

Abstract
An electroretinogram (ERG) screen identified a mouse with a normal a-wave but lacking a b-wave, and as such it was designated no b-wave3 (nob3). The nob3 phenotype mapped to chromosome 11 in a region containing the metabotropic glutamate receptor 6 gene (Grm6). Sequence analyses of cDNA identified a splicing error in Grm6, introducing an insertion and an early stop codon into the mRNA of affected mice (designated Grm6nob3). Immunohistochemistry of the Grm6nob3 retina showed that GRM6 was absent. The ERG and visual behaviour abnormalities of Grm6nob3 mice are similar to Grm6nob4 animals, and similar deficits were seen in compound heterozygotes (Grm6nob4/nob3), indicating that Grm6nob3 is allelic to Grm6nob4. Visual responses of Grm6nob3 retinal ganglion cells (RGCs) to light onset were abnormal. Grm6nob3 ON RGCs were rarely recorded, but when they were, had ill-defined receptive field (RF) centres and delayed onset latencies. When Grm6nob3 OFF-centre RGC responses were evoked by full-field stimulation, significantly fewer converted that response to OFF/ON compared to Grm6nob4 RGCs. Grm6nob4/nob3 RGC responses verified the conclusion that the two mutants are allelic. We propose that Grm6nob3 is a new model of human autosomal recessive congenital stationary night blindness. However, an allelic difference between Grm6nob3 and Grm6nob4 creates a disparity in inner retinal processing. Because the localization of GRM6 is limited to bipolar cells in the On pathway, the observed difference between RGCs in these mutants is likely to arise from differences in their inputs.
Rod and cone photoreceptors encode variations in light energy with graded changes in neurotransmitter output. At the first synapse, rod and cone photoreceptors provide input to rod and cone bipolar cells (BC), respectively (Wassle, 2004). Then, within the cone pathway there is a further divergence into On and Off pathways, which is based on the postsynaptic glutamate receptor used by the two types of cone BCs. Signalling through the primary rod and the On cone pathways relies on the metabotropic glutamate receptor 6 (GRM6) (Nomura et al. 1994; Masu et al. 1995; Vardi et al. 2000). In contrast, the Off cone pathway utilizes ionotropic glutamate receptors (AMPA/kainate) (DeVries, 2000). At the onset of a light increment, all BCs that subserve the On pathway depolarize, whereas cone BCs in the Off pathway hyperpolarize. As a consequence, BCs in the On pathway have been labelled On or depolarizing (DBCs), while their counterparts in the Off pathway are called Off or hyperpolarizing (HBCs).
Photoreceptor and DBC function can be assessed non-invasively, using the electroretinogram (ERG). This technique has been widely exploited to identify mutations in genes that control the initial stages of visual processing in both mammals and flies. Identification of mutants with an altered ERG a-wave has resulted in the elucidation of many aspects of the photoreceptor signalling cascade (Pak, 1995; Nishina & Naggert, 2003; Pacione et al. 2003). Mutants also have been found where the ERG b-wave was either decreased in amplitude or eliminated. The majority of these mouse mutants result from defects in presynaptic glutamate release (Ball et al. 2002; Haeseleer et al. 2004; Maeda et al. 2005; Mansergh et al. 2005; Chang et al. 2006; Specht et al. 2007). To date, two spontaneous and two genetically engineered mouse mutants with postsynaptic defects have been identified. They are Nyxnob, a defect in nyctalopin, a protein whose exact function is unknown, but is critical to DBC pathway signalling (Pardue et al. 1998; Gregg et al. 2003, 2007); Grm6nob4 (Pinto et al. 2007b), a point mutation of the metabotropic glutamate receptor 6 (Grm6); a knockout (KO) of Grm6 itself (Masu et al. 1995; Renteria et al. 2006); and a KO of GαO, a trimeric G-protein found in DBCs that is a second messenger of GRM6 activation (Vardi et al. 1993; Dhingra et al. 2000, 2002). Although all of these mutants share a negative ERG phenotype, the visual responses of the retinal ganglion cells (RGCs) in the three mutants assayed to date differ significantly (Demas et al. 2006; Renteria et al. 2006; Gregg et al. 2007; Pinto et al. 2007b). These observations suggest that the absence of signalling within the DBCs assessed with the ERG does not translate directly into a simple absence of DBC input to the inner retinal circuit and, ultimately to ON-centre RGCs.
We now describe a new mutant, identified by an ERG screen (The Jackson Laboratory), which we initially designated nob3. The mutant has a normal a-wave but no b-wave, which is associated with altered function primarily within the On pathway of the retina (Chang et al. 2002). In view of the importance of the On pathway in vision, the few postsynaptic mutants that have been identified to date, and the possibility that nob3 represented a mutation in a novel protein in the DBC signalling cascade, we identified the underlying defect. Our data show that the nob3 mouse harbours a mutation in the Grm6 gene and, therefore, should be renamed Grm6nob3. The ERG and visual behaviour phenotypes of Grm6nob3, Grm6nob4 and their compound heterozygous progeny (Grm6nob4/nob3) are the same and differ significantly from controls, confirming that Grm6nob3 and Grm6nob4 are allelic variants of the Grm6 gene. Aspects of the visual response properties of Grm6nob3 RGCs are abnormal compared to control and distinct from those of Grm6nob4 RGCs. First, compared to controls, all Grm6nob3 RGCs that respond to the onset of a bright stimulus (ON RGCs) have ill-defined RFs and require a full-field stimulus to elicit a reliable response. Further, the onset latency of this response is significantly delayed compared to control ON-centre RGCs. This finding is consistent with ON RGC responses in Grm6 KO and Grm6nob4 mice (Renteria et al. 2006; Pinto et al. 2007b). OFF-centre Grm6nob3 RGCs have well-defined RFs similar to control and Grm6nob4 RGCs. However, when stimulated by full-field stimuli, the Grm6nob3 OFF-centre RGCs differ from both Grm6nob4 and control RGCs. Namely, significantly fewer OFF-centre Grm6nob3 RGCs alter their RF centre sign response and become OFF/ON. This difference is likely to result from allelic variance between the Grm6nob3 and Grm6nob4 mutations. We propose that this disparity arises from a difference in the input to the inner retina from DBCs in Grm6nob3 and Grm6nob4 mice. Understanding this difference may provide new clues to the GRM6-mediated DBC signalling cascade and the yet to be identified cation channel that initiates the depolarizing signal to light in the On pathway.
Methods
Mice were treated in accordance with the Animal Care and Use Committees at each of the contributing institutions and in compliance with the statement for ethical care and use of animals of the Association for Research in Vision and Ophthalmology (ARVO). Prior to all experiments in which tissue was harvested or in vivo electrophysiological assessments were performed, anaesthesia was induced with an intraperitoneal injection of a mixture of ketamine/xylazine (127/12 mg kg−1) or alternatively, mice were killed by carbon dioxide inhalation. For in vitro electrophysiological assessments, mice were killed by cervical dislocation. For these experiments, control data were obtained from adult C57BL/6J mice (≥ 3 months of age) and ages are provided in each section below.
Mouse production and mutation mapping
The Grm6nob3 mouse was identified during an ERG screen and arose during the process of moving a transgene (B10.D2-Tg(Igh2k3–83)1Nemz/J) onto a C57BL/10J background. Grm6nob3 mutants were mated for 10 generations to C57BL/6J to remove the transgene and develop a (B6.B10 (D2)-Grm6nob3/Pjn) congenic strain that was utilized for mapping the mutation. A B6.B10 (D2)-Grm6nob3/Pjn X Balb/cJ F2 intercross was generated using The Jackson Laboratory Speed Expansion IVF service (http://jaxmice.jax.org/services/speed_expansion.html). The critical region was refined by genotyping recombinant mice with commercially available MIT markers to simple repeat sequences. Additional single nucleotide polymorphic (SNP) markers were developed to refine the map.
Anatomical assessment
Light microscopy
Retinas from adult animals were fixed in 37.5% methanol–12.5% glacial acetic acid in (1×) phosphate buffered saline and embedded in paraffin, and 6 μm thick sections were cut. Sections were mounted on slides (Superfrost Plus; Fisher Scientific, Pittsburgh, PA, USA) and stained with haematoxylin and eosin (H&E). Light microscopy was performed on a Leitz DMRB microscope (Leica, Germany) and images were collected with a Leica Firecam. Image brightness and contrast were optimized with Adobe Photoshop 7.0.
Immunohistochemistry and fluorescence microscopy
Fresh retinas from adult mice were embedded in Optimum Cutting Temperature (OCT) medium (Sakura Finetek USA Inc., Torrance, CA, USA), frozen, and 10 μm thick sections were cut on a Leica CM3050S cryotome. Sections were mounted onto slides and probed with one of two antibodies derived against the C-terminus of GRM6 at a concentration of 1 : 100 in 1 × phosphate buffered saline plus 4% normal horse serum as previously described (Chang et al. 2006; Pinto et al. 2007b). One antibody was a gift from Dr Catherine Morgans (Casey Eye Institute, Oregon Health and Science University; Morgans et al. 2006), and the other was a commercial antibody (Neuromics, Edina, MN, USA). Appropriate secondary antibodies coupled to either CY3 (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) or Alexa-488 (Molecular Probes, Eugene, OR, USA) were diluted 1 : 500 in 1 × phosphate buffered saline plus 4% normal horse serum. Imaging was performed on a Leica model DMLB microscope (Leica, Germany) and images were collected with a Spot model 2.1.1 digital camera (Avon, MA, USA). Brightness and contrast of images were optimized with Adobe Photoshop 7.0.
Transmission electron microscopy
Eyes from adult mice were fixed by immersion in cold 0.1 m phosphate buffer, pH 7.0 containing 0.8% glutaraldehyde and 1.0% paraformaldehyde for 2 h, cut into small blocks that were postfixed in the same fixative for 12 h, and processed for electron microscopy as previously described (Smith et al. 2002).
Electrophysiological assessment
Electroretinography
Our detailed ERG methods and analysis have been published previously (Pinto et al. 2005, 2007a). Briefly, adult mice were dark-adapted for at least 2 h and all procedures were performed in darkness with the aid of infra-red image converters and safelamps. Body temperature was maintained between 36.5 and 37°C with a temperature-controlled heating pad. Dawson, Trick and Litzkow electrodes (Dawson et al. 1979) were placed on each eye and the corneas were wetted with a solution of 1.2% methylcellulose in 0.9% NaCl and covered with a contact lens. The ERG was recorded differentially between the eyes and the response evoked by a diffuse, full-field flash stimulus that was presented via an LED to one eye. That eye was covered with a clear contact lens, while an opaque lens covered the other eye. The scotopic ERG was assessed using a series of full-field stimuli of increasing intensity (luminance 6.6 × 10−4–300 cd s m−2). The photopic ERG response was evoked by a full-field flash (intensity; 0.2 cd s m−2) presented on and after a 7 min adaptation to a steady rod-desensitizing field (0.5 cd m−2). The duration of the interstimulus interval varied as a function of stimulus intensity from 4 s for low-intensity flashes to 90 s for the highest intensity stimuli. Ten to 20 responses were averaged at the lowest intensities (up to 8.3 × 10−4 cd s m−2) to increase the signal-to-noise ratio. Two responses were averaged for the higher intensity stimuli and a single response used for the highest. All ERG responses were recorded at 1 kHz with a resolution of 0.5 μV and filtered to remove oscillatory potentials to more accurately measure b-wave amplitude. The measurements of the individual components of the ERG have been described in detail (see Fig. 1; Pinto et al. 2007a) and were chosen to objectively measure these amplitudes in WT ERG responses. The a-wave was measured from baseline to its trough within a window of 1–100 ms, and the b-wave was measured from the trough of the a-wave to its peak within a window of 25–100 ms. Similarly, the scotopic threshold response (STR; Saszik et al. 2002) is measured from its peak (positive STR) to ensuing trough (negative STR) (within 300 ms of stimulus onset). Stimulus presentation and data recording were controlled by software available at: http://www.neuromice.org/browseAssays.do or http://www.genome.northwestern.edu/neuro/vision_protocol.cfm, and MATLAB (The Mathworks, Inc., Natick, MA, USA) software was used to analyse the a- and b-wave amplitudes of the ERG.
Figure 1. Grm6nob3 retinal lamination and morphology are normal.

A and B, light microscopic images of transverse retinal sections, stained with haematoxylin and eosin, from adult control (A) and Grm6nob3 mice (B) (3 months of age) show no differences in laminar thickness or cellular organization. Electron microscopic images from Grm6nob3 retina show that outer retinal structures are normal: C, choriocapillaris, Bruchs' membrane (*), retinal pigment epithelium (PE) and photoreceptor outer segments (OS); D, inner segments (IS) and external limiting membrane (arrowheads); E, outer and inner nuclear layers (ONL, INL) and outer plexiform layer (OPL). Scale bar = 15 μm (A and B); 2 μm (C–E).
Extracellular RGC recordings
We recorded the responses of RGCs using both an in vivo (adult mice) and an in vitro (P21–P34 mice) approach. Each technique has advantages that provide complementary features. In vivo, individual RGC responses can be characterized at both dark- and light-adapted levels because the retinal pigment epithelium regenerates critical components of the visual cycle. However, small sample sizes (4–10 cells per mouse) result because RGCs are characterized sequentially. In vitro using a multielectrode array, cell responses to full-field stimuli are characterized simultaneously resulting in large sample sizes (50–100 cells per retina).
In vivo single RGC recordings
Responses were recorded from age-matched Grm6nob3 (n = 7), Grm6nob4 (n = 2) and control mice (n = 4). New data from Grm6nob4 and control RGCs were combined with previously published data to provide the complete data sets used in this study (Sagdullaev & McCall, 2005; Pinto et al. 2007b). Surgical techniques, as well as visual stimulation and recording protocols, have been published previously (Sagdullaev & McCall, 2005; Chang et al. 2006; Pinto et al. 2007b). After inducing anaesthesia, the mouse was mounted in a stereotaxic frame, a craniotomy was performed and suction used to remove tissue to visualize the optic nerve. Action potentials from individual axons were recorded, using a tungsten electrode (impedance, 40–70 MΩ), amplified and digitized at 15 kHz. Eyedrops dilated the pupil and paralysed accommodation and contact lenses covered the corneas (Sagdullaev et al. 2004). When possible, the RF centre of each isolated RGC was mapped onto a tangent screen. Some RGCs in Grm6nob3 and Grm6nob4 had ill-defined RF centres, although an area of the visual field could be defined where a large diameter spot evoked an increase in their firing rate above spontaneous levels. After the RF was mapped, it was centred on a CRT display monitor placed 25 cm from the nodal point of the eye and the display monitor was used to present computer controlled visual stimuli (Vision Works for Electrophysiology; Vision Research Graphics, Durham, NH). Visually evoked responses were recorded from individual RGCs to both spots and full-field stimuli. Spots (100 or 3 cd m−2, for ON- and OFF-centre RGCs, respectively) varied in diameter from 4.5 to 52 deg of visual angle and were presented on a background of 20 cd m−2 for a 2 s duration. Spots and blank trials were presented in random order a total of eight times. Full-field stimuli with a duty cycle of 5 s ON (150 cd m−2) and 5 s OFF (0 cd m−2) were presented 30 times and spontaneous activity estimated over similar intervals to a screen of mean average luminance (75 cd m−2). Responses were collected and displayed (50 ms bin width) as individual raster plots and averaged PSTHs. Analyses of response characteristics were performed from averaged and smoothed PSTHs (Sagdullaev & McCall, 2005). Spontaneous activity was assessed from the average PSTH of the blank trials. The mean spontaneous rate 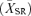 and the standard error of the mean were computed and used to set thresholds defining significant visual responses. A significant response was defined as a response to stimulus onset whose mean peak firing rate was >
and the standard error of the mean were computed and used to set thresholds defining significant visual responses. A significant response was defined as a response to stimulus onset whose mean peak firing rate was > 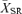 + 3 s.e.m. Peak firing rate (spikes s−1) and time to peak (ms) after stimulus onset were measured. All control RGCs could be defined as ON- or OFF-centre based on their excitatory response evoked by the onset of either a bright or a dark spot matched in size and contrast to their RF centre. Grm6nob3 and Grm6nob4 RGCs did not respond to the onset of bright spot stimuli, but did respond to the onset of the bright phase of the full-field stimulus and were defined as ON RGCs. Using the full-field stimulus, OFF-centre cells, those that responded to the onset of a dark spot, could be subdivided into two groups: sign-conserving (OFF-SC) RGCs that maintained an OFF response and sign-altering (OFF-SA) RGCs that altered their centre sign response and became OFF/ON or occasionally ON only RGCs (Sagdullaev & McCall, 2005).
+ 3 s.e.m. Peak firing rate (spikes s−1) and time to peak (ms) after stimulus onset were measured. All control RGCs could be defined as ON- or OFF-centre based on their excitatory response evoked by the onset of either a bright or a dark spot matched in size and contrast to their RF centre. Grm6nob3 and Grm6nob4 RGCs did not respond to the onset of bright spot stimuli, but did respond to the onset of the bright phase of the full-field stimulus and were defined as ON RGCs. Using the full-field stimulus, OFF-centre cells, those that responded to the onset of a dark spot, could be subdivided into two groups: sign-conserving (OFF-SC) RGCs that maintained an OFF response and sign-altering (OFF-SA) RGCs that altered their centre sign response and became OFF/ON or occasionally ON only RGCs (Sagdullaev & McCall, 2005).
In vitro, multielectrode array retinal recordings
Dissection techniques, as well as visual stimulation and recording protocols, have been previously described (Pinto et al. 2007b). Eyes were enucleated under dim red illumination, placed into a Petri dish containing artificial cerebrospinal fluid (ACSF) of composition (in mm): NaCl, 124; KCl, 2.5; CaCl2, 2; MgCl2, 2; NaH2PO4, 1.25; glucose, 22; NaHCO3, 26; Hepes 20 (pH 7.55). The retina was isolated under infrared illumination using a microscope equipped with infrared converters and placed onto the microelectrode array (MEA) (Multichannel Systems, Reutlingen, Germany), secured with a small manipulator and placed in the preamplifier on an inverted microscope stage. The retina was superfused with oxygenated (95% O2, 5% CO2) ACSF maintained at 34°C with a 2 ml min−1 flow rate. A uniform field, generated using a video card and software interface (VSG, Cambridge Research Systems, England), was projected onto the retina through the objective lens of the microscope and consisted of a 1 s Light-ON period followed by a 1 s Light-OFF period whose luminances on the retina were 3 and 0 cd m−2, respectively. Voltage signals from the MEA electrodes were amplified and recorded using MCRack software (Multi Channel Systems). Noise did not exceed ± 20 μV in the recordings. Signals were high-pass filtered digitally at 25 Hz, and spikes were detected with a −51 μV threshold. Spike sorting was performed in Offline Sorter (Plexon Inc., Dallas, TX, USA) using an automated 2D T-Distribution Expectation Maximization algorithm. Time stamps were exported to MATLAB where custom scripts were used for data analysis. Peristimulus time histograms (PSTHs) and raster plots of individual units were generated. Non-responsive cells and visually responsive cells whose responses were not linked to stimulus presentation (OTHER) were excluded from further analyses. The remaining cells were classified as either ON, OFF, or OFF/ON based on the ratio of mean spike rates during the onset of the bright (RON) and dark (ROFF) phases of the full-field stimulus. ON cells were defined with RON/ROFF ratios > 4, those with ratios < 0.25 were defined as OFF cells, and OFF/ON cells had ratios between 0.25 and 4.0 (Nirenberg & Meister, 1997; Nirenberg et al. 2001).
Assessment of visual behaviour
The visual behaviour of adult mice was measured as previously described (Pinto et al. 2007b), using head movements that tracked rotating and sinusoidally modulated gratings in a virtual optokinetic system (CerebralMechanics, Lethbridge, Alberta, Canada). Spatial frequency and contrast thresholds of tracking were generated using simple staircase protocols that varied the spatial frequency or contrast of gratings rotating at a constant velocity (16 deg s−1; i.e. (McGill et al. 2004). The average spatial frequency cutoff was defined as the highest spatial frequency that elicited a reliable optomotor response to a 100% contrast grating. Mice were tested at two mean luminances (58.5 and 0.032 cd m−2 (photopic and scotopic, respectively). To test at the lower luminance, large neutral density filters covered the surfaces of the liquid crystal displays and a camera sensitive to infrared illumination and two infrared light emitting diodes (940 nm) were used to visualize the mouse (Fire-i board camera; Unibrain, Inc. San Ramon, CA, USA). All measurements were made between 13.00 h and 16.00 h by an observer blind to stimulus condition and mouse genotype.
Results
Grm6nob3 retinal morphology is normal
Examination of H&E stained transverse retinal sections by light microscopy revealed no discernable differences in gross retinal lamination or morphology between control and Grm6nob3 mice at 3 months of age (Fig. 1A and B) or later at 6 or 12 months (data not shown). This result is similar to other mice that harbour mutations in postsynaptic genes involved in the GRM6 signal transduction cascade including GRM6 itself, nyctalopin and GαO (Masu et al. 1995; Pardue et al. 1998; Dhingra et al. 2000). At the ultrastructural level (Fig. 1C–E), the outer retina of 3-month-old Grm6nob3 mice was normal, similar to two other postsynaptic mutants (Pardue et al. 2001; Dhingra et al. 2000).
The nob3 mutation maps to Grm6 and eliminates GRM6 protein expression in the retina
A B6.B10 (D2)-Grm6nob3/Pjn X BALB/cJ F2 intercross of 400 mice, representing 800 meioses, was generated and haplotype mapping defined a critical region on chromosome 11 (Fig. 2A). This region spanned 0.17 Mb between SNP rs6199956 (50.58 Mb) and marker D11PJN2 (50.75Mb) and contained five annotated or predicted genes, including Grm6 (Fig. 2B).
Figure 2. Mapping and cloning narrows the critical region containing the mutant phenotype to a portion of chromosome 11 containing the Grm6 gene.

A, a haplotype map illustrates the location and number of individual crossover events observed in 800 meioses resulting from an F2 intercross of B6.B10(D2)-Grm6nob3/Pjn, the background strain (*) and BALB/cJ. Vertical columns represent a region of chromosome 11 between markers D11Mit20 (top) and D11Mit274 (bottom). Filled boxes represent alleles from the B6.B10(D2)-Grm6nob3/Pjn chromosome and open boxes, alleles from the BALB/cJ chromosome. The numbers at the bottom of each vertical column represent the number of mice with that haplotype. All mice with recombination events in the critical region and carrying BALB/cJ alleles on the other chromosome were progeny tested by backcrossing to Grm6nob3/nob3 mice and determining the phenotype of mice carrying the recombinant chromosome. These experiments refined the critical region containing the B6.B10(D2)-Grm6nob3/Pjn to a 0.17 Mb region between markers rs6199956 and D11Pjn2. B, a representation of the critical region with flanking markers and the 5 annotated genes contained within this region.
Because mutations in Grm6 had previously been shown to produce a no b-wave ERG phenotype, we sequenced cDNA fragments representing the entire coding region of the Grm6 gene. Control sequence was identical to the sequence for Grm6 in Ensembl genome browser http://www.ensembl.org/index.html (data not shown). However, the Grm6nob3 sequence contained a 65 bp insertion (Fig. 3A, expanded view). This insertion was confirmed when the control and Grm6nob3 amplicons, produced with primers whose sequence was located near the 5′ end of the cDNA were size separated by agarose gel electrophoresis and compared (Fig. 3D). The size of the amplicon from Grm6nob3 cDNA was larger than that of control. Analysis of genomic DNA from Grm6nob3 mice (n = 5) identified a C to T transition at bp 648 of intron 1 of the Grm6 gene (Fig. 3B). This point mutation created a new splice donor site, which altered splicing and resulted in the insertion of a new 65 bp exon into the mRNA between exons 1 and 2. Further analyses, predicted that this insertion would cause a frameshift mutation that would truncate the protein and produce a null allele (Fig. 3C).
Figure 3. Sequence analysis indicates an insertion, resulting in a null Grm6nob3 allele that underlies the Grm6 phenotype.

A, chromatogram and sequence analysis of a control amplicon from the 5′ end of Grm6, which matches the published Grm6 sequence in Ensembl (OTTMUST0000001792). The black arrow and grey bar in the chromatogram (bottom row) indicate the position of the 65 bp insertion (expanded view below) in the Grm6nob3 cDNA fragment. B, genomic Grm6nob3 sequence indicates a ‘C’ (red) to ‘T’ (blue) transition at bp 648 of intron 1 of the Grm6 gene, which creates a new splice donor site (http://www.fruitfly.org/seqtools/splice.html). The use of a novel cryptic splice acceptance site (blue) and this new splice donor site leads to the insertion of a new exon (underlined sequence) into the coding sequence of Grm6nob3 cDNA. C, protein alignments for control (top), Grm6nob4 (middle) and Grm6nob3 (bottom). The Grm6nob4 mutation is a substitution of proline for serine 207 (S207P). In Grm6nob3 the insertion truncates the normal sequence at alanine 163 and encodes an additional 21 amino acids (green). The stop codon at position 265 is indicated by an asterisk. D, image of a stained agarose gel of RT-PCR amplicons from control and Grm6nob3 retina RNA generated using the same Grm6 primer set. mm – DNA size ladder. The Grm6nob3 amplicon is larger than control, confirming an insertion. E, images of transverse sections of control and Grm6nob3 retinas probed with a primary antibody against the C-terminus of GRM6 and visualized with a fluorescent secondary antibody (see Methods). Punctate label is abundant in the control OPL and absent in Grm6nob3 OPL. Scale bar = 15 μm.
To assess the effect of this putative null mutation on GRM6 expression in the OPL, we performed immunohistochemistry on transverse sections from control and Grm6nob3 retinas (Fig. 3E and F). Control retinal sections showed the characteristic punctuate expression of GRM6 in the OPL regardless of the antibody used (NB both are C-terminal peptide antibodies) (Morgans et al. 2005; Chang et al. 2006; Pinto et al. 2007b). In contrast, no detectable GRM6 signal was found anywhere in the Grm6nob3 retina, using the same antibodies. This result is similar to our observations in Grm6nob4 retina (Pinto et al. 2007b) and consistent with our prediction that Grm6nob3 is a null allele.
Grm6nob3 ERG responses are consistent with a mutation in Grm6 and that nob3 is allelic to Grm6nob4
We compared ERG responses of control, Grm6nob3, Grm6nob4 and Grm6nob4/nob3 mice under scotopic and photopic conditions and with several different full-field flash luminances (Fig. 4). Representative ERG responses and average responses (+s.e.m.) are shown for control and the three Grm6 mutants under scotopic conditions to examine rod-generated responses (Fig. 4A and B) and the scotopic threshold response (STR) (Fig. 4C and D) and under photopic conditions (Fig. 4E and F). All three Grm6 mutants have a normal a-wave whose amplitude increases with increasing flash luminance (indicated on the left of each ERG trace). All Grm6 mutants lack both a scotopic (Fig. 4A and B, open symbols) and a photopic b-wave (Fig. 4E and F). The a-wave (Fig. 4B; filled symbols) in Grm6 mutants differs slightly in amplitude and timing from control due to the absence of their positive going b-wave. While accurate for WT ERG b-wave responses, our quantitative estimate (Fig. 4B) is less informative for Grm6 mutants than a simple inspection of their ERG traces (Fig. 4A). Namely, our algorithm returns (see Methods) positive values for the b-wave because the ERG response remains below baseline through the end of our 200 ms window.
Figure 4. ERG analyses show similar no b-wave phenotypes in Grm6nob3, Grm6nob4 and Grm6nob4/nob3 compound heterozygote mice.

Representative ERG responses from adult control, Grm6nob3, Grm6nob4 and Grm6nob4/nob3 compound heterozygous progeny evoked under scotopic (A and C) and photopic (E) conditions using the flash intensities indicated to the left of each graph. A and B, under scotopic conditions all mice show a normal a-wave and only controls show a b-wave. B, average (± s.e.m.) a- and b-wave amplitudes (filled and open symbols, respectively) are plotted as a function of flash luminance. C and D, only control mice show a scotopic threshold response (positive or negative STR). D, average (+ s.e.m.) STR total amplitude evoked by a 2.8 × 10−4 cd s m−2 flash is plotted for control and all Grm6 mutants. E and F, under photopic conditions (rod desensitizing background, 0.5 cd m−2) only control mice show a b-wave. F, average (+ s.e.m.) photopic b-wave amplitude evoked by a 0.2 cd s m−2 flash is plotted for control and all Grm6 mutant mice. (C57BL/6J, n = 5; Grm6nob3, n = 7; Grm6nob4, n = 5; Grm6nob4/nob3, n = 4.)
All three Grm6 mutants lack an STR (Fig. 4C and D), indicating a reduction in inner retinal activity under rod isolating conditions (Saszik et al. 2002). Similar to our morphological analysis, longitudinal studies of Grm6nob3 ERGs (> 1 years) showed no additional changes (data not shown). The similarities across Grm6nob3, Grm6nob4 and Grm6nob4/nob3 ERGs, together with previously published studies of the Grm6 KO (Masu et al. 1995; Renteria et al. 2006; Pinto et al. 2007b), indicate that Grm6nob3 is allelic to Grm6nob4 and that photoreceptors in these Grm6 mutants function normally but the DBC response is compromised.
Visual function in Grm6nob3 is significantly reduced
We evaluated reflexive tracking thresholds in Grm6nob3, Grm6nob4 and Grm6nob4/nob3 mice, and compared them to controls. The measures for our controls are similar to those published previously for C57Bl/6J mice (Prusky et al. 2004). The contrast sensitivity of Grm6nob3 mice was significantly lower than that in control mice at all spatial frequencies, under both scotopic (Fig. 5A) and photopic (Fig. 5B) conditions. There was no significant difference in peak contrast sensitivity among Grm6 mutant mice, but there were significant differences between each of these mutants and control mice (photopic and scotopic: all P-values ≤ 0.0002). Spatial frequency thresholds were similar in Grm6nob3 and Grm6nob4 mutants, and were significantly lower than in controls (Fig. 5C; photopic all P-values ≤ 0.03; scotopic: Grm6nob4 P = 0.02). In addition, the spatial frequency thresholds of Grm6nob4/nob3 mice were similar to both Grm6nob3 and Grm6nob4. It should be noted that the spatial frequency cutoffs of Grmnob3 and Grmnob4/nob3 mice, defined as the highest spatial frequency that elicited a reliable optomotor response to a 100% contrast grating, are lower than controls although not statistically different (P = 0.17 and 0.06, respectively). In addition, contrast sensitivity is lower under scotopic conditions for controls as well as mutants, making it more difficult to demonstrate a significant decline in this behaviour. Taken together, these results indicate that each of the mutations resulted in reduced visual thresholds.
Figure 5. Visually mediated optomotor responses show significant deficits in Grm6 mutant mice.

A, scotopic spatial frequency tuning curves plot average contrast (± s.e.m.) sensitivity as a function of grating spatial frequency. Mean screen luminance is shown at the top of the graph. Tuning curves for control mice differ significantly from all three Grm6 mutants, which are all similar. Curves for the mutants have clear peaks, but all mutants have reduced contrast sensitivity across spatial frequencies. B, average photopic (± s.e.m.) spatial frequency tuning curves (same conventions as in A) for control mice differ significantly from the three Grm6 mutants, which are all similar. C, the average spatial frequency cutoff (+ s.e.m.), the highest spatial frequency that elicits a reliable optomotor response to a 100% contrast grating, is significantly) different between control and all three Grm6 mutants under photopic conditions. Under scotopic conditions the spatial frequency cutoff is lower in all three Grm6 mutants, but only Grm6nob4 mice reach statistical significance compared to controls. (A–C C57BL/6J, n = 5; Grm6nob3, n = 5; Grm6nob4, n = 5; Grm6nob4/nob3, n = 4)
The Grm6nob3 mutation alters the responses of ON RGCs and the distribution of RGC types
The absence of scotopic and photopic ERG b-waves and the defects in visual behaviour in Grm6nob3 mice are similar to data reported for the Grm6 KO mouse (Ueda et al. 2006; Renteria et al. 2006) and the Grm6nob4 mouse (Pinto et al. 2007b). Taken together these results indicate that On pathway signalling via both the rod and cone pathways is compromised in Grm6nob3 retina. Given our previous characterization of Cacna1fnob2 and Grm6nob4 RGC responses (Chang et al. 2006; Pinto et al. 2007b), as well as those reported for Grm6 KOs (Renteria et al. 2006), we knew that similarities in ERG phenotype do not always predict similarities at the level of the RGC response. If Grm6nob3 was similar to Grm6nob4 and the Grm6 KO, we hypothesized that the mutation would reduce the percentage of Grm6nob3 RGCs that exhibited an excitatory response to the onset of the bright phase of a full-field stimulus. To test this hypothesis, we evaluated visually evoked responses of single RGCs to full-field stimuli in vivo and in vitro.
Control and Grm6nob3 RGCs were classified by their excitatory response to the onset of the bright (ON) and/or dark (OFF) phase of a full-field stimulus in vivo and in vitro (Fig. 6Aa and b, respectively). Responses to full-field stimuli allowed us to compare in vivo with in vitro MEA responses, which were only evoked with full-field stimuli. In vivo, both control and Grm6nob3 RGCs also were characterized to define their RF centre sign, using spots whose diameter matched their RF centre. All control RGCs in vivo responded to the onset of either a bright (ON-centre RGCs) or a dark (OFF-centre RGCs) spot stimulus. Full-field stimulation of control RGCs in vivo also evoked robust responses with short onset latency, regardless of RF centre sign (Fig. 6Aa and Ba). With this stimulus, control OFF-centre RGCs split evenly into two groups, sign-conserving (22/43; OFF-SC) RGCs, which retained their RF centre response sign, and sign-altering (21/43; OFF-SA), which changed their OFF-centre response to OFF/ON (Fig. 6Ba). In vitro, full-field stimulation of control RGCs also evoked robust responses with short onset latencies that we grouped into ON, OFF or OFF/ON responses (Fig. 6Ab and Bb).
Figure 6. Altered Grm6nob3 RGC visual response properties.

Representative responses to full-field stimuli in control and Grm6nob3 RGCs recorded either in vivo (Aa) or in vitro (Ab). Aa, in vivo, RGCs were classified by their RF centre sign, using spot stimuli whose diameter matched the RGC RF centre size. Based on their full-field response, they were then classified as ON (top), OFF-SC (middle) or OFF-SA (bottom). Ab, in vitro RGCs were classified by their full-field response as ON (top), OFF (middle) or OFF/ON (bottom). Schematic diagrams shown below the PSTHs indicate the dark phase (filled bars) and the bright phase (open bars) of full-field stimuli; 5/5 s and 1/1 s in vivo and in vitro, respectively. B, distribution of RGC types in vivo (Ba) and in vitro (Bb). Ba, in vivo, all control RGCs had either ON- or OFF-centre responses and OFF-centre RGCs could be divided by the change in their response to the full-field stimulus. In control retina, about 50% of OFF-centre RGCs conserve their RF centre sign (OFF-SC), while the other half alter their centre sign (OFF-SA) and show OFF/ON responses. Among Grm6nob4 OFF-centre RGCs, a similar division was found. The division of OFF-centre RGCs differed in Grm6nob3 compared to either control or Grm6nob4, with OFF-SCs significantly increased. Bb, because full-field stimuli are only used in vitro, RGCs were divided into ON, OFF and OFF/ON. No ON RGCs were recorded in Grm6nob3 retinas. The distributions of OFF and OFF/ON response types are similar for control and Grm6nob4 RGCs, but differ significantly in Grm6nob3, with significantly more OFF RGCs.
In the Grm6nob3 retina, only a small number of RGCs responded to the onset of the bright phase of the full-field stimulus in vivo (ON RGCs, n = 8/40 visually responsive RGCs). When tested with spot stimuli, all Grm6nob3 RGCs had ill-defined RFs, with no clear surrounds, as reported previously for Grm6nob4 RGCs (Pinto et al. 2007b). Similarly, in vitro among the visually responsive Grm6nob3 RGCs, we failed to detect any ON RGCs (0/85), although we previously reported a small number of Grm6nob4 ON RGCs (4/118). With both in vivo and in vitro approaches, we found Grm6nob3 RGCs that we classified as non-responsive. These were RGCs that were spontaneously active, but changes in full-field illumination failed to elicit a response (3/43 RGCs in vivo; 78/197 in vitro). In control retinas, non-responsive RGCs were never encountered in vivo (0/113) and comprised only 15% of RGCs in vitro (30/197). In vitro, we also found a small number (10/193) of OTHER Grm6nob3 RGCs (see Methods) that were rarely encountered in control retinas, and never observed in vivo. The reduction in ON RGCs coupled with the increase in non-responsive and OTHER RGCs in both Grm6nob3 and Grm6nob4 retina suggests that many RGCs have lost their primary excitatory drive through the On pathway.
When OFF-centre RGCs were assessed in vivo, using full-field stimuli, 88% (28/32) of OFF-centre Grm6nob3 RGCs conserved their RF centre sign response (OFF-SC RGCs), leaving only 12% OFF-SA RGCs (Fig. 6Ba). In contrast, Grm6nob4 OFF-centre RGCs split evenly into OFF-SC (48%; 12/29) and OFF-SA (52%; 17/29), which is identical to the percentages found in control OFF-centre RGCs. The distribution of Grm6nob3 OFF-SC and SA RGCs differs significantly from both control and Grm6nob4 RGCs (2-tailed Fischer'exact probability test; P = 0.002 for both). Similarly, there were significant differences in the distribution of Grm6nob3 OFF and OFF/ON RGCs recorded in vitro compared to control and Grm6nob4 RGCs (Fig. 6Bb; P = 0.00003 for both). In control and Grm6nob4 retinas, 48% (75/156) and 64% (75/118) of RGCs show OFF/ON responses to full-field stimulation. In contrast, only 34% (29/85) Grm6nob3 RGCs responded with an OFF/ON response, similar to the decrease in OFF-SA RGCs in vivo. Further, there was an increase in Grm6nob3 OFF RGCs compared to Grm6nob4 or control RGCs (66%versus 27 and 33%, respectively), mirroring the increase in OFF-SC Grm6nob3 RGCs in vivo. Thus, even though different recording techniques and response classifications were used, the changes from WT controls between the two Grm6 mutants is very similar; namely, there are significantly more RGCs with purely OFF responses in Grm6nob3 than in Grm6nob4 mice. Concomitantly, there are fewer RGCs with OFF/ON responses in Grm6nob3 than in Grm6nob4 mice.
Because we had observed changes in the timing of the excitatory response of Grm6nob4 ON RGCs, we assessed the response onset latency of Grm6nob3 RGCs to full-field stimuli. Among Grm6nob3 RGCs with both OFF and ON response components, OFF-SA RGCs (in vivo) and the OFF/ON RGCs (in vitro), the mean onset latency for the ON component was significantly longer than its matched OFF component (Fig. 7A and B; paired t test; P = 0.0008 for both). This was different from control RGCs in vivo and in vitro where onset latency of either ON and OFF RGCs or the ON and OFF components of OFF-SA or OFF/ON RGCs were similar (data not shown). A long-latency ON response component has been reported in Grm6 KO RGCs and emerges in control retinas in the presence of l-2-amino-4-phosphonobutyric acid (l-APB), an agonist of GRM6 (Renteria et al. 2006). Thus, the results are consistent with the emergence or the unmasking of a long latency ON input to WT OFF-centre RGCs that is independent of the l-APB-sensitive On pathway and in Grm6 KO mice is not developmental in origin. Whether this delayed ON component in Grm6 mutants is related to the ON response in OFF-SA RGCs requires identification of its underlying current and sensitivity to l-APB.
Figure 7. Delayed ON responses are present in Grm6nob3 RGCs.

A, in vivo the average onset latency (± s.e.m.) for control ON and OFF responses in OFF-SA RGCs is short and similar. In Grm6nob3 and Grm6nob4 retinas, ON responses have significantly longer onset latency than their OFF counterparts or any of the responses in controls. B, in vitro, the average onset latency (± s.e.m.) for control ON and OFF responses in OFF/ON RGCs also is similar and significant differences also are seen in the ON component of OFF/ON Grm6nob3 and Grm6nob4 RGCs.
Grm6nob4/nob3 RGC responses verify that Grm6nob3 and Grm6nob4 are allelic
We also characterized the responses of Grm6nob4/nob3 RGCs in vitro (Fig. 8; n = 2 retinas, 120 RGCs) to verify our ERG and behaviour results that indicated the two mutants were allelic. The distributions of Grm6nob4/nob3 ON, OFF and OFF/ON RGCs are shown in Fig. 8B. First, no Grm6nob4/nob3 ON RGCs were observed. Further, there was a disproportionate representation of Grm6nob4/nob3 OFF versus OFF/ON RGCs (87%versus 13%), which is more similar to Grm6nob3 than to Grm6nob4. Similar to both Grm6 mutants, the onset latencies of the ON component of Grm6nob4/nob3 OFF/ON RGCs were significantly slower than their OFF component (Fig. 8C; P = 6 × 10−13). These results verify that the two Grm6 mutants are allelic and may suggest that the Grm6nob3 mutation is the dominant phenotype.
Figure 8. Grm6nob4/nob3 RGC responses verify that Grm6nob3 and Grm6nob4 are allelic.

A, representative raster plots and average PSTHs evoked by full-field stimulation and recorded in vitro show robust responses in Grm6nob4/nob3 retinas, which are similar in character to Grm6nob3 RGCs (see Fig. 6Ab). B, using the same classifications as in Fig. 6Bb, many OFF, few OFF/ON and no ON RGCs are recorded in Grm6nob4/nob3 retinas, similar to Grm6nob3 and different from Grm6nob4. C, Grm6nob4/nob3 ON RGC responses in OFF/ON cells have significantly longer average response onset latencies (± s.e.m.) than their OFF counterparts, while OFF response latencies are similar to control (Fig. 7B).
Discussion
We have identified and characterized a mouse with a new mutation in the metabotropic glutamate receptor 6 (Grm6) gene. We have determined the impact of this mutation on visual function and compared the abnormalities to another previously published allele, Grm6nob4 (Pinto et al. 2007b). Our results show that Grm6nob3 and Grm6nob4 mice are allelic and their mutations produce very similar deficits in the retinal On pathway, leading to significant reductions in overall visual function. Notably, we also observed a significant and unexpected difference in the visually evoked responses of Grm6nob3 and Grm6nob4 OFF-centre RGCs, a result inconsistent with the idea that a mutation in the Grm6 gene only affects the visual signal conveyed through the On pathway.
The location of the mutations in Grm6 differ among mouse models and human patients
Grm6nob3 is a mouse mutant that arose spontaneously and was identified by an ERG screen. The phenotype is caused by a ‘T’ to ‘C’ transition at bp 648 of intron 1. This results in the insertion of a new exon 65 bp in length into the mRNA causing a frameshift in the protein at residue 163 that also introduces an additional 101 amino acids before a stop codon at position 265. This truncation of the GRM6 protein occurs within the N-terminal extracellular domain that contains the glutamate binding pocket (Zeitz et al. 2007). The two other Grm6 mutant mice that have been identified or created have different underlying changes. The Grm6nob4 mutation, generated by N-ethyl-N-nitrosourea mutagenesis, is a T709C transition at codon 85 and results in a missense mutation S85P. The Grm6 KO mouse has a deletion of 1.2 kb, which includes part of an exon and should truncate the protein at amino acid 494, within one of its seven transmembrane domains. This alters protein function (Masu et al. 1995). In the ensuing text, we will use the term Grm6 mouse mutants when referring collectively to all three Grm6 mutant mouse models.
Mutations in human GRM6 give rise to autosomal recessive congenital stationary night blindness (arCSNB) (Dryja et al. 2005; Zeitz et al. 2005; O'Connor et al. 2006). A large constellation of mutations, which include missense and nonsense mutations, have been identified throughout the GRM6 gene. The impact of several of these mutations have been investigated by expressing them in vitro in HEK-293 cells (Zeitz et al. 2007). In all cases, the mutation significantly alters trafficking of the protein to the cell surface. This is consistent with data from the Grm6 mutant mice, which all lack GRM6 protein expression in the OPL by immmunohistochemistry (Tagawa et al. 1999; Pinto et al. 2007b). Because the available antibodies are made against the C-terminus of the protein, the possibility remains that a truncated protein could be present in some of the mouse or human mutants.
Similar phenotypic defects within On pathway signalling among mouse mutants and patients with Grm6 mutations
In addition to the absence of GRM6 protein, the Grm6 mouse mutants share other phenotypic similarities to humans with GRM6 arCSNB (Dryja et al. 2005; Zeitz et al. 2005; O'Connor et al. 2006; Zeitz et al. 2007). ERG studies of Grm6 mouse mutants and Grm6 arCSNB patients all define a selective reduction in the ERG b-wave, with a preserved a-wave component. The preserved a-wave indicates that photoreceptors respond normally to light, and the marked reduction of the b-wave localizes the defect to one of synaptic transmission to and/or signalling within the DBCs. Further, both mouse and humans show no progressive functional changes. These functional results are consistent with our mapping, sequencing and protein localization results, indicating that the primary defect in Grm6nob3 is in the Grm6 gene. All Grm6 mouse mutants share similar functional changes in On pathway signalling at the level of the RGCs and beyond. Specifically, there is a decrease in the proportion of RGCs that respond to the onset of the bright phase of a full-field stimulus (ON RGCs), and in RGCs where an ON response is present, it has a significantly longer onset latency compared to control (Renteria et al. 2006; Pinto et al. 2007b). In Grm6 KO mice a decrease in the proportion of cells with ON responses has been reported in more central visual structures, e.g. the superior colliculus, which receives direct retinal input (Sugihara et al. 1997) and the visual cortex (Renteria et al. 2006). In addition, these ON responses also have significantly longer onset latencies. A similar outcome has been created in both adult primate and control mouse retina by pharmacological application of the metabotropic glutamate receptor type III agonist, l-APB (Schiller et al. 1986; Renteria et al. 2006). As expected, in the presence of l-APB the proportion of ON RGCs is reduced and the ON responses that are evoked have abnormally long onset latencies. This ability to pharmacologically manipulate wild-type retinas to yield responses similar to those observed in Grm6 mutant mice makes it unlikely that their On pathway defect results from abnormal retinal development.
In Grm6nob3 and Grm6nob4 retinas, ON RGCs recorded in vivo have ill-defined RFs and require full-field stimuli to evoke a consistent response. In some of the Grm6 KO ON RGCs, spatial tuning has been demonstrated, using the multielectrode array in vitro (Renteria et al. 2006). The simplest explanation for this difference is that different RGC populations are likely to be sampled with the two techniques.
Significant deficits in visual function occur in all Grm6 mutant mice (Iwakabe et al. 1997; Takao et al. 2000), as well as in Grm6 arCSNB patients (Dryja et al. 2005; Zeitz et al. 2005; O'Connor et al. 2006). However, neither the Grm6 mutant mice nor the patients are completely blind at either scotopic or photopic levels. Generally, these findings indicate that a mutation in Grm6 markedly reduces the number of cells that respond to the onset of a bright stimulus throughout the visual system, but that there may be compensatory mechanisms that underlie the difference in retinal output and visual behaviour.
Phenotypic differences between Grm6nob3 and Grm6nob4 OFF-centre RGCs indicates allelic variance
Grm6nob3 and Grm6nob4 OFF-centre RGCs have RFs with defined centres and response onset latencies to the dark phase of a full-field stimulus that are similar to controls. Similar results have been reported for Grm6 KO OFF RGCs (Renteria et al. 2006). However, there is a difference in the OFF-centre RGC responses between Grm6nob3 and Grm6nob4 mice. Namely, there are significantly fewer Grm6nob3 OFF-centre RGCs that manifest an ON response component to a full-field stimulus (OFF-SA) compared to Grm6nob4 and control mice (Sagdullaev & McCall, 2005). We also recorded a significantly smaller proportion of Grm6nob3 OFF/ON RGCs compared to Grm6nob4 when we used the multielectrode array. We believe this result is consistent with the change we see in vivo. While a disparity in the phenotypes caused by mutant alleles of the same gene are common, the mechanism underlying the difference between Grm6nob3 and Grm6nob4 requires further study. The mutation in Grm6nob3 mice is expected to truncate the protein and disrupt the glutamate binding domain. It is possible that the missense mutation in Grm6nob4 mice, which is slightly more carboxy, may produce a soluble albeit altered form of the protein with the potential to bind to some of its intracellular partners. Soluble glutamate receptor variants have been created by alternative splicing using rat and human sequence (Valerio et al. 2001a,b), although these truncated proteins would be considerably larger than in Grm6nob4.
Etiology of the ON response
We do not yet know what generates the short latency ON response component in OFF-SA control RGCs or whether it is related to the long latency ON responses in Grm6 mutant RGCs. In control RGCs, evidence suggests that the short latency ON component of the OFF-SA RGC arises from an additional surround mechanism superimposed on the classical surround (Sagdullaev & McCall, 2005). Long-latency ON responses can be created pharmacologically in some control RGCs by blocking all inhibitory inputs, and in other RGCs by eliminating both metabotropic glutamate receptor type III-mediated inputs and all inhibitory input (Renteria et al. 2006). However, the long latency ON response is resistant to pharmacological elimination by either excitation or inhibition or both in Grm6 KO retinas (Renteria et al. 2006). Our data also suggest that there is a difference in this mechanism between Grm6nob3 and Grm6nob4 mutants. If a truncated Grm6nob4 protein is expressed, it may be sufficient to interact with other intracellular elements of the GRM6 signalling pathway and create an altered DBC output, as opposed to a complete absence of output. Characterization and comparisons of the excitatory and inhibitory currents in Grm6 mutant RGCs will be required to understand the underlying differences and whether they are related to the short latency ON response component of OFF-SA control RGCs. Regardless, of the exact mechanism, these mice represent a system in which new insights into the GRM6-mediated DBC signalling cascade can be gained. A further important implication of these results is that a no b-wave ERG phenotype does not directly correlate with the RGC visual response phenotype and, presumably, with upstream processing of the visual signal.
Implications for retinal function in mouse mutants and CSNB patients
The results from the analyses of these Grm6 mutants have advanced our understanding of the potential role of allelic variability and its impact on the responses of RGCs. The changes in the distribution of RGC response types between the Grm6 mutants indicate that a subtle difference in a mutation in the same gene may give rise to the same ERG phenotype, but may not be representative of the effect of the mutation on the retinal output. The most likely explanation for the differences between the mutants is that there may be differential levels of altered GRM6 protein, which has an impact on the state of the cation channel, even in the absence of glutamate mediated gating. Empirical testing of the state of the cation channel in these mutants or the output of the DBCs themselves will be required to determine the validity of this hypothesis.
Humans with mutations in the Grm6 gene have clear visual deficits that appear primarily under low light conditions. However, these individuals do have functional vision even in the absence of signalling through their On pathway. If the phenotype at the level of the RGCs in humans is similar to those we describe in Grm6 mouse mutants, this may represent significant plasticity within the visual system, where an ability to construct relatively good vision results even in the absence of responses to light onset. This result may bode well for therapies aimed at restoring light mediated input to the retina in diseases such as RP and macular degeneration.
Acknowledgments
The authors would like to thank Ms. Elaine Gifford, Ms. Jeanie Hansen, Ms. Karen Moore, Ms. Jennifer Torrance and Ms. Barbra Mortimer for excellent technical assistance. This work was supported by National Eye Institute grants: EY11996 (PMN), EY016501 (PMN), EY014701, (MAMc), EY012354, (RGG), EY007758 (BC), EY06669 (JBT), EY016313 (DMM), a National Defense Science & Training Fellowship (DRC), NIH Cooperative Research Agreement U01MH61915 (LHP), an Institutional Core Grant to The Jackson Laboratory (CA34196), and R24EY015636 and an unrestricted from Research to Prevent Blindness, Inc., NY to the Department of Ophthalmology & Visual Sciences, University of Louisville.
References
- Ball SL, Powers PA, Shin HS, Morgans CW, Peachey NS, Gregg RG. Role of the β2 subunit of voltage-dependent calcium channels in the retinal outer plexiform layer. Invest Ophthalmol Vis Sci. 2002;43:1595–1603. [PubMed] [Google Scholar]
- Chang B, Hawes NL, Hurd RE, Davisson MT, Nusinowitz S, Heckenlively JR. Retinal degeneration mutants in the mouse. Vision Res. 2002;42:517–525. doi: 10.1016/s0042-6989(01)00146-8. [DOI] [PubMed] [Google Scholar]
- Chang B, Heckenlively JR, Bayley PR, Brecha NC, Davisson MT, Hawes NL, Hirano AA, Hurd RE, Ikeda A, Johnson BA, McCall MA, Morgans CW, Nusinowitz S, Peachey NS, Rice DS, Vessey KA, Gregg RG. The nob2 mouse, a null mutation in Cacna1f: Anatomical and functional abnormalities in the outer retina and their consequences on ganglion cell visual responses. Vis Neurosci. 2006;23:11–24. doi: 10.1017/S095252380623102X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson WW, Trick GL, Litzkow CA. Improved electrode for electroretinography. Invest Ophthalmol Vis Sci. 1979;18:988–991. [PubMed] [Google Scholar]
- Demas J, Sagdullaev BT, Green E, Jaubert-Miazza L, McCall MA, Gregg RG, Wong RO, Guido W. Failure to maintain eye-specific segregation in nob, a mutant with abnormally patterned retinal activity. Neuron. 2006;50:247–259. doi: 10.1016/j.neuron.2006.03.033. [DOI] [PubMed] [Google Scholar]
- DeVries SH. Bipolar cells use kainate and AMPA receptors to filter visual information into separate channels. Neuron. 2000;28:847–856. doi: 10.1016/s0896-6273(00)00158-6. [DOI] [PubMed] [Google Scholar]
- Dhingra A, Jiang M, Wang TL, Lyubarsky A, Savchenko A, Bar-Yehuda T, Sterling P, Birnbaumer L, Vardi N. Light response of retinal ON bipolar cells requires a specific splice variant of Gαo. J Neurosci. 2002;22:4878–4884. doi: 10.1523/JNEUROSCI.22-12-04878.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhingra A, Lyubarsky A, Jiang M, Pugh EN, Jr, Birnbaumer L, Sterling P, Vardi N. The light response of ON bipolar neurons requires Gαo. J Neurosci. 2000;20:9053–9058. doi: 10.1523/JNEUROSCI.20-24-09053.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryja TP, McGee TL, Berson EL, Fishman GA, Sandberg MA, Alexander KR, Derlacki DJ, Rajagopalan AS. Night blindness and abnormal cone electroretinogram ON responses in patients with mutations in the GRM6 gene encoding mGluR6. Proc Natl Acad Sci U S A. 2005;102:4884–4889. doi: 10.1073/pnas.0501233102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg RG, Kamermans M, Klooster J, Lukasiewicz PD, Peachey NS, Vessey KA, McCall MA. Nyctalopin expression in retinal bipolar cells restores visual function in a mouse model of complete X-linked congenital stationary night blindness. J Neurophysiol. 2007;98:3023–3033. doi: 10.1152/jn.00608.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg RG, Mukhopadhyay S, Candille SI, Ball SL, Pardue MT, McCall MA, Peachey NS. Identification of the gene and the mutation responsible for the mouse nob phenotype. Invest Ophthalmol Vis Sci. 2003;44:378–384. doi: 10.1167/iovs.02-0501. [DOI] [PubMed] [Google Scholar]
- Haeseleer F, Imanishi Y, Maeda T, Possin DE, Maeda A, Lee A, Rieke F, Palczewski K. Essential role of Ca2+-binding protein 4, a Cav1.4 channel regulator, in photoreceptor synaptic function. Nat Neurosci. 2004;7:1079–1087. doi: 10.1038/nn1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwakabe H, Katsuura G, Ishibashi C, Nakanishi S. Impairment of pupillary responses and optokinetic nystagmus in the mGluR6-deficient mouse. Neuropharmacology. 1997;36:135–143. doi: 10.1016/s0028-3908(96)00167-0. [DOI] [PubMed] [Google Scholar]
- Maeda T, Lem J, Palczewski K, Haeseleer F. A critical role of CaBP4 in the cone synapse. Invest Ophthalmol Vis Sci. 2005;46:4320–4327. doi: 10.1167/iovs.05-0478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansergh F, Orton NC, Vessey JP, Lalonde MR, Stell WK, Tremblay F, Barnes S, Rancourt DE, Bech-Hansen NT. Mutation of the calcium channel gene Cacna1f disrupts calcium signaling, synaptic transmission and cellular organization in mouse retina. Hum Mol Genet. 2005;14:3035–3046. doi: 10.1093/hmg/ddi336. [DOI] [PubMed] [Google Scholar]
- Masu M, Iwakabe H, Tagawa Y, Miyoshi T, Yamashita J, Fukuda Y, Sasaki H, Hiroi K, Nakamura Y, Shigemoto R, Takada M, Nakamura K, Nakao K, Katsuki M, Nakanishi S. Specific deficit of the ON response in visual transmission by targeted disruption of the mGluR6 gene. Cell. 1995;80:757–765. doi: 10.1016/0092-8674(95)90354-2. [DOI] [PubMed] [Google Scholar]
- McGill TJ, Douglas RM, Lund RD, Prusky GT. Quantification of spatial vision in the Royal College of Surgeons rat. Invest Ophthalmol Vis Sci. 2004;45:932–936. doi: 10.1167/iovs.03-0964. [DOI] [PubMed] [Google Scholar]
- Morgans CW, Bayley PR, Oesch NW, Ren G, Akileswaran L, Taylor WR. Photoreceptor calcium channels: insight from night blindness. Vis Neurosci. 2005;22:561–568. doi: 10.1017/S0952523805225038. [DOI] [PubMed] [Google Scholar]
- Morgans CW, Ren G, Akileswaran L. Localization of nyctalopin in the mammalian retina. Eur J Neurosci. 2006;23:1163–1171. doi: 10.1111/j.1460-9568.2006.04647.x. [DOI] [PubMed] [Google Scholar]
- Nirenberg S, Carcieri SM, Jacobs AL, Latham PE. Retinal ganglion cells act largely as independent encoders. Nature. 2001;411:698–701. doi: 10.1038/35079612. [DOI] [PubMed] [Google Scholar]
- Nirenberg S, Meister M. The light response of retinal ganglion cells is truncated by a displaced amacrine circuit. Neuron. 1997;18:637–650. doi: 10.1016/s0896-6273(00)80304-9. [DOI] [PubMed] [Google Scholar]
- Nishina PM, Naggert JK. Mouse genetic approaches to access pathways important in retinal function. Adv Exp Med Biol. 2003;533:29–34. doi: 10.1007/978-1-4615-0067-4_4. [DOI] [PubMed] [Google Scholar]
- Nomura A, Shigemoto R, Nakamura Y, Okamoto N, Mizuno N, Nakanishi S. Developmentally regulated postsynaptic localization of a metabotropic glutamate receptor in rat rod bipolar cells. Cell. 1994;77:361–369. doi: 10.1016/0092-8674(94)90151-1. [DOI] [PubMed] [Google Scholar]
- O'Connor E, Allen LE, Bradshaw K, Boylan J, Moore AT, Trump D. Congenital stationary night blindness associated with mutations in GRM6 encoding glutamate receptor MGluR6. Br J Ophthalmol. 2006;90:653–654. doi: 10.1136/bjo.2005.086678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacione LR, Szego MJ, Ikeda S, Nishina PM, McInnes RR. Progress toward understanding the genetic and biochemical mechanisms of inherited photoreceptor degenerations. Annu Rev Neurosci. 2003;26:657–700. doi: 10.1146/annurev.neuro.26.041002.131416. [DOI] [PubMed] [Google Scholar]
- Pak WL. Drosophila in vision research. The Friedenwald Lecture. Invest Ophthalmol Vis Sci. 1995;36:2340–2357. [PubMed] [Google Scholar]
- Pardue MT, Ball SL, Mukhopadhyay S, Candille SI, McCall MA, Gregg RG, Peachey NS. nob: A mouse model of CSNB1. In: Anderson RE, LaVail MM, Hollyfield JG, editors. New Insights Into Retinal Degenertive Diseases. London: Kluwer Academic/Plenum Publishers; 2001. [Google Scholar]
- Pardue MT, McCall MA, LaVail MM, Gregg RG, Peachey NS. A naturally occurring mouse model of X-linked congenital stationary night blindness. Invest Ophthalmol Vis Sci. 1998;39:2443–2449. [PubMed] [Google Scholar]
- Pinto LH, Invergo B, Shimomura K, Takahashi JS, Troy JB. Interpretation of the mouse electroretinogram. Doc Ophthalmol. 2007a;115:127–136. doi: 10.1007/s10633-007-9064-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto LH, Vitaterna MH, Shimomura K, Siepka SM, Balannik V, McDearmon EL, Omura C, Lumayag S, Invergo BM, Glawe B, Cantrell DR, Inayat S, Olvera MA, Vessey KA, McCall MA, Maddox D, Morgans CW, Young B, Pletcher MT, Mullins RF, Troy JB, Takahashi JS. Generation, identification and functional characterization of the nob4 mutation of Grm6 in the mouse. Vis Neurosci. 2007b;24:111–123. doi: 10.1017/S0952523807070149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto LH, Vitaterna MH, Shimomura K, Siepka SM, McDearmon EL, Fenner D, Lumayag SL, Omura C, Andrews AW, Baker M, Invergo BM, Olvera MA, Heffron E, Mullins RF, Sheffield VC, Stone EM, Takahashi JS. Generation, characterization, and molecular cloning of the Noerg-1 mutation of rhodopsin in the mouse. Vis Neurosci. 2005;22:619–629. doi: 10.1017/S0952523805225117. [DOI] [PubMed] [Google Scholar]
- Prusky GT, Alam NM, Beekman S, Douglas RM. Rapid quantification of adult and developing mouse spatial vision using a virtual optomotor system. Invest Ophthalmol Vis Sci. 2004;45:4611–4616. doi: 10.1167/iovs.04-0541. [DOI] [PubMed] [Google Scholar]
- Renteria RC, Tian N, Cang J, Nakanishi S, Stryker MP, Copenhagen DR. Intrinsic ON responses of the retinal OFF pathway are suppressed by the ON pathway. J Neurosci. 2006;26:11857–11869. doi: 10.1523/JNEUROSCI.1718-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagdullaev BT, DeMarco PJ, McCall MA. Improved contact lens electrode for corneal ERG recordings in mice. Doc Ophthalmol. 2004;108:181–184. doi: 10.1007/s10633-004-5734-1. [DOI] [PubMed] [Google Scholar]
- Sagdullaev BT, McCall MA. Stimulus size and intensity alter fundamental receptive-field properties of mouse retinal ganglion cells in vivo. Vis Neurosci. 2005;22:649–659. doi: 10.1017/S0952523805225142. [DOI] [PubMed] [Google Scholar]
- Saszik SM, Robson JG, Frishman LJ. The scotopic threshold response of the dark-adapted electroretinogram of the mouse. J Physiol. 2002;543:899–916. doi: 10.1113/jphysiol.2002.019703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller PH, Sandell JH, Maunsell JH. Functions of the ON and OFF channels of the visual system. Nature. 1986;322:824–825. doi: 10.1038/322824a0. [DOI] [PubMed] [Google Scholar]
- Smith RS, Zabaleta A, Savinova OV, John SW. The mouse anterior chamber angle and trabecular meshwork develop without cell death. BMC Dev Biol. 2001;1:3. doi: 10.1186/1471-213X-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specht D, Tom Dieck S, Ammermuller J, Regus-Leidig H, Gundelfinger ED, Brandstatter JH. Structural and functional remodeling in the retina of a mouse with a photoreceptor synaptopathy: plasticity in the rod and degeneration in the cone system. Eur J Neurosci. 2007;26:2506–2515. doi: 10.1111/j.1460-9568.2007.05886.x. [DOI] [PubMed] [Google Scholar]
- Sugihara H, Inoue T, Nakanishi S, Fukuda Y. A late ON response remains in visual response of the mGluR6-deficient mouse. Neurosci Lett. 1997;233:137–140. doi: 10.1016/s0304-3940(97)00656-3. [DOI] [PubMed] [Google Scholar]
- Tagawa Y, Sawai H, Ueda Y, Tauchi M, Nakanishi S. Immunohistological studies of metabotropic glutamate receptor subtype 6-deficient mice show no abnormality of retinal cell organization and ganglion cell maturation. J Neurosci. 1999;19:2568–2579. doi: 10.1523/JNEUROSCI.19-07-02568.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takao M, Morigiwa K, Sasaki H, Miyoshi T, Shima T, Nakanishi S, Nagai K, Fukuda Y. Impaired behavioral suppression by light in metabotropic glutamate receptor subtype 6-deficient mice. Neuroscience. 2000;97:779–787. doi: 10.1016/s0306-4522(00)00053-1. [DOI] [PubMed] [Google Scholar]
- Ueda Y, Tammitsu N, Imai H, Honda Y, Shichida Y. Recovery of rod-mediated a-wave during light-adaptation in mGluR6-deficient mice. Vision Res. 2006;46:1655–1664. doi: 10.1016/j.visres.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Valerio A, Ferraboli S, Paterlini M, Spano P, Barlati S. Identification of novel alternatively-spliced mRNA isoforms of metabotropic glutamate receptor 6 gene in rat and human retina. Gene. 2001a;262:99–106. doi: 10.1016/s0378-1119(00)00547-3. [DOI] [PubMed] [Google Scholar]
- Valerio A, Zoppi N, Ferraboli S, Paterlini M, Ferrario M, Barlati S, Spano P. Alternative splicing of mGlu6 gene generates a truncated glutamate receptor in rat retina. Neuroreport. 2001b;12:2711–2715. doi: 10.1097/00001756-200108280-00024. [DOI] [PubMed] [Google Scholar]
- Vardi N, Duvoisin R, Wu G, Sterling P. Localization of mGluR6 to dendrites of ON bipolar cells in primate retina. J Comp Neurol. 2000;423:402–412. doi: 10.1002/1096-9861(20000731)423:3<402::aid-cne4>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Vardi N, Matesic DF, Manning DR, Liebman PA, Sterling P. Identification of a G-protein in depolarizing rod bipolar cells. Vis Neurosci. 1993;10:473–478. doi: 10.1017/s0952523800004697. [DOI] [PubMed] [Google Scholar]
- Wassle H. Parallel processing in the mammalian retina. Nat Rev Neurosci. 2004;5:747–757. doi: 10.1038/nrn1497. [DOI] [PubMed] [Google Scholar]
- Zeitz C, Forster U, Neidhardt J, Feil S, Kalin S, Leifert D, Flor PJ, Berger W. Night blindness-associated mutations in the ligand-binding, cysteine-rich, and intracellular domains of the metabotropic glutamate receptor 6 abolish protein trafficking. Hum Mutat. 2007;28:771–780. doi: 10.1002/humu.20499. [DOI] [PubMed] [Google Scholar]
- Zeitz C, van Genderen M, Neidhardt J, Luhmann UF, Hoeben F, Forster U, Wycisk K, Matyas G, Hoyng CB, Riemslag F, Meire F, Cremers FP, Berger W. Mutations in GRM6 cause autosomal recessive congenital stationary night blindness with a distinctive scotopic 15-Hz flicker electroretionogram. Invest Ophthalmol Vis Sci. 2005;46:4328–4335. doi: 10.1167/iovs.05-0526. [DOI] [PubMed] [Google Scholar]