

Abstract
Focal Adhesion Kinase (FAK) is a non-receptor tyrosine kinase that plays a key role in cellular processes such as cell adhesion, migration, proliferation and survival. Recent studies have also implicated FAK in the regulation of cell-cell adhesion. Here, evidence is presented showing that siRNA-mediated suppression of FAK levels in NBT-II cells and expression of dominant negative mutants of FAK caused loss of epithelial cell morphology and inhibited the formation of cell-cell adhesions. Rac and Rho have been implicated in the regulation of cell-cell adhesions and can be regulated by FAK signaling. Expression of active Rac or Rho in NBT-II cells disrupted formation of cell-cell contacts, thus promoting a phenotype similar to FAK-depleted cells. The loss of intercellular contacts in FAK-depleted cells is prevented upon expression of a dominant negative Rho mutant, but not a dominant negative Rac mutant. Inhibition of FAK decreased tyrosine phosphorylation of p190RhoGAP and elevated the level of GTP-bound Rho. This suggests that FAK regulates cell-cell contact formation by regulation of Rho.
Keywords: FAK, adherens junctions, Rho, tyrosine kinase
The regulation of cell adhesion, both with the extracellular matrix and between cells, is fundamental to a number of biological properties including cell migration and the maintenance of tissue architecture. The establishment of cell-cell contacts in epithelial cells is mediated by a transmembrane family of proteins called the cadherins, of which E-, P-, VE- and N-cadherin are members. In the presence of calcium, homophilic interactions occur between the extracellular domains of cadherins expressed on the lateral membrane domains of adjacent cells. The cytoplasmic domain of the cadherins establishes a linkage to the actin cytoskeleton by binding to multiple actin-binding proteins including α-catenin, the tight junction component ZO-1 and a component of focal adhesions, vinculin [1]. These sites of cell-cell contact containing cadherins are termed adherens junctions. The importance of cadherin-mediated cellular contacts in the differentiated phenotype and other functions such as an environmental barrier has been well established [2].
Dramatic changes in the actin cytoskeleton occur upon cell-cell adhesion suggesting a role for the Rho family of GTPases in regulating junction assembly. Indeed, the activity of RhoA and Rac is required for the formation and maintenance of cadherin dependent cell-cell contacts [3-5]. However, the activity of these GTPases must be properly regulated as constitutive activation of RhoA and Rac can impair junction assembly and/or disrupt cell-cell contacts in some scenarios [3,6,7]. Interestingly, one stimulus that triggers activation of Rac is the homophilic interaction between E-cadherins on adjacent cells [8-11]. In some cell types this also triggers Cdc42 activation, but notably homophilic E-cadherin interactions cause a reduction in the activation of RhoA. The activation of Rac at an early stage of nascent cell-cell contacts is regulated via a signaling pathway containing c-Src, Rap and PI3-kinase culminating in activation of the guanine nucleotide exchange factor, Vav2 [12,13]. In contrast, RhoA activity is regulated via a GTPase activating protein, p190RhoGAP, which becomes tyrosine phosphorylated and activated upon E-cadherin ligation [8,9]. Presumably the regulation of Rac and RhoA upon homophilic E-cadherin interactions is a feedback mechanism to promote strengthening of the adhesion following initial contact.
FAK is a focal adhesion-associated protein tyrosine kinase that transmits signals downstream of integrins and is a well established as a regulator of the actin cytoskeleton and cell motility [14]. FAK-mediated effects on the actin cytoskeleton occur via regulation of Rho family GTPases. FAK apparently activates Rac via the p130Cas adaptor protein. Tyrosine phosphorylation of p130Cas recruits into complex the Crk adaptor protein, which binds the Dock180/Elmo complex. These function as a RacGEF promoting the activation of Rac [15-17]. Rho is also regulated by FAK [18]. Fak-/- fibroblasts exhibit a Rho-dependent phenotype, i.e. large focal adhesions [19]. Inhibition of Rho reverts the phenotype of the focal adhesions of fak-/- cells to wild-type, whereas introduction of active Rho into normal fibroblasts induces a focal adhesion morphology resembling fak-/- cells [18]. Upon cell adhesion to fibronectin, fibroblasts exhibit a transient reduction in the activity of Rho. Under similar conditions fak-/- fibroblasts exhibit no reduction in Rho activity [18]. This suppression of Rho activity has been linked to tyrosine phosphorylation and activation of p190RhoGAP [20], suggesting FAK might regulate Rho activity through this mechanism. FAK might also regulate Rho activity through an associated Rho GAP called GRAF and p190RhoGEF, which binds to the C-terminal domain of FAK [21,22]. These studies demonstrate that FAK utilizes multiple mechanisms to regulate the activity of Rho family GTPases.
A role for FAK in the regulation of cell-cell contacts is beginning to emerge. Firstly, elevated Src activity in KM12C colon cancer cells was found to inhibit adherens junction assembly [23]. Normal junction assembly could be restored in cells expressing a FAK mutant lacking all Src-dependent phosphorylation sites [23]. This suggests that FAK functions downstream of activated Src to impair the formation of adherens junctions and may promote EMT (epithelial-mesenchymal transition). Secondly, depletion of FAK from Hela cells impairs the formation of N-cadherin-mediated cell-cell contacts and increases cell protrusiveness [24]. This phenotype is dependent on cell density. In confluent cultures, where cell protrusions cannot form, the loss of FAK did not affect the presence of cell-cell contacts. In subconfluent cultures, where cells may form cellular protrusions and migrate, N-cadherin mediated cell-cell contacts are lost in FAK depleted cells [24]. Thus in different scenarios, FAK may promote the formation of cell-cell contacts and control the disassembly of cell-cell junctions. To further explore the role of FAK in regulation of cell-cell junctions, activated FAK was expressed in NBT-II rat bladder adenocarcinoma cells, which are an attractive model for the study of EMT [25]. Expression of an activated form of FAK did not induce EMT as adherens junction disassembly was not observed. However, in cells expressing a kinase inactive form of FAK, cell-cell adhesions were vastly diminished. This observation suggested that the activity of FAK may be important for the formation/stabilization of intercellular adhesions and prompted further investigation of FAK function in the assembly of cell-cell junctions.
MATERIALS AND METHODS
Cell culture and transfection
NBT-II were maintained in Dulbecco’s modified Eagle’s medium containing 10% Fetal Bovine Serum (FBS). GFP/YFP-fusion constructs were transfected into cells using LipofectAMINE™ Plus (Invitrogen) following the manufacturer’s recommended protocol. FAK expression was inhibited using RNA interference technology [26]. The FAK siRNA target sequence was 5′-AAGAUUACCAAUGCCUCCAAA-3′. Both FAK and control #1 siRNA duplexes were purchased from Dharmacon. Duplexes were transfected into cells using Oligofectamine™ (Invitrogen) following the manufacturer’s instructions. Routinely, 2 × 105 cells were transfected with 200 nM siRNA in a 6-well tissue culture dish. For retroviral infection of NBT-II cells, avian SuperFAK (a FAK variant with activation loop point mutations that elevate catalytic activity [27]) and kinase-inactive SuperFAK (K454M) were subcloned into a modified retroviral bicistronic GFP vector, MIGR1 [28]. Retroviruses encoding SuperFAK, SuperFAK K454M and vector alone were introduced into NBT-II cells. GFP-positive cells were selected by FACs cell sorting. Experiments were performed using cells at passage 8 or less.
Molecular Biology
Plasmids encoding GFP fusion proteins of active Rac (Q61L) and dominant negative Rac (N17) have been previously described [29]. Active Rho (Q63L) and dominant negative Rho (N19) were amplified by PCR and subcloned into pEYFP-C1 in frame with the YFP coding sequence. The FERM domain of FAK was amplified by PCR and subcloned into pEYFPc1 in frame with YFP. GFP-FAK has been described previously [30]. Point mutations were engineered into GFP-FAK using the QuikChange strategy (Stratagene, La Jolla CA). Sequence analysis was performed on each mutant to verify the intended point mutations/deletions and that no unintended mutations were present. These analyses were performed in the UNC-CH Genome Analysis Facility on a model 3730 DNA Analyzer (Perkin Elmer, Applied Biosystems Division) using the ABI PRISM™ Dye Terminator Cycle Sequencing Ready Reaction Kit with AmpliTaq DNA Polymerase, FS (Perkin Elmer, Applied Biosystems Division).
Fluorescence Microscopy
NBT-II were fixed in 3.7% formaldehyde prepared in phosphate-buffered saline (PBS) for 10 minutes, followed by permeabilization with PBS containing 0.25% Triton-X-100 for 5 minutes. The slides were blocked in PBS containing 3% BSA for 1 hour at 37°C. Samples were stained with an E-cadherin antibody (1:200 BD Biosciences) by incubation overnight at 4°C. Alternatively, junctions were visualized by staining with a β-catenin antibody (1:200 BD Biosciences). Primary antibody was visualized by incubation with rhodamine-conjugated anti-mouse immunoglobulin G (1:200) for 1 hour at 37°C. Endogenous FAK was detected by immunostaining with the 4.4.7 monoclonal antibody (Upstate Biotechnology), a rabbit anti-mouse secondary and a donkey anti-rabbit tertiary antibody conjugated to fluorescein. Phosphorylated FAK was detected by immunostaining with the PY397 phospho-specific antibody (Biosource International). The cells were mounted in a mixture of 10% Tris pH 7.5, 90% glycerol, and 25 mg/ml 1,4-diazabicyclo (2,2,2) octane (DABCO) (Sigma, St Louis MO). Confocal microscopy was performed on Leica SP2 AOBS confocal laser scanning microscope at 63x magnification.
Protein Analysis
Cells were lysed in 1% Triton X-100 lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10% glycerol, 1% Triton X-100) or 0.2% Triton X-100 lysis buffer (0.2% triton, 0.3 M sucrose, 50 mM Tris-HCl (pH 7.5), 100 mM KCl, 1 mM CaCl2, 2.5 mM MgCl2) containing protease and phosphatase inhibitors and the protein concentration determined using the bicinchoninic acid assay (Pierce, Rockford IL). Immunoprecipitations were performed using 1 mg of protein lysate and 15 μl of FAK (BC4) polyclonal antiserum [30]. Immune complexes were precipitated using protein A Sepharose beads. The beads were washed three times in lysis buffer and boiled in sample buffer. Immune complexes and were analyzed by Western blotting using the following antibodies; FAK (clone 4.47 1:1000, Upstate Biotechnology), phospho-FAK Y861, phospho-FAK Y397, phospho-FAK Y407 (all 1:500, Biosource International) and visualized using secondary antibodies conjugated to horseradish peroxidase and enhanced chemiluminescence. To confirm equal loading for Western blotting, antibodies against E-cadherin (1:1000), ERK1 (1:1000, Santa Cruz Biotechnology) or actin (1:1000-Chemicon) were used. The Rho activity assay was performed as previously described [31].
RESULTS
Inhibition of FAK inhibits E-cadherin mediated cell-cell contact formation
To further investigate the role of FAK in controlling cell-cell junctions an activated form of FAK (SuperFAK) and a catalytically inactive variant (SuperFAK K454M) were moderately overexpressed in NBT-II cells (Fig. 1A). Confluent cultures were treated with 2 mM EGTA for two hours to remove extracellular calcium. Under these conditions, interactions between cadherins on opposing cells are lost and the cells lack E-cadherin at the cell periphery (Fig. 1B, top row). Restoration of 2 mM calcium to the growth media for four hours allows reassembly of cell-cell junctions and E-cadherin becomes localized to cell-cell contacts. This was observed in both the vector expressing control cells and in SuperFAK expressing cells (Fig. 1B, second row). In contrast, SuperFAK K454M expressing cells exhibited only the initial stages of cell-cell contact, where cell protrusions from one cell contact a neighboring cell and diffuse localization of E-cadherin is observed (Fig. 1B, second row). Twenty-four hours after calcium addition, the cell-cell contacts formed by control cells, SuperFAK expressing cells and SuperFAK K454M expressing cells were similar (Fig. 1B, bottom row). This indicated that expression of SuperFAK K454M induced a delay and not a complete block of the assembly of cell-cell adhesions. SuperFAK K454M may be acting as a dominant-negative mutant to inhibit endogenous FAK and impairing nascent cell-cell contact formation in NBT-II cells.
Figure 1. FAK is required for the formation of cell-cell contacts.

A) Stable NBT-II lines containing vector alone (lane 1), SuperFAK (lane 2) or SuperFAK K454M (lane 3) were generated by retroviral infection. Protein expression was assessed by Western blotting with a FAK antibody (top row). An ERK antibody was used to demonstrate equal loading. B) NBT-II cells containing empty vector, SuperFAK or SuperFAK K454M cells were grown to confluence, then treated with cell-culture media supplemented with 2mM EGTA for two-hours to disrupt E-cadherin positive cell-cell contacts (top row). Fresh calcium-containing media was added and the cells were stained 4 and 24-hours later with an E-cadherin antibody to mark cell-cell contacts. C) NBT-II cells were transiently transfected with plasmids encoding GFP fused to FAK (row 1), FAK Y397F (row 2) or FAK Y861F (row 3). Forty-eight hours later the cells were subjected to a two-hour calcium switch. Expressing cells were identified as GFP positive cells (right column). Cell-cell contacts were marked by E-cadherin antibody staining using a rhodamine conjugated secondary antibody (left panels). Asterisks denote transfected cells. D) NBT-II cells were transiently transfected with plasmids encoding GFP (top panels) or GFP-FRNK (bottom panels) and analyzed as in C, except cell-cell contacts were stained with a β-catenin antibody. E) Quantification of the percentage of GFP positive cells containing one or more cell-cell contacts. Results shown correspond to at least three independent experiments (+/−S.E.M) with over 100 GFP positive cells scored per experiment. The data were analyzed using a one-way analysis of variance (p<0.005) and Dunnett’s multiple comparison post test (*, p<0.05).
The ability of other mutants of FAK to inhibit the formation of cell-cell contacts was addressed by transient expression of GFP fusion proteins in NBT-II cells. Forty-eight hours post transfection, the cells were subjected to the calcium switch protocol and cell-cell contacts were visualized by immunofluorescence using an E-cadherin or β-catenin antibody. Transfected cells were identified as GFP-positive. Expression of GFP alone or GFP-FAK had no detrimental effect on the formation of cell-cell contacts (Fig. 1C and D). Similarly, contact formation was unaffected in cells expressing a mutant of FAK, where tyrosine residue 861 is replaced with phenylalanine (Fig. 1C). However, many cells expressing GFP-FAK Y397F lacked typical epithelial morphology and intercellular contacts, and the cells appeared more spread (Fig. 1C). Similarly, cells expressing an established dominant negative mutant of FAK, GFP-FRNK, also exhibited impaired junction formation under these conditions with an apparent increase in cytosolic β-catenin (Fig 1D). Expression of the N-terminal FERM domain of FAK fused to YFP had no effect upon the formation of E-cadherin positive, cell-cell junctions following the calcium switch (data not shown). The proportion of GFP-positive cells exhibiting E-cadherin localization at points of contact between opposing cells was quantified (Fig. 1E). Whereas over 90% of GFP or GFP-FAK expressing cells exhibited E-cadherin localization at cell-cell contacts, fewer than 45% of cells expressing GFP-FRNK or GFP-FAK Y397F showed similar E-cadherin localization (Fig. 1E). GFP-FAK Y861F, GFP-FAK Y407F and YFP-FERM expressing cells exhibited no significant difference in the formation of E-cadherin positive cell-cell contacts relative to GFP expressing control cells. Taken together, FAK kinase activity and tyrosine 397 are important for the formation of cell-cell contacts in NBT-II cells.
The observation that mutants of FAK can disrupt cell-cell contacts, suggested that FAK plays an important role in the maintenance of the epithelial phenotype. To further substantiate these claims, FAK-specific small interfering RNA duplexes were transfected into NBT-II cells. The effect of FAK depletion on formation of cell-cell contacts was assessed. Routinely, over 70% suppression of FAK protein levels was achieved at seventy hours post-transfection (Fig. 2A, top row, lane 2). ERK and E-cadherin expression levels were unaltered (Fig. 2A, row 2 and 3). At seventy-two hours post-transfection, cells were plated to approximately 50% confluence and subjected to calcium switch. Two hours after calcium restoration, approximately 41% of cells transfected with control siRNA formed cell-cell contacts (Fig. 2B, left panel and Fig. 2C). In contrast, FAK-depleted cells exhibited loss of epithelial cell morphology, and lacked cell-cell contacts in all but 15% of cells (Fig. 2B, middle panel and Fig. 2C). This phenotype resembled NBT-II cells transfected with FRNK or FAK Y397F as described above.
Figure 2. Suppression of FAK protein levels impairs cell-cell contact formation in NBT-II cells.

A) Stable cell lines containing vector alone (lanes 1 and 2) or expressing avian-FAK (lane 3) were generated by retroviral infection. These cells were transiently transfected with control siRNA (lanes 1) or a FAK siRNA targeting the endogenous rat protein (lanes 2 and 3). Note that avian FAK is resistant to knockdown by this FAK siRNA. Seventy-two hours post transfection the cells were lysed and blotted for FAK (top row), ERK (middle row) and E-cadherin (bottom row). B) Seventy-two hours post transfection, vector expressing cells transfected with control siRNA (control) or transfected with FAK siRNA (control + FAK siRNA) and avian FAK expressing cells transfected with FAK siRNA (FAK + FAK siRNA) were subjected to a two-hour calcium switch and stained with an E-cadherin antibody to identify cell-cell contacts. C) the percentage of cells containing one or more contact sites was quantified. Results shown correspond to three independent experiments (+/− S.E.M) with over one hundred cells scored per experiment. The data was analyzed using a one-way ANOVA (p<0.02) and a Dunnett’s multiple comparison post test (*, p <0.05).
The FAK siRNA used for these experiments was specific for rat, mouse and human, but not avian FAK. Stable NBT-II cells expressing avian FAK and control cells containing empty vector were generated to test whether the phenotype observed in cells lacking endogenous FAK could be rescued by re-expression of FAK. NBT-II avian-FAK cells continued to express FAK even upon treatment with the FAK siRNA targeting the endogenous rat FAK (Fig. 2A, top panel, lane 3). Furthermore, NBT-II-avian FAK cells treated with FAK siRNA exhibited E-cadherin positive contacts in approximately 52% of the cells following calcium switch (Fig. 2B, right panel and Fig. 2C). This proportion was not statistically different from vector containing NBT-II cells treated with control siRNA (Fig. 2C). In addition, most NBT-II-avian FAK cells treated with the FAK siRNA retained epithelial cell morphology, in contrast to vector containing cells treated with the FAK siRNA (Fig. 2B). These findings support a role for FAK in regulating the assembly of E-cadherin positive cell-cell contacts.
Cell-cell contact formation regulates FAK phosphorylation
The formation of homophilic contacts between cadherin molecules on adjacent cells triggers activation of cytoplasmic signaling molecules. To assess whether FAK is regulated upon cell-cell adhesion its tyrosine phosphorylation was monitored during cell-cell interaction following calcium switch. The cells were lysed at various times, FAK was immunoprecipitated and the immune complexes immunoblotted for phospho-FAK 407 and phospho-FAK 861 (Fig. 3). Alternatively, cell lysates were directly blotted using phosphospecific antibodies recognizing FAK tyrosine residues 397, 576, 577 and 925. Little or no change in the level of phosphorylation of most of these residues relative to untreated confluent cultures was detected (Fig. 3 A and C). However, elevated phosphorylation of tyrosine 861 was detected after two-hours of calcium replenishment (Fig. 3A). Phosphorylation of tyrosine 861 was dependent upon E-cadherin interactions since the response was blocked in the presence of a function blocking E-cadherin antibody (Fig. 3B).
Figure 3. Formation of nascent cell-cell contacts increases phosphorylation at FAK Y861.

NBT-II cells were cultured to confluence. One plate, indicative of untreated conditions, was lysed (control). The cells were subjected to a calcium switch and lysed at the indicated time after the re-addition of calcium. A) FAK was immunoprecipiated and the immune complexes blotted for phospho-FAK Y861 and phospho-FAK Y407. The immunoblot in row 2 was reprobed with a FAK antibody to demonstrate equal loading. B) Cells were subjected to a calcium switch in the absence (−) or presence (+) of 5 μg/ml SHE78-7, an E-cadherin function blocking antibody. Cells were lysed 2 hours after the addition of calcium, FAK was immunoprecipitated and blotted for PY861 or FAK. C) Equal amounts of cell lysate were directly blotted for phospho-FAK Y397, phospho-FAK Y576, phospho-FAK 577 and phospho-FAK Y925. Lysates were blotted for FAK as a loading control. D) After calcium switch, NBT-II cells were stained for endogenous FAK using a monoclonal antibody, a rabbit anti-mouse secondary antibody and a FITC-labeled donkey anti-rabbit tertiary antibody. Two examples are shown (panel ii and iii). The controls lack the primary antibody (i). E) Following calcium switch, NBT-II cells were immunostained to examine the cellular localization of phospho-FAK Y397. Cells were co-stained with E-cadherin to mark cell-cell contacts.
Previous studies have suggested that FAK can localize at sites of cell-cell contact in several cell types [32,33]. In confluent cultures of NBT-II cells, GFP-FAK constructs did not localize to cell-cell contacts, however during the reassembly of junctions following calcium switch these constructs could be detected at a low frequency at cell junctions (data not shown). The localization of endogenous FAK following calcium switch was examined by immunofluorescence. A small fraction of endogenous FAK was localized to cell-cell junctions (Fig. 3D, panels ii and iii). This staining was specific, since it was not observed in immunofluorescence controls (Fig. 3D, panel i). Interestingly, the phospho-specific antibody recognizing PY397 weakly immunostained cell-cell junctions in NBT-II cells following calcium switch, suggesting that the population of FAK recruited to this site was phosphorylated at tyrosine 397 (Fig. 3E). This could be significant since tyrosine 397 is a critical phosphorylation site in FAK and expression of the FAK 397F mutant blocks junction formation.
FAK regulation of cell-cell contact formation: role of Rho proteins
Both Rho and Rac are downstream targets of FAK signaling upon adhesion to the extracellular matrix. Furthermore, the activity of both Rho and Rac are altered upon cadherin signaling, and each has been implicated in regulating adherens junction formation. To validate the role of Rho and Rac in the formation of E-cadherin positive cell-cell contacts in NBT-II cells, constitutively active and inactive forms of both GTPases (Rho Q63L/N19 and Rac Q61L/N17) were transiently expressed as YFP and GFP fusion proteins. Forty-eight hours post-transfection the cells were subjected to a two-hour calcium switch. The cells were stained for E-cadherin to identify cell-cell contacts and Rho GTPase expressing cells were identified as GFP- or YFP-positive by fluorescent microscopy (Fig. 4A). GFP-Rac N17 and YFP-Rho N19 expressing cells formed E-cadherin positive cell-cell contacts as well as control cells (Fig. 4A first and third rows). Quantification revealed that greater than 82% of cells expressing GFP-RacN17, YFP-RhoN19 or GFP alone formed E-cadherin positive cell-cell contacts (Fig. 4B). In contrast, most cells expressing GFP-Rac Q61L or YFP-Rho Q63L were devoid of intercellular contacts and exhibited a loss of epithelial cell morphology (Fig. 4A row 2 and 4). Only 28 and 35% of cells expressing activated Rac and Rho respectively contained E-cadherin positive cell-cell contacts (Fig. 4B). This result is consistent with the literature describing a detrimental effect of expression of constitutively activated Rac and Rho upon the formation of cell-cell junctions [3,6,7]. Further, this phenotype resembled that of NBT-II cells with suppressed FAK expression or expressing dominant negative FAK mutants.
Figure 4. Expression of activated Rho (Q63L) or Rac (Q61L) impairs formation of cell-cell contacts in NBT-II cells.
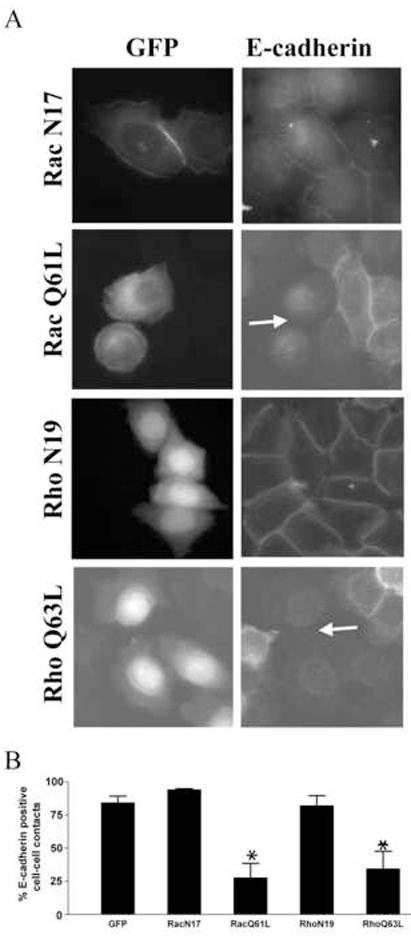
A) NBT-II cells were transiently transfected with the empty pEGFP vector or plasmids encoding GFP-fusion proteins containing dominant negative/constitutively active Rac (N17/Q61L) or YFP-fusion proteins containing dominant negative/constitutively active Rho (N19/Q63L). Forty-eight hours post-transfection the cells were subjected to a two-hour calcium switch. Expressing cells were identified as GFP positive (left column) and cell-cell contacts were visualized by staining with an E-cadherin antibody and a rhodamine-conjugated secondary antibody (right column). The arrows indicate examples of Rac Q61L and RhoQ63L expressing cells that exhibit loss of cell-cell contacts. B) The proportion of GFP/YFP positive cells containing more than one intercellular contact was quantified. Results shown correspond to at least three independent experiments (+/− S.E.M) with over one hundred GFP/YFP positive cells scored per experiment. The data were analyzed using a one-way analysis of variance (p<0.0005) and the Dunnett’s multiple comparison post test (*, p<0.01).
Given that active Rac and Rho induced a phenotype similar to suppression of FAK expression in NBT-II cells, the effect of inhibiting Rac or Rho in NBT-II cells with suppressed FAK expression was assessed. Populations of NBT-II cells stably expressing GFP, GFP-avian FAK, GFP-RacN17 and YFP-RhoN19 were generated. These cells were transfected with FAK siRNA and 72 hours post-transfection the cells were subjected to a calcium switch and the reformation of cell-cell contacts quantified. Approximately 33% of vector expressing cells transfected with FAK siRNA exhibited E-cadherin positive cell-cell contacts (Fig. 5A). This defect was rescued by re-expression of FAK since FAK depleted cells expressing GFP-avian FAK exhibited E-cadherin positive cell-cell contacts in approximately 79% of the cells. A similar proportion of GFP-RacN17- and GFP-expressing cells transfected with FAK siRNA exhibited E-cadherin positive cell-cell contacts (Fig. 5A). This suggested that inhibition of Rac with this dominant-negative mutant could not prevent the defect in cell-cell contact formation observed in FAK deficient cells. In contrast, this cell-cell contact defect was rescued by expression of a dominant negative Rho mutant. Approximately 80% of the YFP-RhoN19 expressing, FAK siRNA treated cells exhibited E-cadherin positive cell-cell contacts (Fig. 5A). This observation suggested that dominant negative Rho could prevent the loss of epithelial cell-cell contacts in cells where FAK signaling is impaired. This finding is consistent with the observation that Rho activity is elevated in fak-/- fibroblasts [18]. To validate that inhibition of FAK function resulted in increased Rho activity in NBT-II cells, the amount of GTP-bound Rho was measured using an effector pulldown assay [31]. NBT-II cells exhibited low Rho activity under low calcium conditions, which increased by two hours following the addition of calcium (Fig. 5B). In contrast, cells expressing FRNK exhibited higher levels of Rho activity under low calcium conditions and following the addition of calcium (Fig. 5B). The level of active Rho from multiple experiments was quantified by Image J analysis. While FRNK expressing cells exhibited a reproducible increase in Rho activity under these conditions, the increase did not reach statistical significance (Fig. 5C). Therefore FAK’s affect upon total cellular Rho activity was modest. To further explore the role of FAK in Rho regulation, the effect of inhibiting FAK expression upon established Rho regulatory proteins was assessed. One potential regulatory mechanism is via regulation of p190RhoGAP. Tyrosine phosphorylation of RhoGAP results in its activation and consequently a reduction in the level of active Rho. Tyrosine phosphorylation of p190RhoGAP in control and FAK siRNA transfected NBT-II cells was compared by immunoprecipitation and Western blotting. Knockdown of FAK expression reduced p190RhoGAP tyrosine phosphorylation suggesting that this mechanism of regulation might be operative in NBT-II cells (Fig. 5D).
Figure 5. FAK regulates cell-cell contact formation by inhibition of Rho.

A) NBT-II cells stably expressing EGFP (GFP), GFP-avian FAK (FAK), GFP-Rac N17 (RacN17) or YFP-RhoN19 (RhoN19) were generated. These cells were transfected with control siRNA or FAK siRNA. Seventy-two hours post transfection cells were subjected to a two-hour calcium switch, and cells stained with an E-cadherin antibody to identify cell-cell contacts. Cells with E-cadherin positive cell-cell contacts were scored as described above. Results shown correspond to at least three independent experiments (+/− S.E.M) with over one hundred GFP/YFP positive cells scored per experiment. The data were analyzed using a one-way analysis of variance (p<0.001) and the Dunnett’s multiple comparison post test (*, p<0.01). B) Control cells and GFP-FRNK expressing cells were subjected to calcium switch and lysed at the indicated times following re-addition of calcium. GTP-bound Rho was identified using an effector pulldown assay and Western blotting (top panel). Rho expression was examined by Western blotting equal amounts of lysate (bottom panel). C) Control or FRNK expressing cells were subjected to calcium switch and at 0, 1 and 2 hours following re-addition of calcium the cells were lysed. The levels of active Rho was determined as in B. Images were quantified using ImageJ and the ratio of active Rho/total Rho was determined for each sample. The relative levels of active Rho, with the level seen in control cells at t=0 normalized to 1, is shown (n=7). D) NBT-II cells treated with control or FAK siRNA were lysed and p190RhoGAP immunoprecipitated. Phosphotyrosine was examined by Western blotting (top panel) and the amount of p190RhoGAP protein determined as a loading control (bottom panel).
DISCUSSION
In this study disruption of endogenous FAK using several different strategies resulted in impaired cell-cell junction formation. These findings demonstrate that endogenous FAK functions in regulating the assembly of E-cadherin containing junctions in NBT-II cells. In addition, FAK was shown to localize to sites of cell-cell contact. This likely represents a transient association, since only ∼10% of cell contacts contained detectable FAK by immunofluorescence and FAK was not detected in the cell-cell junctions in confluent cultures of NBT-II cells. Insight into the mechanism through which FAK regulates cell-cell adhesions in NBT-II cells was also obtained. Inhibition of FAK resulted in reduced phosphorylation of p190RhoGAP and a modest elevation in total Rho activity. Since inhibition of Rho signaling rescues the effect of FAK suppression on cell-cell junction formation, FAK seems to control cell-cell junction formation in NBT-II cells by regulating Rho activity.
The association of FAK with cell-cell junctions is intriguing. A recent finding also suggests that FAK localizes to cell-cell contact sites in the Panc-1 pancreatic cancer cell line when the cells are plated on collagen [33]. FAK is reported to interact with a number of transmembrane tyrosine kinase receptors including Met, PDGFR and EGFR [34-36]. As EGFR has been shown to localize at sites of cell-cell contact FAK might be recruited to these sites through interaction with this receptor tyrosine kinase [37]. FAK also associates with integrins and integrins have been localized to sites of cell-cell contact in some cell types, including several epithelial cell lines [38-41]. In fact, oligomerized E-cadherin may serve as a ligand for some integrins, e.g. α2β1 [42]. Hence, it is plausible that FAK may be recruited to cell-cell contact sites via integrins which are localized to the adherens junction complex. FAK is reported to associate with E-cadherin in Panc-1 cells [33], and to associate with α- and β-catenin in normal cervical tissue and cervical carcinomas [43]. While these are potential mechanisms of localization, we have been unable to demonstrate specific interactions between full length FAK and E-cadherin or α-/β-catenin in NBT-II cells by co-immunoprecipitation (data not shown).
While phosphorylation of other tyrosine residues did not change appreciably, Y861 did become strikingly phosphorylated upon the formation of cell-cell contacts in NBT-II cells. Elevated Y861 phosphorylation has been observed in Hela cells plated on collagen [24]. Under these conditions HeLa cells form N-cadherin positive cell-cell contacts and the formation of these contacts is impaired in cells treated with FAK-specific siRNAs [24]. Re-expression of wild type FAK restored N-cadherin positive cell-cell adhesions, but re-expression of a FAK Y861F mutant failed to rescue the phenotype, implicating Y861 phosphorylation in the control of junction assembly. In our study, exogenous expression of the Y861F mutant had no detrimental effect on formation of cell-cell contacts, in contrast to expression of other dominant negative mutants of FAK. Further, expression of the Y861F mutant was able to rescue the cell-cell junction defect induced by FAK siRNA (data not shown). Thus, Y861 phosphorylation was not required to promote E-cadherin positive cell-cell junctions in NBT-II cells. The difference between these studies may be explained by the use of different cell types, different extracellular matrix proteins or unique mechanisms of regulation of cell-cell contacts containing different cadherins.
Expression of FAK Y397F in NBT-II cells induced a dramatic change of epithelial polarity and impaired cell-cell contact formation following calcium switch. This suggests phosphorylation of Y397 is required to promote cell-cell contact formation, a conclusion that is supported by the findings of Yano et al. [24]. However, in response to the calcium switch in NBT-II cells, very modest changes in Y397 phosphorylation occurred. There are two possible explanations to reconcile these observations. Either E-cadherin dependent adhesion promotes phosphorylation at Y397 in a small fraction of the total cellular FAK or cell-extracellular matrix adhesion provides sufficient autophosphorylated FAK to function in regulating cell-cell adhesion. In either event it appears that some autophosphorylated FAK is recruited to cell-cell junctions during the calcium switch since junctions exhibit PY397 staining. Autophosphorylation of FAK-Y397 recruits Src and PI3-kinase into complex [44,45]. Both Src and PI-3 kinase have been implicated in regulation of cell-cell adhesions. Exogenous expression of c-Src or activated Src variants in various cell lines, including NBT-II, leads to the disassembly of cell-cell junctions as EMT is induced [23,46,47]. However, c-Src is activated upon E-cadherin mediated adhesion and is required for activation of Rac [12]. To further examine a role for Src, NBT-II cells were treated with the Src inhibitor PP2 and the cells observed to increase E-cadherin positive cell-cell contacts (data not shown). As this phenotype is opposite that observed upon inhibition of FAK, the result does not support Src as a key component downstream of FAK, although the complex role of Src in the assembly/disassembly of junctions makes it difficult to drawn an absolute conclusion. Inhibition of PI3-kinase activity causes a decrease in E-cadherin at cell-cell contacts in monolayers of scp2 mouse mammary epithelial cells [12,48]. Hence, it is plausible that the effects on cell-cell adhesion in cells expressing FAK Y397F may be mediated by lack of PI3-kinase binding.
To further address the downstream signaling pathways by which FAK regulates NBT-II cell-cell contacts, the Rho family of GTPases was examined since Rho and Rac can regulate cell-cell adhesions and are components of FAK signaling pathways [4,18,24,49]. Expression of both active Rac and Rho in NBT-II cells increased the frequency of cells lacking E-cadherin positive cell-cell contacts, a phenotype resembling that of cells with suppressed levels of FAK. Subsequent experiments indicated that dominant negative Rho, prevents the defect in intercellular contact formation observed in cells with suppressed FAK expression and cells expressing FRNK exhibited elevated levels of Rho activity. This result suggested that FAK promotes cell-cell contact formation by dampening the activity of Rho. This model is consistent with previous studies demonstrating the role of FAK in preventing hyperactivation of Rho in fibroblasts [18]. However, the effect of FAK expression upon Rho activation levels in NBT-II cells was quite modest. These disparate observations can be reconciled if FAK functions to regulate Rho activity in a spatially restricted manner, and thus only affects a small fraction of the total cellular Rho. There is precedent for this hypothesis since FAK regulates Rac activity in a spatially restricted manner in HeLa cells, which impacts the formation of N-cadherin containing cell-cell junctions [24]. The mechanism of Rho regulation by FAK is possibly via phosphorylation and activation of a GTPase activating protein such as p190RhoGAP since reduced p190RhoGAP tyrosine phosphorylation was observed upon knock down of FAK expression. A similar mechanism of FAK dependent regulation of Rho via p190RhoGAP in endothelial cells has been proposed [50]. Interestingly, in HeLa cells FAK suppression impaired the formation of N-cadherin containing cell-cell contacts and this effect was rescued by expression of dominant negative Rac, suggesting FAK functioned to impair excessive Rac signaling [24]. Thus while suppression of FAK expression produced a similar phenotype in NBT-II and HeLa cells, even to the extent that the defect in each case could be rescued by plating cells at very high density (data not shown)[24], the molecular mechanisms utilized appear distinct. Whether these distinct mechanisms are due to the use of different cells or reflect different modes of regulation of these two types of cadherins remains to be established.
ACKNOWLEDGEMENTS
We would like to thanks Zenon Rajfur, Wendy Salmon and Stephen Wincovitich (NHGRI, NIH) for their microscopy expertise and for valuable discussions during the course of this project. Thanks also to Adi Dubash for his assistance with the Rho activity assays and to Lindsey Buckingham for technical assistance. This study was supported by funding by NIH grant HL45100 (to M.D.S. and K.B).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Gates J, Peifer M. Can 1000 reviews be wrong? Actin, alpha-Catenin, and adherens junctions. Cell. 2005;123:769–772. doi: 10.1016/j.cell.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 2.Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nat.Rev.Mol.Cell Biol. 2005;6:622–634. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- 3.Sahai E, Marshall CJ. ROCK and Dia have opposing effects on adherens junctions downstream of Rho. Nat.Cell Biol. 2002;4:408–415. doi: 10.1038/ncb796. [DOI] [PubMed] [Google Scholar]
- 4.Braga VM. Small GTPases and regulation of cadherin dependent cell-cell adhesion. Mol.Pathol. 1999;52:197–202. doi: 10.1136/mp.52.4.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takaishi K, Sasaki T, Kotani H, Nishioka H, Takai Y. Regulation of cell-cell adhesion by rac and rho small G proteins in MDCK cells. J.Cell Biol. 1997;139:1047–1059. doi: 10.1083/jcb.139.4.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zondag GC, Evers EE, ten Klooster JP, Janssen L, van der Kammen RA, Collard JG. Oncogenic Ras downregulates Rac activity, which leads to increased Rho activity and epithelial-mesenchymal transition. J.Cell Biol. 2000;149:775–782. doi: 10.1083/jcb.149.4.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braga VM, Betson M, Li X, Lamarche-Vane N. Activation of the small GTPase Rac is sufficient to disrupt cadherin-dependent cell-cell adhesion in normal human keratinocytes. Mol.Biol.Cell. 2000;11:3703–3721. doi: 10.1091/mbc.11.11.3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noren NK, Arthur WT, Burridge K. Cadherin engagement inhibits RhoA via p190RhoGAP. J.Biol.Chem. 2003;278:13615–13618. doi: 10.1074/jbc.C200657200. [DOI] [PubMed] [Google Scholar]
- 9.Noren NK, Niessen CM, Gumbiner BM, Burridge K. Cadherin engagement regulates Rho family GTPases. J.Biol.Chem. 2001;276:33305–33308. doi: 10.1074/jbc.C100306200. [DOI] [PubMed] [Google Scholar]
- 10.Kim SH, Li Z, Sacks DB. E-cadherin-mediated cell-cell attachment activates Cdc42. J.Biol.Chem. 2000;275:36999–37005. doi: 10.1074/jbc.M003430200. [DOI] [PubMed] [Google Scholar]
- 11.Nakagawa M, Fukata M, Yamaga M, Itoh N, Kaibuchi K. Recruitment and activation of Rac1 by the formation of E-cadherin-mediated cell-cell adhesion sites. J.Cell Sci. 2001;114:1829–1838. doi: 10.1242/jcs.114.10.1829. [DOI] [PubMed] [Google Scholar]
- 12.Kovacs EM, Ali RG, McCormack AJ, Yap AS. E-cadherin homophilic ligation directly signals through Rac and phosphatidylinositol 3-kinase to regulate adhesive contacts. J.Biol.Chem. 2002;277:6708–6718. doi: 10.1074/jbc.M109640200. [DOI] [PubMed] [Google Scholar]
- 13.Fukuyama T, Ogita H, Kawakatsu T, Inagaki M, Takai Y. Activation of Rac by cadherin through the c-Src-Rap1-phosphatidylinositol 3-kinase-Vav2 pathway. Oncogene. 2006;25:8–19. doi: 10.1038/sj.onc.1209010. [DOI] [PubMed] [Google Scholar]
- 14.Schaller MD. Biochemical signals and biological responses elicited by the focal adhesion kinase. Biochim.Biophys.Acta. 2001;1540:1–21. doi: 10.1016/s0167-4889(01)00123-9. [DOI] [PubMed] [Google Scholar]
- 15.Klemke RL, Leng J, Molander R, Brooks PC, Vuori K, Cheresh DA. CAS/Crk coupling serves as a “molecular switch” for induction of cell migration. J.Cell Biol. 1998;140:961–972. doi: 10.1083/jcb.140.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valles AM, Beuvin M, Boyer B. Activation of Rac1 by paxillin-Crk-DOCK180 signaling complex is antagonized by Rap1 in migrating NBT-II cells. J.Biol.Chem. 2004;279:44490–44496. doi: 10.1074/jbc.M405144200. [DOI] [PubMed] [Google Scholar]
- 17.Playford MP, Schaller MD. The interplay between Src and integrins in normal and tumor biology. Oncogene. 2004;23:7928–7946. doi: 10.1038/sj.onc.1208080. [DOI] [PubMed] [Google Scholar]
- 18.Ren X, Kiosses WB, Sieg DJ, Otey CA, Schlaepfer DD, Schwartz MA. Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J.Cell Sci. 2000;113(Pt 20):3673–3678. doi: 10.1242/jcs.113.20.3673. [DOI] [PubMed] [Google Scholar]
- 19.Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- 20.Arthur WT, Burridge K. RhoA inactivation by p190RhoGAP regulates cell spreading and migration by promoting membrane protrusion and polarity. Mol.Biol.Cell. 2001;12:2711–2720. doi: 10.1091/mbc.12.9.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hildebrand JD, Taylor JM, Parsons JT. An SH3 domain-containing GTPase-activating protein for Rho and Cdc42 associates with focal adhesion kinase. Mol.Cell Biol. 1996;16:3169–3178. doi: 10.1128/mcb.16.6.3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhai J, Lin H, Nie Z, Wu J, Canete-Soler R, Schlaepfer WW, Schlaepfer DD. Direct interaction of focal adhesion kinase with p190RhoGEF. J.Biol.Chem. 2003;278:24865–24873. doi: 10.1074/jbc.M302381200. [DOI] [PubMed] [Google Scholar]
- 23.Avizienyte E, Wyke AW, Jones RJ, McLean GW, Westhoff MA, Brunton VG, Frame MC. Src-induced de-regulation of E-cadherin in colon cancer cells requires integrin signalling. Nat.Cell Biol. 2002;4:632–638. doi: 10.1038/ncb829. [DOI] [PubMed] [Google Scholar]
- 24.Yano H, Mazaki Y, Kurokawa K, Hanks SK, Matsuda M, Sabe H. Roles played by a subset of integrin-signaling molecules in cadherin based cell-cell adhesion. J.Cell Biol. 2004;166:283–295. doi: 10.1083/jcb.200312013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat.Rev.Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 26.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 27.Gabarra-Niecko V, Keely PJ, Schaller MD. Characterization of an Activated Mutant of Focal Adhesion Kinase: SuperFAK. Biochem.J. 2002;365:591–603. doi: 10.1042/BJ20020065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pear WS, Miller JP, Xu L, Pui JC, Soffer B, Quackenbush RC, Pendergast AM, Bronson R, Aster JC, Scott ML, Baltimore D. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood. 1998;92:3780–3792. [PubMed] [Google Scholar]
- 29.Wennerberg K, Ellerbroek SM, Liu RY, Karnoub AE, Burridge K, Der CJ. RhoG signals in parallel with Rac1 and Cdc42. J.Biol.Chem. 2002;277:47810–47817. doi: 10.1074/jbc.M203816200. [DOI] [PubMed] [Google Scholar]
- 30.Cooley MA, Broome JM, Ohngemach C, Romer LH, Schaller MD. Paxillin binding is not the sole determinant of focal adhesion localization or dominant-negative activity of focal adhesion kinase/focal adhesion kinase-related nonkinase. Mol.Biol.Cell. 2000;11:3247–3263. doi: 10.1091/mbc.11.9.3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 1999;18:578–585. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stewart A, Ham C, Zachary I. The focal adhesion kinase amino-terminal domain localises to nuclei and intercellular junctions in HEK 293 and MDCK cells independently of tyrosine 397 and the carboxy-terminal domain. Biochem.Biophys.Res.Commun. 2002;299:62–73. doi: 10.1016/s0006-291x(02)02547-0. [DOI] [PubMed] [Google Scholar]
- 33.Koenig A, Mueller C, Hasel C, Adler G, Menke A. Collagen type I induces disruption of E-cadherin-mediated cell-cell contacts and promotes proliferation of pancreatic carcinoma cells. Cancer Res. 2006;66:4662–4671. doi: 10.1158/0008-5472.CAN-05-2804. [DOI] [PubMed] [Google Scholar]
- 34.Chen SY, Chen HC. Direct interaction of focal adhesion kinase (FAK) with Met is required for FAK to promote hepatocyte growth factor-induced cell invasion. Mol.Cell Biol. 2006;26:5155–5167. doi: 10.1128/MCB.02186-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, Schlaepfer DD. FAK integrates growth-factor and integrin signals to promote cell migration. Nat.Cell Biol. 2000;2:249–256. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- 36.Golubovskaya V, Beviglia L, Xu LH, Earp HS, III, Craven R, Cance W. Dual Inhibition of Focal Adhesion Kinase and Epidermal Growth Factor Receptor Pathways Cooperatively Induces Death Receptor-mediated Apoptosis in Human Breast Cancer Cells. J.Biol.Chem. 2002;277:38978–38987. doi: 10.1074/jbc.M205002200. [DOI] [PubMed] [Google Scholar]
- 37.Hoschuetzky H, Aberle H, Kemler R. Beta-catenin mediates the interaction of the cadherin-catenin complex with epidermal growth factor receptor. J.Cell Biol. 1994;127:1375–1380. doi: 10.1083/jcb.127.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schaller MD, Otey CA, Hildebrand JD, Parsons JT. Focal adhesion kinase and paxillin bind to peptides mimicking beta integrin cytoplasmic domains. J.Cell Biol. 1995;130:1181–1187. doi: 10.1083/jcb.130.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schoenenberger CA, Zuk A, Zinkl GM, Kendall D, Matlin KS. Integrin expression and localization in normal MDCK cells and transformed MDCK cells lacking apical polarity. J.Cell Sci. 1994;107(Pt 2):527–541. doi: 10.1242/jcs.107.2.527. [DOI] [PubMed] [Google Scholar]
- 40.Shigeta M, Sanzen N, Ozawa M, Gu J, Hasegawa H, Sekiguchi K. CD151 regulates epithelial cell-cell adhesion through PKC- and Cdc42-dependent actin cytoskeletal reorganization. J.Cell Biol. 2003;163:165–176. doi: 10.1083/jcb.200301075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chattopadhyay N, Wang Z, Ashman LK, Brady-Kalnay SM, Kreidberg JA. alpha3beta1 integrin-CD151, a component of the cadherin-catenin complex, regulates PTPmu expression and cell-cell adhesion. J.Cell Biol. 2003;163:1351–1362. doi: 10.1083/jcb.200306067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Whittard JD, Craig SE, Mould AP, Koch A, Pertz O, Engel J, Humphries MJ. E-cadherin is a ligand for integrin alpha2beta1. Matrix Biol. 2002;21:525–532. doi: 10.1016/s0945-053x(02)00037-9. [DOI] [PubMed] [Google Scholar]
- 43.Moon HS, Park WI, Choi EA, Chung HW, Kim SC. The expression and tyrosine phosphorylation of E-cadherin/catenin adhesion complex, and focal adhesion kinase in invasive cervical carcinomas. Int.J.Gynecol.Cancer. 2003;13:640–646. doi: 10.1046/j.1525-1438.2003.13396.x. [DOI] [PubMed] [Google Scholar]
- 44.Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2- dependent binding of pp60src. Mol.Cell Biol. 1994;14:1680–1688. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen HC, Appeddu PA, Isoda H, Guan JL. Phosphorylation of tyrosine 397 in focal adhesion kinase is required for binding phosphatidylinositol 3-kinase. J.Biol.Chem. 1996;271:26329–26334. doi: 10.1074/jbc.271.42.26329. [DOI] [PubMed] [Google Scholar]
- 46.Rodier JM, Valles AM, Denoyelle M, Thiery JP, Boyer B. pp60c-src is a positive regulator of growth factor-induced cell scattering in a rat bladder carcinoma cell line. J.Cell Biol. 1995;131:761–773. doi: 10.1083/jcb.131.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Irby RB, Yeatman TJ. Increased Src activity disrupts cadherin/catenin-mediated homotypic adhesion in human colon cancer and transformed rodent cells. Cancer Res. 2002;62:2669–2674. [PubMed] [Google Scholar]
- 48.Somasiri A, Wu C, Ellchuk T, Turley S, Roskelley CD. Phosphatidylinositol 3-kinase is required for adherens junction-dependent mammary epithelial cell spheroid formation. Differentiation. 2000;66:116–125. doi: 10.1046/j.1432-0436.2000.660206.x. [DOI] [PubMed] [Google Scholar]
- 49.Tokman MG, Porter RA, Williams CL. Regulation of cadherin-mediated adhesion by the small GTP-binding protein Rho in small cell lung carcinoma cells. Cancer Res. 1997;57:1785–1793. [PubMed] [Google Scholar]
- 50.Holinstat M, Knezevic N, Broman M, Samarel AM, Malik AB, Mehta D. Suppression of RhoA activity by focal adhesion kinase-induced activation of p190RhoGAP: role in regulation of endothelial permeability. J.Biol.Chem. 2006;281:2296–2305. doi: 10.1074/jbc.M511248200. [DOI] [PubMed] [Google Scholar]