedure for hydrazone formation, 33 was obtained as a yellow solid (10 mg, 14%). mp 86-88 °C. 1H NMR (400 MHz, methanol-d4) δ: 2.23 (d, J=32.96 Hz, 3 H) 2.39 - 2.61 (m, 1 H) 2.63 - 2.72 (m, 1 H) 2.82 - 3.09 (m, 2 H) 7.14 - 7.34 (m, 4 H) 7.58 - 7.70 (m, 1 H). 13C NMR (125 MHz, methanol-d4) δ: 21.54, 27.18, 28.21, 28.81, 29.98, 125.82, 126.07, 128.01, 128.23, 128.49, 128.82, 130.27, 130.96. HRMS: Calc'd for C13H16N4Cl [M+H]+: 263.1063. Found: 263.1052.
Luciferase Reporter Gene Assay
Human endometrial cancer (HEC-1) cells were maintained in culture as described and transfected in 24 well plates.20 A mixture of HBSS (50 μL/well), Holo-transferrin (Sigma T1408) (20 μL/well), and lipofectin (Invitrogen #18292-011) (5 μL/well) were incubated at room temperature for 5 minutes. The DNA mixture was made by adding 200 ng of pCMVβ-galactosidase as internal control, 500 ng of the estrogen responsive reporter gene plasmid 2ERE Luc, and 100 ng of full-length ER alpha expression vector with 75 μL HBSS per well and, after addition to the first mixture, allowed to incubate for 20 minutes at room temperature. The cell media was changed to Opti-MEM (350 μL/well) and 150 μL of the transfection mixture was added to each well. The cells were incubated at 37 °C in a 5% CO2 containing incubator for 6 h. The medium was then replaced with fresh medium containing 5% charcoal-dextran-treated calf serum and the desired concentrations of ligands. Reporter gene activity was assayed at 24 h after ligand addition. Luciferase activity, normalized for the internal control β-galactosidase activity, was assayed as described.20 In the initial screen, antagonist activity was determined at four concentrations, ranging from 20 μM to 0.6 μM, in the presence of 10−9 M estradiol (E2). Upon validation that compounds acted as antagonists, mechanism of action was examined by repeating the compound titration in the presence of both 10−7 and 10−9 M E2.
Mammalian Two-Hybrid Assay
Human endometrial cancer (HEC-1) cells were maintained in culture and transfected in 24-well plates using lipofectin as described previously.23 Briefly, HEC-1 cells were plated at 2 ×104 cells per well in 24-well plates and transfected 24 h later with 1 μg of pFR-Luc (Stratagene), 0.2 μg of pCMV β-Gal, 0.2 μg of pM-SRC-1NRD, and 0.2 μg of pVP16-ERDEF (plasmids prepared as previously described).23 At 8 h after transfection, cells were treated with ligand or control vehicle. Cells were harvested 24 h after ligand treatment, and cell extracts were prepared. Both β-Galactosidase and luciferase activity were assayed as described previously.23
Estrogen Receptor Relative Binding Affinity Assays
Relative binding affinities (RBA) were determined by a competitive radiometric binding assay as previously described20 using 2 nM [3H]estradiol as tracer ([2,4,6,7-3H]estra-1,3,5,(10)-triene-3,17β-diol, 89 Ci/mmol, Amersham/GE Healthcare Bio-Sciences Corp, Piscataway, NJ) and purified full-length human ERα and ERβ receptors (PanVera/Invitrogen, Carlsbad, CA). Incubations were for 18-24 h at 0 °C. Hydroxyapaptite (BioRad, Hercules, CA) was used to absorb the receptor-ligand complexes, and free ligand was washed away. The binding affinities are expressed as relative binding affinity (RBA) values with that of estradiol set to 100%. Estradiol binds to ERα with a Kd of 0.2 nM and to ERβ with a Kd of 0.5 nM.
Scheme 6.

Figure 1.
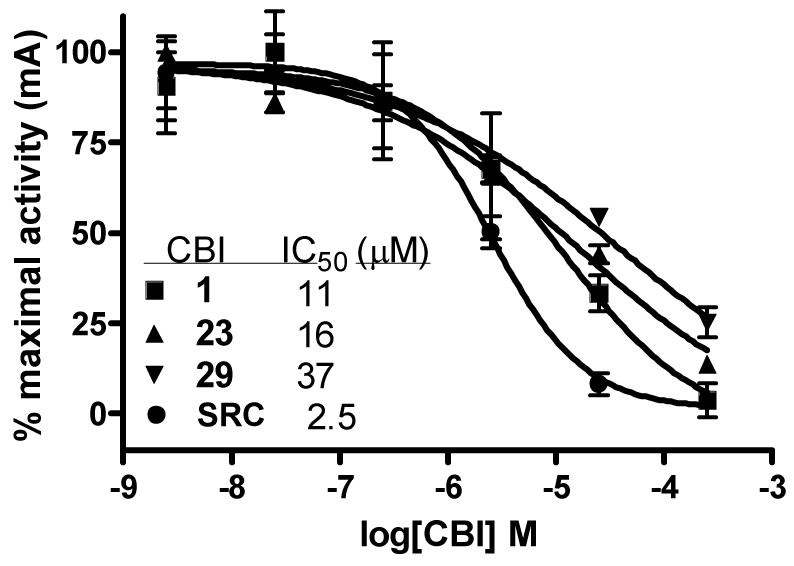
Dose-dependent inhibition of ER/coactivator interaction for selected guanylhydrazone CBIs (1, 23, 29), and positive control 15-mer SRC1-Box II peptide (SRC), as evaluated with a fluorescence polarization assay.7
Acknowledgments
We are grateful for the support of this research by grants from the National Institutes of Health (PHS 5R37 DK15556) and J.R.G. received support from a David Robertson Fellowship and the National Institutes of Health (NRSA 1 F30 ES016484-01 and NRSA 5 T32 GM070421).
References
- 1.Biggins JB, Koh JT. Current Opinion in Chem. Bio. 2007;11:99–110. doi: 10.1016/j.cbpa.2006.10.042. [DOI] [PubMed] [Google Scholar]
- 2.Ring A, Dowsett M. Endocrine-Related Cancer. 2004;11:643–658. doi: 10.1677/erc.1.00776. [DOI] [PubMed] [Google Scholar]
- 3.Gronemyer H, Gustafsson J, Lauder V. Nature Rev. Drug Disc. 2004;3:950–964. doi: 10.1038/nrd1551. [DOI] [PubMed] [Google Scholar]
- 4.Meegan MJ, Lloyd DG. Current. Med. Chem. 2003;10:181–210. doi: 10.2174/0929867033368501. [DOI] [PubMed] [Google Scholar]
- 5.Carlson KE, Choi I, Gee A, Katzenellenbogen BS, Katzenellenbogen JA. Biochemistry. 1997;36:14897–14905. doi: 10.1021/bi971746l. [DOI] [PubMed] [Google Scholar]
- 6.Katzenellenbogen JA, Johnson HJ, Jr., Myers HN. Biochemistry. 1973;12:4085–4092. doi: 10.1021/bi00745a010. [DOI] [PubMed] [Google Scholar]
- 7.Rodriguez AL, Tamrazi A, Collins ML, Katzenellenbogen JA. J. Med. Chem. 2004;47:600–611. doi: 10.1021/jm030404c. [DOI] [PubMed] [Google Scholar]
- 8.Geistlinger TR, McReynolds AC, Guy RK. Chemistry & Biology. 2004;11:273–281. doi: 10.1016/j.chembiol.2004.01.016. [DOI] [PubMed] [Google Scholar]
- 9.Galande AK, Bramlett KS, Burris TP, Wittliff JL, Spatola AF. J. of Peptide Res. 2004;63:297–302. doi: 10.1111/j.1399-3011.2004.00152.x. [DOI] [PubMed] [Google Scholar]
- 10.Gunther JR, Moore TW, Collins ML, Katzenellenbogen JA. ACS Chem. Biol. 2008;3:282–286. doi: 10.1021/cb800056r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shao D, Berrodin TJ, Manas E, Hauze D, Powers R, Bapat A, Gonder D, Winneker RC, Frail DE. J. Steroid Biochem. Mol. Biol. 2004;88:351–360. doi: 10.1016/j.jsbmb.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 12.Virgilio JA, Heilweil E. Org. Prep. Proced. Int. 1982;14:9–20. [Google Scholar]
- 13.Karmakar AC, Sharma S, Chatterjee BG, Ray JK. Indian J. Chem., Sect A. 1988;27:364–365. [Google Scholar]
- 14.Rappaport Z, Gazit A. J. Org. Chem. 1985;50:3184–3194. [Google Scholar]
- 15.Rappaport Z, Gazit A. J. Org. Chem. 1986;51:4112–4131. [Google Scholar]
- 16.Duan J, Dolbier WR, Chen Q. J. Org. Chem. 1998;63:9486–9489. [Google Scholar]
- 17.Guo X, Chen G. J. Fluor. Chem. 1999;97:149–156. [Google Scholar]
- 18.Ichikawa J, Yokota N, Kobayahsi M, Minami T. Synlett. 1993:186–188. [Google Scholar]
- 19.House HO, Respess WL, Whitesides GM. J. Org. Chem. 1966;31:3128–3141. [Google Scholar]
- 20.Dess DB, Martin JC. J. Org. Chem. 1983;48:4155–4156. [Google Scholar]
- 21.Almeida M, Boman A, Lundstedt T. J. Labelled Compd. Radiopharm. 2002;45:371–377. [Google Scholar]
- 22.Sun J, Meyers MJ, Fink BE, Rajendran R, Katzenellenbogen JA, Katzenellenbogen BS. Endocrinology. 1999;140:800–804. doi: 10.1210/endo.140.2.6480. [DOI] [PubMed] [Google Scholar]
- 23.Delage-Mourroux R, Martini PG, Choi I, Kraichely DM, Hoeksema J, Katzenellenbogen BS. J. Biol. Chem. 2000;275:35848–35856. doi: 10.1074/jbc.M001327200. [DOI] [PubMed] [Google Scholar]
- 24.Arnold LA, Estébanez-Perpiñá E, Togashi M, Jouravel N, Shelat A, McReynolds AC, Mar E, Nguyen P, Baxter JD, Fletterick RJ, Webb P, Guy RK. J. Biol. Chem. 2005;280:43048–43055. doi: 10.1074/jbc.M506693200. [DOI] [PubMed] [Google Scholar]