

Abstract
Factor VIIa (VIIa), the serine protease that initiates the coagulation pathways, is catalytically activated upon binding to its cell surface receptor and cofactor tissue factor (TF). This study provides a comprehensive analysis of the functional surface of VIIa by alanine scanning mutagenesis of 112 residues. Residue side chains were defined which contribute to TF binding and factor X hydrolysis. Energetically important binding contacts at the interface with TF were identified in the first epidermal growth factor domain of VIIa (Gln-64, Ile-69, Phe-71, Arg-79) and in the protease domain (Arg-277, Met-306, Asp-309). The observed energetic defects are in good agreement with the corresponding residues in TF, suggesting that the VIIa light chain plays a prominent role in high affinity binding of cofactor. Mutation of protease domain interface residues indicated that TF allosterically influences the active site of VIIa. Stabilization of a labile zymogen to enzyme transition could explain the activating effect of TF on VIIa catalytic function. Residues important for factor X hydrolysis were found in three regions of the protease domain: (i) specificity determinants in the catalytic cleft and adjacent loops, (ii) an exosite near the TF binding site, and (iii) a large electronegative exosite which is in a position analogous to the basic exosite I of thrombin. TF regions involved in factor X activation are positioned on the same face of the TF·VIIa complex as the two exosites identified on the protease domain surface, providing evidence for an extended interaction of TF·VIIa with macromolecular substrate.
Keywords: coagulation cascade, alanine scanning mutagenesis
Coagulation factor VIIa (VIIa) is the initiating protease of the coagulation pathways (1). VIIa binds to its cellular receptor and catalytic cofactor, the transmembrane glycoprotein tissue factor (TF). Interaction of VIIa with TF markedly enhances the catalytic activity of the serine protease which is a poor enzyme when free in solution. However, the mechanism by which the cofactor achieves this activation is unknown. VIIa is a multidomain enzyme characterized by an amino-terminal γ-carboxyglutamic acid-rich (Gla) domain, two epidermal growth factor (EGF)-like modules and a trypsin-like serine protease domain. Various experimental approaches indicate that the Gla-domain, the first EGF domain (EGF1), and the protease domain provide the most significant contributions to the interaction with TF (reviewed in ref. 2). The recently determined three-dimensional structure of the TF·VIIa complex revealed a discontinuous interaction involving each domain of VIIa and the two fibronectin type III modules that constitute the cytokine receptor-like extracellular domain of TF (3).
While extensive mutagenesis of TF has defined the functional and energetic importance of residues that interface with VIIa, there is little information on the contributions of specific amino acid side chains in VIIa to TF binding or proteolytic function. The following surface-exposed residues in VIIa are known to play a functionally important role: Arg-79 in EGF1 (4, 5) and Arg-304 [c162] (6, 7) in the protease domain are implicated in binding to TF; whereas Arg-290 [c148] (8) and Lys-341 [c192] (9) are critical for proteolytic function and substrate specificity of VIIa. In the present study, we used the structurally conservative approach of alanine scanning mutagenesis of surface residues (10) to establish an extensive list of residues that are functionally involved in cofactor interaction and substrate factor X (X) activation. We provide evidence that EGF1 predominantly tethers the protease to its receptor, whereas specific interactions of protease domain residues with TF are required for full catalytic function of the bound protease. The location of catalytically important residues in the VIIa protease domain provides further insight into the potential mechanisms of protease activation through cofactor interaction.
MATERIALS AND METHODS
Homology Modeling.
Homology models for the three-dimensional structure of VIIa domains were generated using Homology in InsightII (Molecular Simulations, Waltham, MA). Xa (11) was the primary reference source for the structure of the catalytic domain docked with the EGF2 domain. Certain nonconserved loops were built using coordinates from neutrophil elastase (12) (residues 16–21 for VIIa residues 153–158 [c16–21], 33–41 for 170–177 [c33–41], and 144–153 for 287–295 [c144–153]), streptomyces trypsin (13) (residues 59–67 for VIIa residues 195–203 [c59–63]), human trypsinogen (14) (residues 69–81 for VIIa residues 209–221 [c69–81]), and citrate synthase (15) (residues 190–198 for VIIa residues 313–321 [c171–174]). According to the classification of Morris et al. (16), the overall structure quality of the model was rated class 2 for phi-psi distribution, class 1 for chi-1 standard deviation, and class 3 for hydrogen bonding energy. Using procheck (17) there are no bad contacts and only Asn-240 [c100] was found in a unfavorable region of the Ramachandran plot. The EGF1 domain was modeled upon the respective module of factor IX (18) and rated class 1, 1, and 2, respectively, with no bad contacts.
Proteins.
Full-length recombinant human TF from insect cells was reconstituted into 30% phosphatidylserine/70% phosphatidylcholine as described (8). Plasma X was purified according to Fair et al. (19), followed by immunoaffinity chromatography on mAb f21-4.2 to reduce the levels of contamination by VII. After adsorption and washes with 1 M NaCl/10 mM EDTA (pH 8.0), X was eluted with 2 M guanidine·HCl and immediately dialyzed against TBS (10 mM Tris/150 mM NaCl, pH 7.4). The preparation contained <1 pM VII/100 nM X.
Mutagenesis and VII Expression.
The VII coding sequence in pED4 (8) was used for oligonucleotide-directed mutagenesis (20). All mutations were confirmed by DNA sequencing. Mutated proteins were transiently expressed by transfecting Chinese hamster ovary (CHO-K1) cells using lipofectamine (BRL). Cells were maintained in serum-free Excel 301 (JRH Scientific, Lenexa, KS) for 48 h to collect VII containing supernatant which was concentrated with a Centricon 30 (Amicon), if necessary. VII in serum-free culture supernatant from stably transfected cells was isolated by a Ca2+-dependent anti-VII antibody followed by ion exchange chromatography with elution by Ca2+ gradient to select for fully γ-carboxylated VII (8). VIIAla-287 did not spontaneously activate during purification and this mutant was activated with factor IXa following repurification by ion exchange chromatography. All other mutants and wild-type VIIa converted >90% to the active enzyme during the purification.
Functional Characterization of Mutants.
Serum-free supernatants from transiently transfected cells were analyzed by ELISA to determine the concentration of secreted wild-type or mutant VII. The assay used two light chain reactive mAbs (F5-8A1 and F4-2.1B) with the biotinylated detection antibody directed to the Ca2+-dependent conformation of the VII Gla domain. The standard curve was generated with purified VII, and interassay variability was assessed by an internal standard. For all light chain mutants, the concentration was confirmed by an alternative ELISA employing antibodies reactive with the VII protease domain. The binding of mutant VII to TF and the catalytic function of VII when in complex with TF was analyzed in a functional assay, which had previously been used to characterize mutants of TF (21, 22). A fixed concentration of TF (5 pM) was saturated with increasing concentrations of VII in the serum-free culture supernatant, followed by measuring activation of 100 nM X to Xa. Rates of Xa generation versus the VII concentration were fitted to the single site binding equation, as described by Krishnaswamy (23). This calculation yielded apparent dissociation constants (KDapp) as well as the maximum rate of Xa generation as a measure of the proteolytic function of the mutant VIIa in complex with TF.
Characterization of Peptidyl Substrate Hydrolysis by VIIa in Complex with Soluble TF1–218.
A fixed concentration of mutant or wild-type VIIa (5 nM) was saturated with increasing concentrations of TF1–218, followed by monitoring of the hydrolysis of 1 mM chromogenic substrate Chromozym tPA (Boehringer Mannheim), S2366 (Chromogenix, Molndal, Sweden), or Spectrozyme FXa (American Diagnostica, Greenwich, CT) at 405 nM. Apparent dissociation constants for these experiments were calculated based on the saturation curves as described above.
Surface Plasmon Resonance Analysis of VIIa Binding to TF.
In a BIAcore 2000 (Pharmacia Biosensor AB) the flow cell was equilibrated with 10 mM Hepes, 150 mM NaCl (pH 7.4), 5 mM CaCl2, 1 mM 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), and 0.005% surfactant P20 and the same buffer was used for all washes at a flow rate of 10 μl/min. Full-length TF1–263 solubilized in 1 mM CHAPS was captured by a non-inhibitory anti-TF mAb (10H10) (24) which was immobilized to the sensor chip surface by amine coupling. Association of wild-type or mutant VIIa was monitored during injection of 12 nM to 2 μM VIIa onto the TF-saturated sensor chip. VIIa dissociation was monitored over 5 min after return to buffer flow. After each analysis, bound VIIa was completely eluted by injection of 100 mM EDTA, and the antibody was saturated by a new injection of TF1–263. The kinetic binding constants (ka, kd, and KD) were determined by nonlinear regression analysis as described previously for the TF·VIIa interaction (25) with software from the manufacturer. The calculation of the association rate constant (ka) was based on multiple association sensograms with at least five different VIIa concentrations. The dissociation rate constant (kd) was calculated from the initial dissociation phase of the binding curves, and the equilibrium dissociation constant KD equals the ratio of kd/ka.
RESULTS
Mutational Analysis of VII.
Alanine scanning mutagenesis of solvent-exposed residue side chains is a proven strategy to define functional regions in proteins (10). We generated models for the structural domains of VII to predict solvent-accessible residue side chains in the VII EGF1, EGF2, and protease domains. One hundred and twelve residues of VII were individually replaced with Ala, and these mutants were tested for both TF binding and proteolytic function in a linked functional assay. This assay has proven reliable in identifying functional defects resulting from mutations in TF (21, 22). The analysis (Fig. 1) of transiently expressed Ala mutants at positions known to affect TF binding (Arg-79) (4, 5), proteolytic function {Arg-290 [c148] (8) and Lys-341 [c192] (9)}, or to produce a combined defect in TF binding and proteolytic function {Arg-304 [c162] (6, 7)} demonstrated that the functional assay has the sensitivity to discriminate between specific functional defects resulting from single residue replacements in VII.
Figure 1.

Affinity for phospholipid reconstituted TF (5 pM) (Left) and the maximum rate of Xa formation (Right) are shown for individual Ala replacement mutants at residue positions in the light chain (A) and the protease domain (B) of VII. Solid and dashed lines indicate mean ± 2 SD, respectively, for values obtained with wild-type VII transiently expressed in parallel with mutant VII. Protease domain mutants are grouped according to the functional defects: (I) combined decrease in TF binding and proteolytic function, (II) selective loss of proteolytic function to >3 SD of control, (III) subtle loss of proteolytic function (<3 and >2 SD), and (IV) normal function. Mean ± SD (n ≥ 3) are shown. In the right margin, chymotrypsin numbering is given for the protease domain. Replacement of the following residues resulted in maximum rates <0.1 s−1, and these essentially inactive mutants are not included in the figure: Leu-177 [c41], Leu-287 [c144], Ser-344 [c195], Val-362 [c213], and Trp-364 [c215].
Light Chain Mutations That Reduced Affinity for TF.
We hypothesized that a hydrophobic face on the EGF1 homology model was a likely docking site for TF. Ala replacements of Lys-62, Gln-64, Ile-69, Phe-71, and Arg-79 had a significant impact on the binding of TF. The largest reduction in the calculated free energy of binding was 1.9 kcal/mol for the mutant at Ile-69. Mutants in EGF1 which were defective in TF binding generally appeared to have normal proteolytic activity (Fig. 1). We attribute the slightly reduced maximal rate of Xa generation by the Ile-69 mutant to technical difficulties in achieving full saturation due to the severe binding defect. Other selected mutations in EGF1 and EGF2 showed no defect in the functional assay.
Mutations in the Protease Domain That Affect Proteolytic Function.
Out of 97 Ala mutations in the protease domain, 33 substitutions resulted in a reduction of proteolytic function without affecting TF binding, whereas 5 mutant proteins had essentially unmeasurable activity in the functional assay (Fig. 1). Mutations affecting proteolytic function are mapped to the face of the VIIa protease domain displayed in Fig. 2. Among the residue replacements within the catalytic cleft, the most significant functional defects were observed for (from left to right): Gln-366 [c217], Trp-364 [c215] (inactive), Leu-177 [c41] (inactive), Gln-176 [c40], Thr-293 [c151], and Leu-213 [c73]. The catalytic triad (in red) is flanked by two functionally important regions. In the loop 236–239 [c96-99] (Fig. 2 Left), residue Thr-239 [c99] contributes to X activation. We also observed a similar mutational effect at an adjacent residue, Asn-240 [c100], which is more buried (not visible in Fig. 2), and attribute the loss of function to conformational effects on Thr-239 [c99]. Secondly, Lys-341 [c192] and Arg-290 [c148], previously shown to be important for catalytic function (8, 9), constitute a prominent basic cluster just below the catalytic triad.
Figure 2.
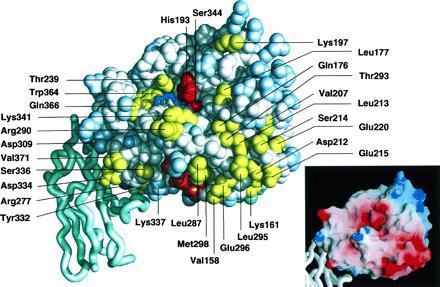
Space-filling representation (midas-plus) (26) of the VIIa EGF2/protease domain homology model docked with the amino-terminal module of TF (ribbon, carboxyl-terminal Tyr-103 at the bottom) based on distance constraints from Banner et al. (3). Functionally important residue side chains in VIIa are colored yellow (selective loss in proteolysis, Fig. 1BII) or magenta (combined defect in TF binding and proteolysis, Fig. 1BI). Residues not (Fig. 1BIV) or only marginally (Fig. 1BIII) involved in function are colored light blue, and backbone atoms and untested residues are white. The catalytic cleft is visible running horizontally with catalytic triad residues Ser-344 and His-193 in red. The first five residues of the amino terminus of the protease domain are in orange. The backbone of a tripeptide substrate (blue) is placed in the active site, based on ppack-thrombin (27). Some atoms of EGF2 (light green) are visible behind the protease domain. The Inset shows a grasp display (28) of the surface charge distribution with red for negative and blue for positive charge.
These basic residues and the amino terminus ([c16–22]) separate two potential exosite regions located below the cleft. (i) Positioned near the TF interface (Fig. 2 Left), substitutions at Val-371 [c223], Ser-336 [c187], and Asp-334 [c185B] caused functional defects, whereas mutation of the adjacent Lys-337 [c188] actually enhanced proteolytic function. (ii) To the right, a large cluster of mutations which caused catalytic defects extended from the exit of the catalytic cleft downwards to where the amino-terminal Ile-153 [c16] inserts to form the activating salt-bridge with Asp-343 [c194]. This exosite also includes the trypsin-analogous Ca2+ binding site at its right perimeter. Consistent with previous analysis (29), replacements of the Ca2+-coordinating residue side chains Glu-210 [c70] and Glu-220 [c80] affected catalytic function of VIIa. The mutational effect at these positions is likely due to structural effects on other residues in this loop, most notably Asp-212 [c72] and Leu-213 [c73], the latter being located in the catalytic cleft. Marked defects were further observed for mutations of Glu-296 [c154], which may contribute to the exosite, and Met-298 [c156] and Leu-287 [c144] (inactive) which may affect stabilization of the activating insertion of the protease domain amino terminus. We exclude overall conformational defects of the Leu-287 [c144] mutant, based on the normal binding to TF (Table 1). Most functionally important residues are located in or below the catalytic cleft. We observed only a modest defect for mutation of Lys-197 [c60A], located above the catalytic triad. Moreover, only subtle effects on X activation were detected upon mutations in the Gln-313 [c171] to Glu-325 [c178] loop which has a uniquely extended conformation in VIIa (3) and is located at the left perimeter of the display in Fig. 2.
Table 1.
Characterization of TF binding by selected mutants of VIIa
| Mutant | Functional assay
|
Surface
plasmon resonance analysis
|
|||||
|---|---|---|---|---|---|---|---|
| KDapp, pM | ΔΔG, kcal/mol | Maximal rate, mol Xa/mol TF·VIIa/s | kon, M−1·s−1 | koff, s−1 | KD, nM | ΔΔG, kcal/mol | |
| Wild type | 3.8 ± 0.7 | 2.01 ± 0.16 | 1.6 ± 0.3 × 105 | 5.7 ± 1.5 × 10−4 | 3.7 ± 0.8 | ||
| Arg-277 [c134] | 12.3 ± 1.4 | 0.68 | 0.65 ± 0.04 | 1.4 ± 0.2 × 105 | 18 ± 0 × 10−4 | 13.1 ± 1.8 | 0.74 |
| Met-306 [c164] | 20.2 ± 1.9 | 0.98 | 0.82 ± 0.09 | 1.2 ± 0.2 × 105 | 15 ± 2 × 10−4 | 13.0 ± 1.5 | 0.74 |
| Lys-341 [c192] | 3.3 ± 1.1 | −0.13 | 0.24 ± 0.01 | 1.5 ± 0.3 × 105 | 3.9 ± 0.9 × 10−4 | 2.7 ± 0.1 | −0.18 |
| Leu-287 [c144] | 1.5 ± 0.3 × 105 | 5.7 ± 0.3 × 10−4 | 3.8 ± 0.9 | 0.01 | |||
Binding of purified VIIa to phospholipid reconstituted TF was analyzed by functional assay at 37°C (see Fig. 1), and binding to full-length, detergent solubilized TF was analyzed by surface plasmon resonance at 22°C. Free energies of binding were calculated based on ΔG = −RT ln (1/KD), ΔΔG is the change in ΔG for mutants as compared to wild-type VIIa. KDapp from the functional assay are in pM, whereas KD from BIAcore analysis are in nM. It is unclear whether these differences result from the presence of substrate. However, calculated ΔΔG values demonstrate consistent results in assessing the functional defect of mutations.
Protease Domain Mutations Which Reduced Affinity for TF.
A limited number of protease domain residue mutants—i.e., Arg-277 [c134], Pro-303 [c161], Arg-304 [c162], Leu-305 [c163], Met-306 [c164], Asp-309 [c167], and Arg-379 [c230]—had reduced affinity for TF with modest losses in the calculated free energy of binding ranging from 0.4 to 0.8 kcal/mol. In contrast to residues in EGF1, all protease domain substitutions that caused cofactor binding defects were also defective in proteolytic function. To exclude the possibility that the increase in KDapp resulted from limitations of the functional assay, we purified prototypic mutants and analyzed binding by surface plasmon resonance. We also analyzed the Lys-341 [c192] replacement in the catalytic cleft which had a selective defect in catalytic function (Fig. 1). This mutant displayed TF binding characteristics indistinguishable from wild-type VIIa (Table 1). In contrast, mutants in the TF interface at position Arg-277 [c134] and Met-306 [c164] had concordantly reduced affinity when analyzed by direct binding measurements (BIAcore) or by functional assay. Both mutations selectively affected the dissociation of VIIa from TF, demonstrating that protease domain interactions indeed contribute to the stability of the TF·VIIa complex.
In agreement with results obtained with transiently expressed proteins, VIIaAla-277 and VIIaAla-306 both had reduced proteolytic function (Table 1). To explore the possibility that the mutation of these interface residues interfered with activating allosteric changes of the active site induced by TF binding, both mutants were saturated with increasing concentrations of soluble TF extracellular domain (TF1–218) and analyzed for amidolytic function (Fig. 3). The replacement of Met-306 [c164] reduced amidolytic function of VIIa in complex with TF to an extent which fully accounted for the proteolytic defect (Fig. 1 and Table 1). In contrast, the reductions in hydrolysis of peptidyl p-nitroanilide chromogenic substrates upon Arg-277 [c134] substitution were less pronounced, despite the fact that the proteolytic defect of this mutant exceeded that of the Met-306 [c164] replacement. Thus, these data provide evidence that different residue side chains at the interface of VIIa with TF control the primary specificity site and extended recognition of macromolecular substrate.
Figure 3.
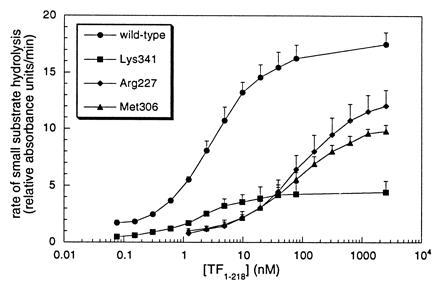
Effect of Ala substitutions of VIIa protease domain residues on amidolytic function of TF·VIIa. The rate of hydrolysis of Chromozym tPA by 5 nM mutant or wild-type VIIa is shown versus the concentration of soluble TF1–218. Binding defects are apparent for mutations of Arg-277 [c134] (KDapp = 73 ± 1 nM) and Met-306 [c164] (KDapp = 77 ± 16 nM), as compared with wild-type VIIa (KDapp = 2.8 ± 0.3 nM) and the mutant at residue Lys-341 [c192] (KDapp = 1.7 ± 0.6 nM).
DISCUSSION
The identification of functionally important residues by the comprehensive mutational analysis of VII presented here complements the recently determined crystal structure of the TF·VIIa complex (3). VIIa contacts TF through an extended interface involving (i) the Gla domain with the carboxyl-terminal module near the membrane insertion, (ii) EGF1 with the TF intermodule interface, and (iii) a protease domain–EGF2 surface contacting the amino-terminal TF module. The energetically most significant contacts were found at the EGF1 interface where Ile-69, Phe-71, and Arg-79 account for a total of 4.2 kcal/mol of the free energy of binding to TF. The respective cofactor contacts, TF residues Lys-20, Ile-22, and Phe-140, together contribute 5.4 kcal/mol (21), thus demonstrating good energetic complementarity of the two interfaces. In contrast to the important energetic contribution of the hydrophobic face of EGF1, the major contact residues in EGF2 (Gln-88, Val-92, Asn-93) had negligible effect on TF binding, consistent with mutational analysis of TF residue Phe-50 (22) which packs into the EGF2 interface. Of several protease domain contacts with TF, only Arg-277 [c134], Met-306 [c164], and Asp-309 [c167] substitutions affected cofactor affinity. The contribution of the protease domain to TF binding was about 2 kcal/mol, in agreement with the mutational effect of reciprocal TF residues Phe-76, Tyr-94, and Trp-45 which contribute 2.9 kcal/mol (30). We have identified protease domain residues (Pro-303 [c161], Leu-305 [c163], Arg-379 [c230]) which displayed binding defects upon substitution but did not contact TF in the crystal structure. The location of these residues suggests that they function indirectly through the local structural presentation of TF binding residues. The protease domain contact is weaker than the hydrophobic EGF1 contact, an observation that fits well with the atypical nature of TF–protease domain interface which contains several water molecules. The role of the EGF1 contact thus appears to be high affinity binding of TF, tethering the protease domain to the cofactor and relieving this interaction of high affinity constraints which could interfere with its activating function. EGF1 residue replacements with TF binding defects were normal in proteolytic function, while all of the protease domain interface mutants with decreased cofactor affinity had substantial reductions in proteolytic function.
The activation of the protease by cofactor binding may involve substantial conformational changes. The flexibility of the light chain of VIIa may allow protease domain interactions that block substrate access, and cofactor binding may release this autoinhibition through reorientation of the light chain and protease domain of VIIa. Because the overall docking of VIIa with TF should be largely unaffected by site-specific mutants in the contacts, the pronounced reduction in proteolytic function of single residue replacement mutants in the protease domain interface with TF indicates more specific cofactor effects on protease conformation. A more localized mechanism of autoinhibition was inferred from the crystal structure of the TF·VIIa complex which showed a flexible, extended loop of residues Gln-313 [c171] to Glu-325 [c178] that in free VIIa might block access to the catalytic cleft or even prevent the activating amino-terminal insertion (3). Since this loop extends from the energetically important Met-306 [c164] and Asp-309 [c167] contact with TF, cofactor interactions may directly influence the loop conformation. Although such a regulatory role of this loop cannot be excluded by our mutational analysis, it is notable that mutations in this loop had only subtle effects on proteolytic function of VIIa.
A potential mechanism by which TF may activate VIIa is through stabilization of the active conformation of the protease domain. This may involve conformational changes of the Cys-329 [c182] to Ser-339 [c190] loop which is disulfide linked to the Thr-307 [c165]–Cys-310 [c168] α-helix at the TF interface. In these segments, partially buried residues Thr-307 [c165] and Tyr-332 [c185] contribute to catalytic function and are thus potentially involved in a TF-induced allosteric switch. The [c182–190] loop is part of the “activation domain,” a region of increased flexibility in zymogens as compared with active enzymes, as defined by structural analysis of trypsinogen and chymotrypsinogen activation (31). In thrombin, the same loop is involved in coordination of a Na+-ion which results in allosteric changes responsible for a switch in substrate specificity (32). If the allosteric change proposed for VIIa activation is analogous to allosteric switches of trypsin and thrombin, TF may ultimately affect Asp-338 [c189] which provides recognition of the basic P1 (33) residue in trypsin-like serine proteases. Consistent with the notion that cofactor interaction influences the active site of VIIa, we found reduced hydrolysis of small peptidyl substrates as a result of replacement of the TF binding residue Met-306 [c164]. However, substitution of another TF binding residue, Arg-277 [c134], caused a less pronounced defect in amidolytic function and a more substantial reduction in macromolecular substrate activation. A similar mutational effect has been found for TF residue Asp-44 (34) which likely has indirect effects on Arg-277 [c134] interactions, based on the spatial arrangement of interface residues in the structure of TF·VIIa (3). These data support the concept that the activating effect of cofactor may include changes in extended recognition of substrate.
TF interactions with the VIIa protease domain appear to stabilize the amino-terminal insertion of Ile-153 [c16] (35). The crystal structure of the TF·VIIa complex demonstrated that the amino terminus of the protease domain does not directly contact TF, and therefore the amino-terminal insertion must be stabilized through indirect effects. Since Ile [c16]–Asp [c194] salt-bridge formation is energetically interdependent with stabilization of the “activation domain” (31), the favored amino-terminal insertion in the presence of cofactor lends support to the concept that TF interactions induce allosteric changes similar to those observed in zymogen-to-enzyme conversion of cofactor-independent serine proteases. Moreover, functionally important residues Leu-287 [c144] and Met-298 [c156] at the insertion site were found at positions which in tissue type plasminogen activator are surface exposed and involved in regulating its zymogenicity (36).
Surface residues which were important for catalytic function are found on the front face of the protease domain as viewed in Fig. 2. Besides residues which form the bottom of the catalytic cleft, determinants for substrate recognition are provided by residues in the rim of the cleft: Thr-239 [c99] is probably involved in P2 recognition, whereas Lys-341 [c192] could contribute to P′ recognition (9). Unlike the Lys-341 → Gln exchange (9), we found a reduction in amidolytic function for the Ala replacement of Lys-341 [c192] (Fig. 3), suggesting that the aliphatic side chain has also some role in forming the P1 and possibly the P3 specificity site.
Macromolecular substrate assembly may further involve two potential exosites, one of which is between the entrance to the catalytic cleft and the TF binding site. Notably, this exosite region is part of the activation domain, indicating that cofactor interactions may influence functionally important surface residue side chains. Sequences amino terminal to the S3 residue in the activation peptide of X have little importance for the interaction with TF·VIIa (37), suggesting that the activation peptide may not directly interact with this exosite located some distance from the P side of the catalytic cleft. The other potential exosite is located at the P′ side of the catalytic cleft and stretches from the groove which accommodates the protease domain amino terminus to the high affinity Ca2+ binding site, known to be important for catalytic function of VIIa (29, 38). The surface of this region, as modeled, was found to be highly electronegative (Fig. 2 Inset). VIIa thus differs from thrombin in which the analogous exosite I is positively charged (39, 40).
Because the zymogen structures of VII, X, and IX are unknown, the precise docking of the macromolecular substrates with TF·VIIa remains unclear. However, the [c22–27] disulfide bond in VII and X imposes constraints on the distance between the zymogen protease domain and the scissile bond which enters the catalytic cleft. Because of these constraints, it is likely that the zymogen protease domain interacts with one or both of the VIIa exosites which are located downwards from the active site cleft to the membrane proximal regions of TF·VIIa. If the zymogens adopt a similarly extended conformation, as demonstrated for VIIa in the TF·VIIa complex (3) or free factor IXa (41), macromolecular substrates may simultaneously contact membrane through their Gla-domains, providing a rational for the rate accelerating effect of charged phospholipid for X activation (42). In this orientation, the substrate EGF and Gla-domains may be positioned in proximity to the face of TF·VIIa which is formed by TF (residues Lys-165, Lys-166) and by the VIIa Gla-domain, both of which are critical for macromolecular substrate activation (24, 43). The detailed analysis of the residues involved in function of VIIa presented here is thus compatible with the concept that macromolecular substrate assembles through an extended interface formed by one face of the TF·VIIa complex.
Acknowledgments
We are grateful for the excellent technical assistance of Justin Shobe, Cindi Biazak, Jennifer Royce, and David Revak. We thank Dr. D. Stuart and Dr. A. Tulinsky for coordinates and Dr. S. Krishnaswamy for curve-fitting software. This work was supported by National Institutes of Health Grant RO1-48752.
Footnotes
Abbreviations: VII/VIIa, coagulation factor VII/VIIa; X/Xa, coagulation factor X/Xa; TF, tissue factor; Gla-domain, γ-carboxyglutamic acid-rich domain; EGF, epidermal growth factor; [c…], chymotrypsin numbering.
References
- 1.Davie E W, Fujikawa K, Kisiel W. Biochemistry. 1991;30:10363–10370. doi: 10.1021/bi00107a001. [DOI] [PubMed] [Google Scholar]
- 2.Martin D M A, Boys C W G, Ruf W. FASEB J. 1995;9:852–859. doi: 10.1096/fasebj.9.10.7615155. [DOI] [PubMed] [Google Scholar]
- 3.Banner D W, D’Arcy A, Chène C, Winkler F K, Guha A, Konigsberg W H, Nemerson Y, Kirchhofer D. Nature (London) 1996;380:41–46. doi: 10.1038/380041a0. [DOI] [PubMed] [Google Scholar]
- 4.Sridhara S, Clarke B J, Blajchman M A. Blood Coagulation Fibrinolysis. 1993;4:505–506. doi: 10.1097/00001721-199306000-00019. [DOI] [PubMed] [Google Scholar]
- 5.O’Brien D P, Kemball-Cook G, Hutchinson A M, Martin D M A, Johnson D J D, Byfield P G H, Takamiya O, Tuddenham E G D, McVey J H. Biochemistry. 1994;33:14162–14169. doi: 10.1021/bi00251a027. [DOI] [PubMed] [Google Scholar]
- 6.O’Brien D P, Gale K M, Anderson J S, McVey J H, Miller G J, Meade T W, Tuddenham E G D. Blood. 1991;78:132–140. [PubMed] [Google Scholar]
- 7.Matsushita T, Kojima T, Emi N, Takahashi I, Saito H. J Biol Chem. 1994;269:7355–7363. [PubMed] [Google Scholar]
- 8.Ruf W. Biochemistry. 1994;33:11631–11636. doi: 10.1021/bi00204a026. [DOI] [PubMed] [Google Scholar]
- 9.Neuenschwander P F, Morrissey J H. Biochemistry. 1995;34:8701–8707. doi: 10.1021/bi00027a020. [DOI] [PubMed] [Google Scholar]
- 10.Wells J A. Biochemistry. 1990;29:8509–8517. doi: 10.1021/bi00489a001. [DOI] [PubMed] [Google Scholar]
- 11.Padmanabhan K, Padmanabhan K P, Tulinsky A, Park C H, Bode W, Huber R, Blankenship D T, Cardin A D, Kisiel W. J Mol Biol. 1993;232:947–966. doi: 10.1006/jmbi.1993.1441. [DOI] [PubMed] [Google Scholar]
- 12.Navia M A, McKeever B M, Springer J P, Lin T Y, Williams H R, Fluder E M, Dorn C P, Hoogsteen K. Proc Natl Acad Sci USA. 1989;86:7–11. doi: 10.1073/pnas.86.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Read R J, James M N. J Mol Biol. 1988;200:523–551. doi: 10.1016/0022-2836(88)90541-4. [DOI] [PubMed] [Google Scholar]
- 14.Bolognesi M, Gatti G, Menagatti E, Guarneri M, Marquart M, Papamokos E, Huber R. J Mol Biol. 1982;162:839–868. doi: 10.1016/0022-2836(82)90550-2. [DOI] [PubMed] [Google Scholar]
- 15.Karpausas M, Holland D, Remington S J. Biochemistry. 1991;30:6024–6031. doi: 10.1021/bi00238a028. [DOI] [PubMed] [Google Scholar]
- 16.Morris A L, MacArthur M W, Hutchinson E G, Thornton J M. Proteins. 1992;12:345–364. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- 17.Laskowski R A, MacArthur M W, Moss D S, Thornton J M. J Appl Crystologr. 1993;26:283–291. [Google Scholar]
- 18.Rao Z, Handford P, Mayhew M, Knott V, Brownlee G G, Stuart D. Cell. 1995;82:131–141. doi: 10.1016/0092-8674(95)90059-4. [DOI] [PubMed] [Google Scholar]
- 19.Fair D S, Plow E F, Edgington T S. J Clin Invest. 1979;64:884–894. doi: 10.1172/JCI109554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deng W P, Nickoloff J A. Anal Biochem. 1992;200:81–88. doi: 10.1016/0003-2697(92)90280-k. [DOI] [PubMed] [Google Scholar]
- 21.Schullek J R, Ruf W, Edgington T S. J Biol Chem. 1994;269:19399–19403. [PubMed] [Google Scholar]
- 22.Ruf W, Schullek J R, Stone M J, Edgington T S. Biochemistry. 1994;33:1565–1572. doi: 10.1021/bi00172a037. [DOI] [PubMed] [Google Scholar]
- 23.Krishnaswamy S. J Biol Chem. 1992;267:23696–23706. [PubMed] [Google Scholar]
- 24.Ruf W, Kalnik M W, Lund-Hansen T, Edgington T S. J Biol Chem. 1991;266:15719–15725. [PubMed] [Google Scholar]
- 25.Kelley R F, Costas K E, O’Connell M P, Lazarus R A. Biochemistry. 1995;34:10383–10392. doi: 10.1021/bi00033a009. [DOI] [PubMed] [Google Scholar]
- 26.Ferrin T E, Huang C C, Jarvis L E, Langridge R. J Mol Graphics. 1988;6:13–27. [Google Scholar]
- 27.Bode W, Mayr I, Baumann U, Huber R, Stone S R, Hofsteenge J. EMBO J. 1989;8:3467–3475. doi: 10.1002/j.1460-2075.1989.tb08511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nicholls, A. (1992) grasp, Graphical Representation and Analysis Surface Properties (Columbia Univ., New York).
- 29.Wildgoose P, Foster D, Schiodt J, Wiberg F C, Birktoft J J, Petersen L C. Biochemistry. 1993;32:114–119. doi: 10.1021/bi00052a016. [DOI] [PubMed] [Google Scholar]
- 30.Ruf W, Kelly C R, Schullek J R, Martin D M A, Polikarpov I, Boys C W G, Tuddenham E G D, Edgington T S. Biochemistry. 1995;34:6310–6315. doi: 10.1021/bi00019a008. [DOI] [PubMed] [Google Scholar]
- 31.Huber R, Bode W. Acc Chem Res. 1978;11:114–122. [Google Scholar]
- 32.Di Cera E, Guinto E R, Vindigni A, Dang Q D, Ayala Y M, Wuyi M, Tulinsky A. J Biol Chem. 1995;270:22089–22092. doi: 10.1074/jbc.270.38.22089. [DOI] [PubMed] [Google Scholar]
- 33.Schechter I, Berger A. Biochem Biophys Res Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 34.Kelly C R, Schullek J R, Ruf W, Edgington T S. Biochem J. 1996;315:145–151. doi: 10.1042/bj3150145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Higashi S, Nishimura H, Aita K, Iwanaga S. J Biol Chem. 1994;269:18891–18898. [PubMed] [Google Scholar]
- 36.Lamba D, Bauer M, Huber R, Fischer S, Rudolph R, Kohnert U, Bode W. J Mol Biol. 1996;258:117–135. doi: 10.1006/jmbi.1996.0238. [DOI] [PubMed] [Google Scholar]
- 37.Baugh R J, Krishnaswamy S. J Biol Chem. 1996;271:16126–16134. doi: 10.1074/jbc.271.27.16126. [DOI] [PubMed] [Google Scholar]
- 38.Sabharwal A K, Birktoft J J, Gorka J, Wildgoose P, Petersen L C, Bajaj S P. J Biol Chem. 1995;270:15523–15530. doi: 10.1074/jbc.270.26.15523. [DOI] [PubMed] [Google Scholar]
- 39.Bode W, Huber R, Rydel T J, Tulinsky A. In: Thrombin: Structure and Function. Berliner L J, editor. New York: Plenum; 1992. pp. 3–61. [Google Scholar]
- 40.Tsiang M, Jain A K, Dunn K E, Rojas M E, Leung L L K, Gibbs C S. J Biol Chem. 1995;270:16854–16863. doi: 10.1074/jbc.270.28.16854. [DOI] [PubMed] [Google Scholar]
- 41.Brandstetter H, Bauer M, Huber R, Lollar P, Bode W. Proc Natl Acad Sci USA. 1995;92:9796–9800. doi: 10.1073/pnas.92.21.9796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruf W, Rehemtulla A, Morrissey J H, Edgington T S. J Biol Chem. 1991;266:2158–2166. [PubMed] [Google Scholar]
- 43.Ruf W, Miles D J, Rehemtulla A, Edgington T S. J Biol Chem. 1992;267:6375–6381. [PubMed] [Google Scholar]