

Abstract
Background & Aims
Resistance to apoptosis is essential for cancer growth. We previously reported that hepatic co-expression of c-Myc and E2F1, two key regulators of proliferation and apoptosis, enhanced HCC development in transgenic mice. Here, we investigated the molecular mechanisms underlying oncogenic cooperation between c-Myc and E2F1 in relationship to human liver cancer.
Methods
Activation of pro- and anti-apoptotic cascades was assessed by immunoblotting in in vivo and in vitro HCC models, and in primary human HCC. Effect of antisense oligodeoxy nucleotides against c-Myc and E2F1 was studied in human HCC cell lines. Suppression of PIK3CA/AKT, mTOR, and COX-2 pathways was achieved by pharmacological inhibitors and specific siRNAs in human and mouse HCC cell lines.
Results
Co-expression with E2F1 did not increase proliferation triggered by c-Myc overexpression but conferred a strong resistance to c-Myc-initiated apoptosis via concomitant induction of PIK3CA/Akt/mTOR and c-Myb/COX-2 survival pathways. COX-2 was not induced in c-Myc and rarely in E2F1 tumors. In human HCC, PIK3CA/Akt/mTOR and c-Myb/COX-2 pathways were similarly activated, with levels of PIK3CA/Akt, mTOR, and c-Myb being inversely associated with patients’ survival length. Knocking down c-Myc and E2F1 oncoproteins reduced PIK3CA/Akt and mTOR and completely abolished c-Myb and COX-2 expression in human HCC cell lines. Finally, simultaneous inhibition of PIK3CA/Akt/mTOR and COX-2 activity in in vitro models caused massive apoptosis of neoplastic hepatocytes.
Conclusion
E2F1 may function as a critical anti-apoptotic factor both in human and rodent liver cancer through its ability to counteract c-Myc-driven apoptosis via activation of PIK3CA/Akt/mTOR and c-Myb/COX-2 pathways.
Deregulation of c-Myc and/or E2F1 protooncogenes is implicated in the development of numerous rodent and human tumors, including hepatocellular carcinoma (HCC)1–5. The importance of c-Myc and E2F1 in carcinogenesis is underscored by their capacity to induce both cell proliferation and cell death. When over-expressed, both c-Myc and E2F1 can trigger proliferation by driving quiescent cells into S phase in the absence of other mitogenic stimuli6–11. Likewise, both transcription factors are capable of sensitizing cells to apoptosis either via p53-dependent or p53-independent mechanisms12–14. In addition to sharing functional properties, increasing evidence suggests that these two protooncogenes can regulate each others activities15–17. Indeed, the requirement for distinct E2F members to mediate Myc-induced proliferation versus apoptosis has been demonstrated16. Furthermore, a recent report indicates that survival of c-Myc-over-expressing cells may depend on E2F activity18, suggesting that E2F1 sustain abnormal c-Myc-driven cell growth via suppression of c-Myc-induced apoptosis. However, the molecular mechanisms whereby E2F1 inhibits c-Myc-dependent apoptosis are unknown. Recent findings underline the role of phosphatidylinositol 3-kinase (PI3K) and it is downstream effector, Akt/PKB serine/threonine kinase, in suppression of E2F apoptotic potential19,20. Once activated, Akt promotes cell survival both by inactivating multiple pro-apoptotic proteins, including Bad, FoxO1, caspase 9, apoptosis signal-regulating kinase-1 (ASK1), and stimulating transcription of BFL1 and cIAP1/2 anti-apoptotic genes21. Furthermore, recent reports indicate that the PI3K/Akt axis suppresses E2F1-dependent apoptosis but not proliferation via induction of topoisomerase (DNA) II beta binding protein (TopBP1)22. In addition to its anti-apoptotic role, Akt sustains cell growth by either direct phosphorylation of the mammalian target of rapamycin (mTOR) or indirectly through inactivation of tuberin (TSC2), an mTOR inhibitor23. Suppression of TSC2 activates the GTP-binding protein Ras homologue enriched in brain (Rheb), which upregulates mTOR24. The latter, in complex with Raptor, mediates cell growth by stimulating protein synthesis via phosphorylation of two key players in translation: p70 ribosomal protein S6 kinase (p70 S6K) and eukaryotic initiation factor 4E binding protein 1 (4E-BP1). p70 S6K phosphorylates the ribosomal protein S6 (rpS6), resulting in increased translation of mRNAs containing a 5’ olygopyrimidine tract, whereas phosphorylation of 4E-BP1 by mTOR relieves inhibition on the initiation factor eIF4E resulting in more efficient cap-dependent translation25.
Previously, we demonstrated that transgenic over-expression of either c-Myc or E2F1 in the liver was sufficient to induce tumor growth, albeit with different latencies5,6. In both transgenic models, there was a reciprocal induction of the other transcription factor further supporting the hypothesis that c-Myc and E2F1 modulate each other’s activity in vivo5,6. Furthermore, c-Myc/E2F1 double transgenic mice displayed a frequent activation of the apoptosis suppressor COX-2 and acceleration of liver carcinogenesis when compared with both parental lines26,27. COX-2 has been shown to upregulate Akt survival pathway in human HCC, and hepatic overexpression of E2F1 transgene greatly increased Akt liver levels28,29. Based on this information, the objective of this study was to understand the molecular basis for synergistic effects of c-Myc/E2F1 co-activation in promoting liver oncogenesis. Using transgenic models of liver cancer and human HCC in combination with mouse and human HCC cell lines, we now demonstrate a key role for E2F1 in restraining Myc-driven apoptosis via concomitant activation of PI3K/Akt and COX-2 pathways.
Materials and Methods
Transgenic Mice
Generation of the Alb/c-Myc (c-Myc), Alb/E2F1 (E2F1), and Alb/c-Myc/Alb/E2F1 (c-Myc/E2F1) transgenic mice has been described5,6,26. Normal, preneoplastic and neoplastic liver tissues were obtained from male mice at different ages (3–20 months). Dissected tissues were divided in two parts: half was stored at –80°C, half was fixed in 10% formalin and embedded in paraffin. Sections were stained with hematoxylin and eosin. Histopathological diagnoses were based upon criteria previously described30. Animal study protocols were conducted according to the National Institutes of Health guidelines for animal care.
Clinical Samples
Five normal livers, 50 HCCs and respective surrounding non-tumorous liver tissues were obtained from patients undergoing surgical treatment for HCC. Patient’s clinicopathological features are shown in Table 1. HCCs were divided in two prognostic subclasses (HCCB and HCCP) based on patient’s survival length, as previously described31. HCCB or HCCP were characterized by a shorter (< 3 years) or longer (> 3 years) survival length following liver partial resection. Liver samples were kindly provided by Dr. Z. Sung (National Laboratory of Molecular Oncology, Beijing, China) and the Liver Tissue Procurement and Distribution System (Minneapolis, MN; Pittsburgh, PA; Richmond, VA), which was funded by NIH Contract #N01-DK-9-2310. Institutional Review Board approval was obtained at participating hospitals and the National Institutes of Health.
Table 1.
Clinicopathological features of HCC patients
| Variable | No. of cases |
|---|---|
| No. of patients | 50 |
| Male | 46 |
| Female | 4 |
| Age | |
| Mean | 55.2 |
| SD | 11.1 |
| Etiology | |
| HBV | 27 |
| HCV | 15 |
| Alcohol | 6 |
| Hemochromatosis | 1 |
| Wilson’s disease | 1 |
| AFP (> 300 ng/ml) | |
| + | 30 |
| − | 20 |
| Cirrhosis | |
| + | 35 |
| − | 15 |
| Tumor size | |
| < 5 cm | 28 |
| > 5 cm | 22 |
| Survival | |
| < 3 years (HCCP) | 25 |
| > 3 years (HCCB) | 25 |
PCNA and Apoptotic Indices
PCNA-labeling and apoptotic indices were determined as reported27 and presented as a percentage of the total cells counted.
Quantitative real-time RT-PCR (QRT-PCR)
Primers for mouse c-Myc, E2F1, GAB2, catalytic subunit p110α of phosphatidylinositol 3-kinase (PIK3CA), and RNR-18 genes were from Applied Biosystems (Foster City, CA). PCR reactions and quantitative evaluation were performed as described31.
Western Blot Analysis
Whole cell lysates were prepared and processed as described27. The complete list of the antibodies used is available upon request. For cytochrome c release, cytoplasmic fractions were obtained using the NE-PER Nuclear and Cytoplasmic Extraction Kit (Pierce Biotechnology Inc., Rockford, IL) following the manufacturer’s protocol.
DNA Extraction and Methylation-specific PCR
High molecular weight DNA was isolated from frozen tissues as described32 and modified by using the EZ DNA methylation kit (Zymo Research, Orange, CA). Promoter methylation status of mouse DAPK was determined with DAPK-unmethylated and - methylated specific primers as previously described33.
Cell Lines, Treatments, Viability assay, and Apoptosis Index
Alexander, HuH1 and HuH7, and two mouse cell lines derived from E2F1 HCC were maintained as monolayer cultures either in DMEM or in RPM1 1640 supplemented with 10% fetal bovine serum. Cells were plated at 2.0 × 103 /well in 96-well plate and grown for 12 h. After 24 h serum deprivation, LY294002 (pan-PI3K inhibitor), Rapamycin (mTOR inhibitor; Cell Signaling Technology), PI-103 (PI3KCA/mTOR inhibitor), and CAY10404 (COX-2 inhibitor; Cayman Chemical, Ann Arbor, MI) were added to the medium at 50 µM, 10 nM, 0.5 µM, and 50 µM, respectively, and cells incubated for 6, 12, and 24h. For silencing experiments, human cell lines plated as described above were serum deprived for 24 h and treated with either antisense oligodeoxy nucleotides (AODN) against c-Myc, E2F1, GAB2, and c-Myb (10 µM, final concentration) following previously described protocols34,35 or siRNA against Akt, mTOR, and COX-2 (Santa Cruz Biotechnology). Cell viability and apoptosis were determined by the WST-1 Cell Proliferation Reagent and the Cell Death Detection Elisa Plus kit (Roche Molecular Biochemicals, Indianapolis, IN), respectively.
Chromatin Immunoprecipitation Analysis
Chromatin immunoprecipitation was performed by the ChIP Assay Kit (Upstate USA Inc.) following the manufacturer’s protocol. Rabbit polyclonal anti-E2F1 and anti-E2F4 (Santa Cruz Biotechnology) antibodies were used to immunoprecipitate chromosomal DNA in crosslinked chromatin prepared from exponentially growing Alexander, HuH1, and HuH7 cell lines. Immunoprecipitated DNA was analyzed by PCR using previously designed primers specific for Gab2 promoter20.
Statistical Analysis
Statistical analyses were performed using Student’s t and Tukey-Kramer tests. P values less than 0.05 were considered statistically significant.
RESULTS
Co-expression of c-Myc and E2F1 Transgenes Enhances Survival of HCC Cells
Recently, we showed acceleration of hepatocarcinogenesis in c-Myc/E2F1 double transgenic mice when compared with parental lines26,27. To address cellular mechanisms responsible for accelerated tumor growth, we compared proliferative and apoptotic rates during hepatocarcinogenesis in all three models. Proliferation was consistently higher in dysplastic livers from transgenic than wild-type mice (Figure 1A). Mitotic indices in c-Myc and Myc/E2F1 mice were comparable and significantly higher than in E2F1 mice at all examined ages. However, apoptosis was constantly higher in c-Myc dysplastic livers than in E2F1 and c-Myc/E2F1 livers (Figure 1B). Consequently, the net growth rate, as judged by PCNA/apoptosis ratios, was highest in double transgenic mice. Co-expression of c-Myc and E2F1 did not produce additive effects on cell proliferation in HCC (Figure 1C). Proliferation indices in c-Myc/E2F1 and c-Myc tumors were equally higher than in E2F1 HCC, suggesting the dominance of c-Myc mitogenic properties. Conversely, apoptotic index was significantly reduced in c-Myc/E2F1 HCC as compared to both c-Myc (about 5-fold) and E2F1 (2-fold) HCC, implying that E2F1 overexpression interfered with c-Myc-mediated apoptosis (Figure 1C). These results suggest that transgenic expression of E2F1 does not further stimulate proliferation of neoplastic hepatocytes driven by c-Myc but reduces apoptosis.
Figure 1.
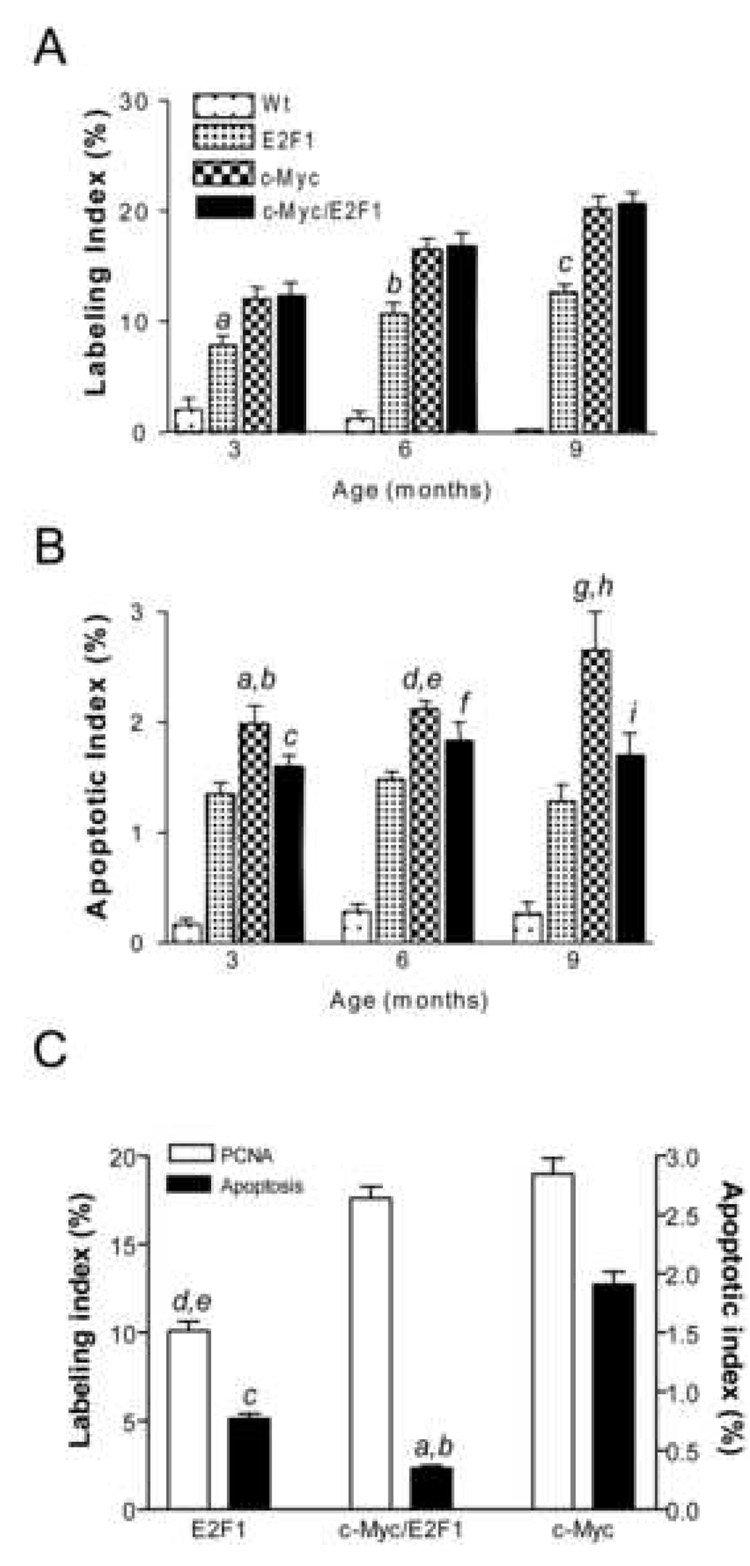
Labeling (A) and apoptotic (B) indices in dysplastic livers and HCC (C) developed in E2F1, c-Myc, and c-Myc/E2F1 mice. The same specimens were analyzed for labeling index (percentage of PCNA-positive cells in 2000 cells) and apoptotic index (percentage of terminal deoxynucleotidyltransferase-mediated UTP end-labeling (TUNEL)-positive cells in 2000 cells). Means and 95% confidence intervals from 10 tumors for each genotype are shown. In (A), a, P=0.002; b, P<2.81E-05; c, P<2.41E-05 versus c-Myc and c-Myc/E2F1; in (B), a, P=0.0001 versus E2F1; b, P=0.004 versus c-Myc/E2F1; c, P=0.007 versus E2F1; d, P=9.50E-07 versus E2F1; e, P=0.01 versus c-Myc/E2F1; f, P=0.004 versus E2F1; g, P=8.97E-05 versus E2F1; h, P=0.001 versus c-Myc/E2F1; i, P=0.009 versus E2F1; in (C); a, P=1.38E-20 versus c-Myc; b, P=7.02E-11 versus E2F1; c, P=1.61E-08 versus c-Myc; d, P<5.44E-06 versus c-Myc; and e, P<3.34E-06 versus c-Myc/E2F1 by two-sided Student’s t test.
Ubiquitous Akt Pathway Activation in E2F1-overexpressing Livers
To investigate the correlation between suppression of apoptosis and E2F1 upregulation, levels of E2F1, its anti-apoptotic effector GAB2 which triggers the Akt survival pathway20, Akt and its upstream inducer, PIK3CA36, were assessed. Among the PI3K subunit, the PIK3CA gene was chosen due to its proven role in hepatocarcinogenesis36. E2F1 was upregulated both at mRNA and protein level in all three transgenic lines when compared with wild-type livers, but at significantly higher levels in dysplastic and neoplastic lesions from E2F1 and c-Myc/E2F1 transgenic mice (Figure 2A&C, Supplemental Figure 1&Supplemental Figure 2). GAB2, Akt, and PIK3CA were induced only in dysplastic and neoplastic livers from E2F1 and c-Myc/E2F1 transgenic mice. Accordingly, phospho-Akt protein levels were progressively increased only in non-tumorous livers and HCC from E2F1 and c-Myc/E2F1 mice in accordance with apoptosis data, and were paralleled by gradual inactivation of Bad, caspase 9, and FOXO1 pro-apoptotic effectors (Figure 2A, Supplemental Figure 2). PTEN phosphorylation levels were also determined, since disruption of PTEN promotes unrestrained Akt activation37. PTEN phosphorylation was just slightly higher in E2F1 and c-Myc/E2F1 mice, suggesting that Akt activation was independent of PTEN silencing (Figure 2A, Supplemental Figure 2). Next, we measured the levels of apoptosis- signaling-regulating kinase 1 (ASK1), another apoptosis effector which is suppressed by Akt38. ASK1 activates SEK1/MKK4 and MKK3 following induction by various stimuli, thus promoting JNK- and p38 MAPK-dependent cell death39. No differences were detected in ASK1 total levels between wild-type and transgenic livers (Figure 2A, Supplemental Figure 2), whereas ASK1 was highly phosphorylated (activated) exclusively in c-Myc hepatic lesions, HCC in particular. Accordingly, ASK1 downstream effectors, including SEK1, p38 MAPK, and JNK were activated only in c-Myc mice (Figure 2A, Supplemental Figure 2). Since recent reports indicate that E2F1-dependent apoptosis may be suppressed by PI3K/Akt via activation of TopBP1 and downregulation of E2F1 pro-apoptotic targets without affecting E2F1-dependent proliferation22,40, we assessed the levels of TopBP1, AMPKα2 and Nr4a3 (E2F1 pro-apoptotic targets), and CDC8A and TACC3 (E2F1 proliferation effectors). TopBP1, CDCA8, and TACC3 were progressively upregulated in E2F1 and c-Myc/E2F1 dysplastic and neoplastic lesions, but not in c-Myc transgenic mice. In contrast, AMPKα2 and Nr4a3 were downregulated in E2F1-overexpressing livers and strongly induced in the lesions of c-Myc transgenic mice (Figure 2A, Supplemental Figure 2). These results indicate that E2F1 hepatic overexpression exerts dominant anti-apoptotic effects through activation of PIK3CA/Akt pathway and suppression of ASK1-mediated cell death driven by c-Myc overexpression.
Figure 2.
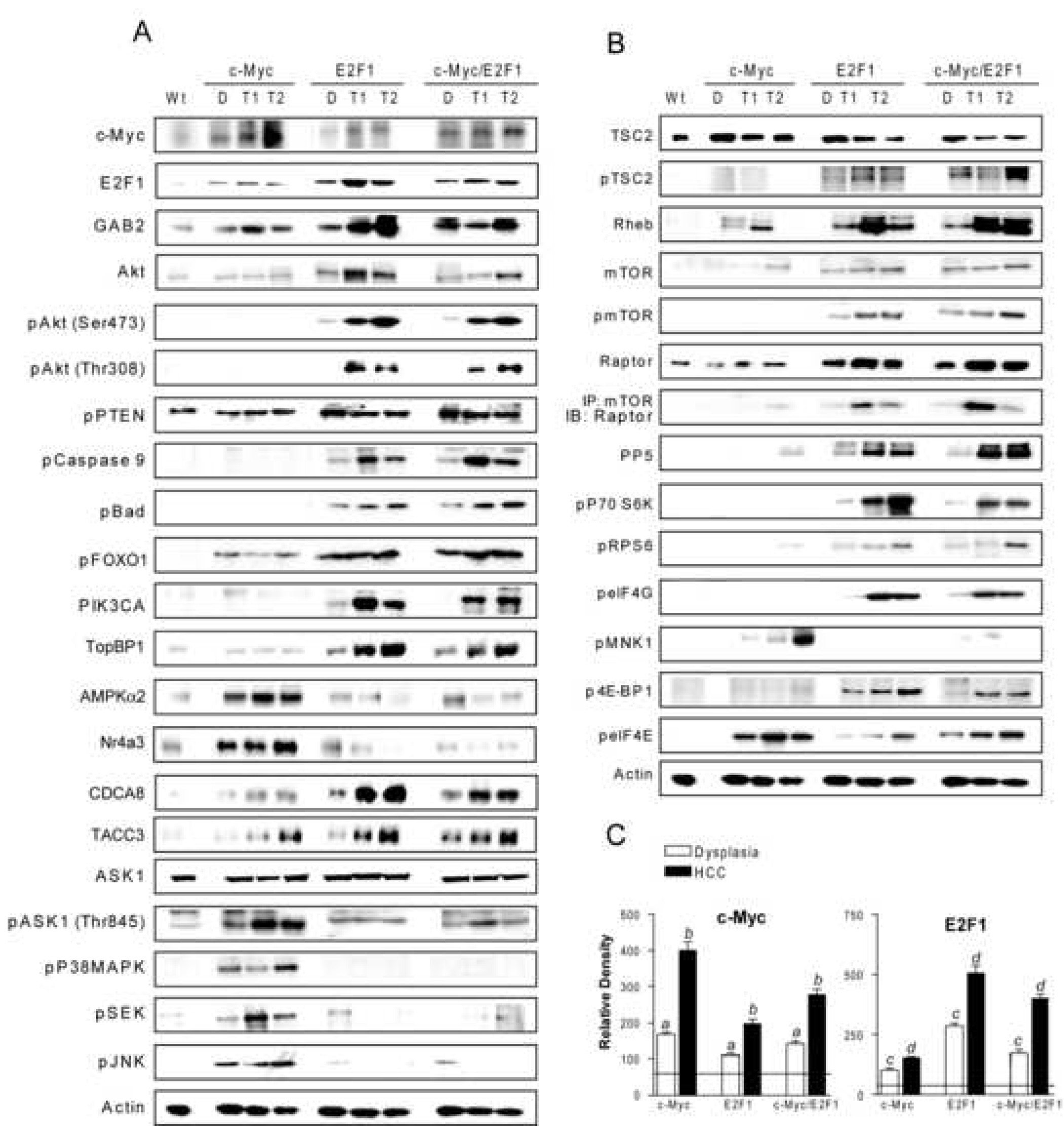
Activation of Akt (A) and mTOR (B) cascades in c-Myc, E2F1, and c-Myc/E2F1 hepatic lesions. Whole cell lysates were prepared from wild-type livers (Wt), dysplastic livers (D) and HCC (T) from at least 15 samples for each transgenic line and immunoblotted with indicated antibodies. Representative western blot is shown. (C) Densitometric analysis of c-Myc and E2F1 levels. Optical densities were normalized to β-actin values and expressed in arbitrary units. Each bar represents mean±SD. Horizontal lines show the levels of c-Myc and E2F1 proteins in wild type mice.
mTOR and p70 S6Kinase are Activated in E2F1 and c-Myc/E2F1 Livers
Next, we addressed whether Akt activation resulted in the induction of mTOR cascade. Upregulation of both total and phosphorylated (activated) mTOR, Raptor, mTOR/Raptor complexes, and PP5, p70 S6K, rpS6, and eIF4G (mTOR targets) occurred mostly in preneoplastic and neoplastic liver lesions from E2F1 and c-Myc/E2F1 mice, and was highest in HCC. Consistent with Akt-dependent activation of mTOR23,24, total levels of TSC2 were reduced, whereas those of phosho-TSC2 and Rheb significantly increased in E2F1 and c-Myc/E2F1 mice (Figure 2B, Supplemental Figure 2). Conversely, c-Myc livers displayed low levels of mTOR and mTOR effectors, and upregulation of the translation inhibitor 4E-BP1. In E2F1 and c-Myc/E2F1 livers, 4E-BP1 was instead inactivated via mTOR phosphorylation (Figure 2B, Supplemental Figure 2). Furthermore, c-Myc livers displayed significantly higher levels of activated eIF4E, suggesting that c-Myc activates translation in mTOR/p70 S6K-independent manner. Upregulation of eIF4E in c-Myc livers was associated with MnK1 induction, implying that p38 MAPK may be responsible for the increased eIF4E expression, as described previously41. These data indicate that Akt-dependent activation of mTOR cascade is a major oncogenic event in E2F1-overexpressing livers.
COX-2 Activation Is Mediated by c-Myb in c-Myc/E2F1 HCCs
Previously, we found a frequent upregulation of COX-2 in HCC from c-Myc/E2F1 transgenic mice27. Since COX-2 has been shown to activate Akt in human HCC28, we investigated the significance of COX-2 upregulation in c-Myc, E2F1 and c-Myc/E2F1 transgenic mice (Figure 3). COX-2 was detected neither in wild-type livers, preneoplastic lesions nor in c-Myc HCC. However, 100% of c-Myc/E2F1 HCC (30/30) and 30% of E2F1 HCC (6/20, 30%; p=1.11E-10) exhibited COX-2 upregulation. Consistent with these findings, c-Myc/E2F1 HCC also displayed the highest expression levels of c-Myb transcription factor shown to be responsible for E2F1-induced COX2 upregulation in prostate cancer42 (Figure 3).
Figure 3.
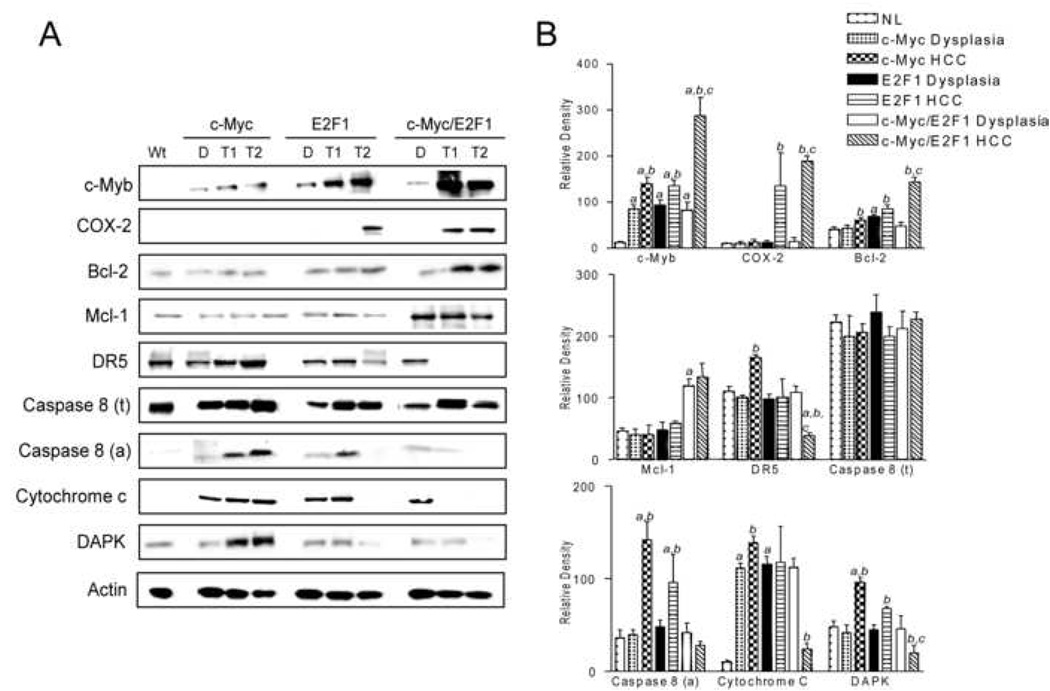
Activation of COX-2 and downstream effectors in wild-type livers (Wt), dysplastic livers (D) and HCC (T) from c-Myc, E2F1, and c-Myc/E2F1 transgenic mice .(A). Whole cell lysates were prepared from at least 15 samples per each stage (dysplasia, HCC) for each transgenic line and immunoblotted with indicated antibodies. Representative western blot is shown. (B) Densitometric analysis of COX-2 and downstream effectors. Optical densities were normalized to β-actin values and expressed in arbitrary units. Each bar represents mean±SD.
COX-2 is able to suppress apoptosis through transactivation of Bcl-2 and Mcl-1 and downregulation of death receptor 5 (DR5), a mediator of Trail-induced apoptosis43. Among the examined mouse models, Bcl-2 and Mcl-1 were upregulated only in c-Myc/E2F1 HCC. Interestingly, Bcl-2 and Mcl-1 levels were not elevated in E2F1 HCC displaying COX-2 activation, suggesting that other genes cooperate with COX-2 to transactivate Bcl-2 and Mcl-1 in c-Myc/E2F1 HCC. On the other hand, all E2F1 and c-Myc/E2F1 HCC exhibiting COX-2 upregulation were characterized by downregulation of DR5 and activated Caspase 8, and reduced cytoplasmic release of cytochrome C. Neoplastic cells can acquire resistance to Trail-mediated apoptosis via multiple mechanisms. Recently, it has been shown that tumor cells can become insensitive to Trail by suppressing the Trail effector death-associated protein-kinase (DAPK) via promoter hypermethylation44. Indeed, DAPK promoter methylation was significantly more frequent in c-Myc/E2F1 HCC (24/30, 80%) when compared with either E2F1 (7/20, 35%; p<0.001) or c-Myc (2/20, 10%; p=3.74E-08) HCC. Accordingly, DAPK protein levels were significantly reduced in HCC displaying DAPK hypermethylation. In contrast, strong DAPK upregulation occurred in c-Myc HCCs with unmethylated DAPK promoter (Figure 3). Collectively, the present data indicate that c-Myc and E2F1 co-expression may confer resistance to Trail- and mitochondria-mediated apoptosis via activation of COX-2, Bcl-2, Mcl-1, and DAPK epigenetic silencing.
Clinicopathological Significance of c-Myc and E2F1 Activation in Human HCC
Next, the prognostic role of c-Myc and E2F1 was assessed in a collection of human HCC previously classified as HCC with better (HCCB) or poor (HCCP) prognosis based on global gene expression profiling31. The c-Myc protein was first detectable in HCCB (8/25, 32%), whereas it was strongly and ubiquitously up-regulated in HCCP (Figure 4A, Supplemental Figure 3). E2F1 expression increased progressively from non-tumorous surrounding liver tissues to HCC, reaching the highest levels in HCCP. Consistent with mouse liver cancer data, E2F1 accumulation was paralleled by induction of phospho-Akt, GAB2, PIK3CA, TopBP1, CDCA8, and TACC3. The Akt inhibitor PTEN was strongly induced in all surrounding livers and HCC, excluding the possibility that Akt activation depends on deregulated PTEN activity. Induction of Rheb, mTOR, Raptor, PP5, p70 S6K, and rpS6 was present only in HCC, HCCP in particular (Figure 4A, Supplemental Figure 3). Furthermore, E2F1 pro-apoptotic effectors, including AMPKα2 and Nr4a3, were upregulated in HCCB, implying an intact E2F1-dependent apoptosis, whereas they were downregulated in HCCP. Mcl-1 and c-Myb levels progressively increased from non-neoplastic lesions to HCCP (Figure 4B, Supplemental Figure 3). In contrast, COX-2 levels were equally higher in all HCCs when compared with matching non-neoplastic livers. COX-2 expression positively correlated with increase in Bcl-2 and inversely with levels of DR5, Caspase 8, cytoplasmic cytochrome c, and DAPK. These data indicate that increased expression of c-Myc and E2F1 correlates with upregulation of some (Akt, PIK3CA, TopBP1, mTOR, Raptor, c-Myb, Mcl-1) but not all (COX-2 and downstream targets) effectors, and is associated with a poor outcome in human HCC.
Figure 4.
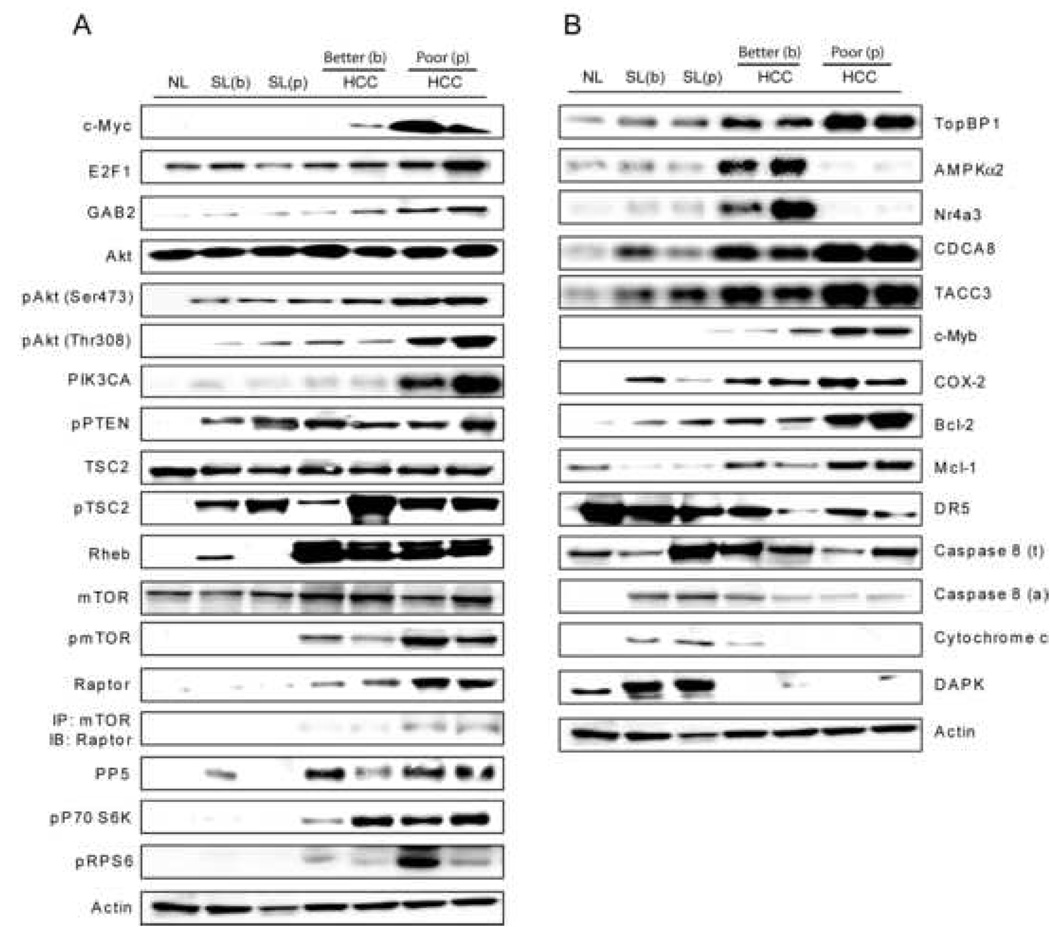
Activation of Akt and mTOR (A) and COX-2 (B) in human liver samples. Whole cell lysates were prepared from normal livers (NL), surrounding livers (SL) and HCC with better (b) or poor (p) prognosis and immunoblotted with indicated antibodies. Equal protein loading was verified by β-actin expression. 25 tissue samples were analyzed from each HCC group and matching surrounding livers, and 5 tissue samples from normal livers. Representative western blot analysis is shown.
E2F1 Silencing Suppresses AKT and mTOR while Inhibition of both E2F1 and c-Myc Suppresses c-Myb and COX-2 in Human HCC Cell Lines
To further investigate whether Akt, mTOR, c-Myb, and COX-2 were c-Myc and/or E2F1 targets, we treated the Alexander, HuH1, and HuH7 human HCC cell lines with AODN against c-Myc and E2F1. Knockdown of c-Myc protein effectively decreased the amount of E2F1, COX-2 and c-Myb proteins but had a very small effect on PIK3CA, Akt, and mTOR protein levels (Figure 5A, Supplemental Figure 4A). In contrast, E2F1 suppression caused a dramatic reduction in PIK3CA, Akt, and mTOR proteins, whereas downregulation of c-Myc, c-Myb and COX2 proteins was less remarkable (Figure 5B, Supplemental Figure 4B). We next tested whether Akt was induced by E2F1 via GAB2, as reported in osteosarcoma cell lines20 (Supplemental Figure 5). The role for GAB2 in E2F1-mediated Akt activation was substantiated by the detection of E2F1-GAB2 immunocomplexes in chromatin lysates, and about 50% suppression of Akt activation following GAB2 silencing by AODN (Supplemental Figure 5). PIK3CA expression was instead unaffected by GAB2 suppression, implying that PIK3CA induction by E2F1 occurs via GAB2-independent mechanisms in human HCC. In accordance with this hypothesis, simultaneous suppression of GAB2 and PIK3CA resulted in complete inhibition of Akt (Supplemental Figure 5).
Figure 5.
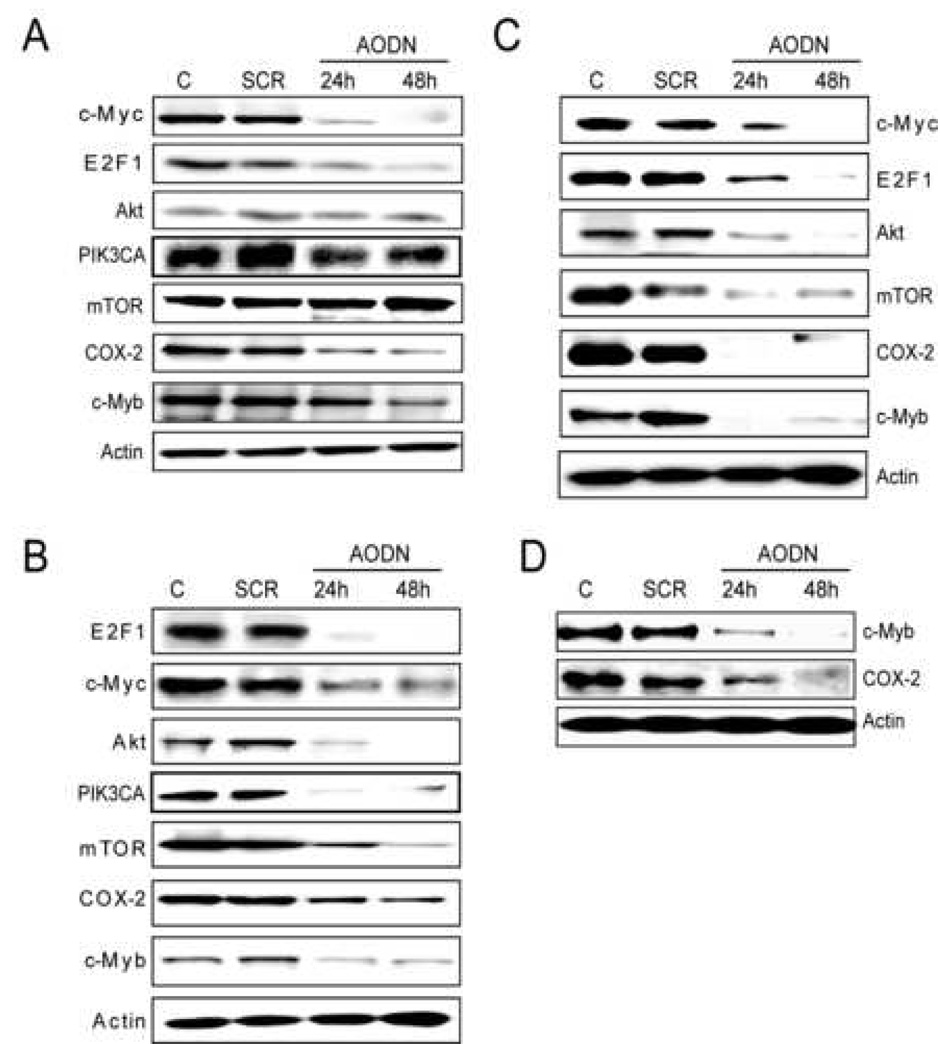
Effect of antisense oligodeoxy nucleotides against c-Myc (A), E2F1 (B), c-Myc and E2F1 (C), and c-Myb (D) on the expression of Akt, mTOR, c-Myb, and COX-2 in Alexander HCC cell line. Cells were subjected to serum deprivation for 24 h, followed by treatment with antisense oligodeoxy nucleotides (AODN) against c-Myc, E2F1, and c-Myb. Experiments were repeated at least 3 times, the results were reproduced in HuH1 and HuH7 HCC cell lines. AODN, antisense oligodeoxy nucleotides; SCR, scramble.
Combined inactivation of c-Myc and E2F1 totally suppressed c-Myb and COX-2 expression with no further decrease in Akt and mTOR protein levels (Figure 5C, Supplemental Figure 4C). Furthermore, c-Myb knockdown caused downregulation of COX-2, confirming that COX-2 is c-Myb target (Figure 5D, Supplemental Figure 4D). These observations underline the pivotal role of E2F1 in promoting PIK3CA, Akt and mTOR activation, and requirement for a concomitant induction of c-Myc and E2F1 for sustaining c-Myb/COX-2 upregulation.
Combined Inhibition of Akt, mTOR and COX-2 Pathways Suppresses HCC Cell Growth in vitro and in vivo
To test the involvement of Akt, mTOR and COX-2 pathways in HCC cell growth, Alexander, HuH1, and HuH7 human HCC cell lines and mouse cell lines established from E2F1 HCC were treated with pharmacological inhibitors of PI3Ks (LY294002), mTOR (rapamycin), and COX-2 (CAY10404). Although each single treatment decreased HCC cell growth, a dramatic synergistic effect leading to growth arrest and massive apoptosis was observed only after combining all three inhibitors (Figure 6A–D). Importantly, a more rapid and remarkable growth suppression was obtained with very low doses of PI-103, a PIK3CA/mTOR inhibitor. PI-103 anti-neoplastic efficacy was reinforced by the COX-2 inhibitor CAY10404. Furthermore, siRNA against Akt, mTOR, and COX-2 genes reduced growth of both human and mouse HCC cell lines, and combined suppression of the three genes resulted in remarkable growth restraint and massive apoptosis (Supplemental Figure 6A, B). These results argue that activation of Akt, mTOR, and COX-2 is required for human and mouse HCC cell survival.
Figure 6.
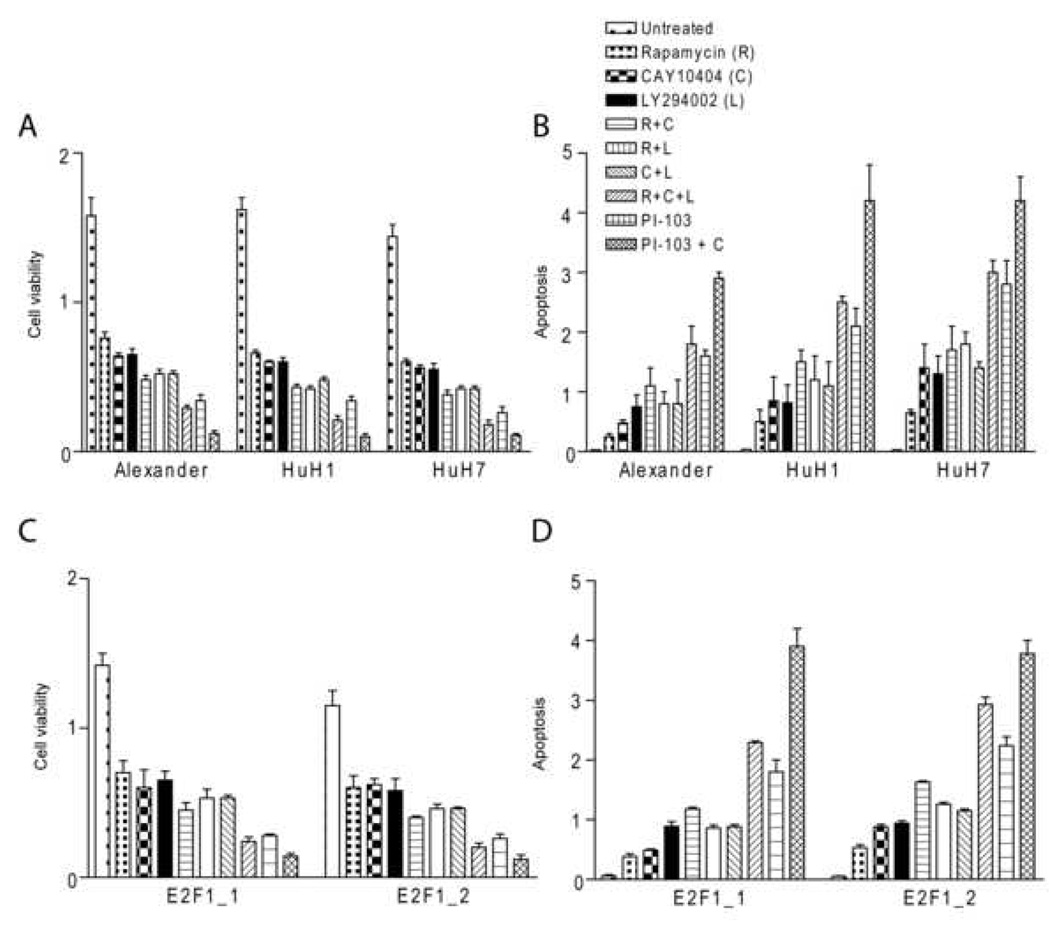
Effect of inhibition of Akt, mTOR and COX-2 pathways in vitro and in vivo. Human (Alexander, HuH1, and HuH7) and mouse (E2F1_1 and E2F1_2) HCC cell lines were treated with Akt inhibitor LY294002 (50 µM), mTOR inhibitor Rapamycin (10 nM), PIK3CA/mTOR inhibitor PI-103 (0.5 µM), and COX-2 inhibitor CAY10404 (50 µM) for 24 h. (A and C) Cell viability was determined by WST-1 assay performed in a 96-well format as described in the Materials and Methods (B and D). Apoptosis was determined by Cell Death Detection Elisa Plus kit performed in a 96-well format as described in the Materials and Methods. Values for apoptosis and cell viability represent the mean and 95% confidence interval from 3 independent duplicate experiments.
Discussion
Dysregulation of c-Myc and/or E2F1 protooncogenes has been described in a wide range of rodent and human neoplasms1–5. Here, we used transgenic mice overexpressing either E2F1, c-Myc or both selectively in the hepatocytes, to address cellular and molecular mechanisms whereby these two transcription factors promote malignant transformation and/or tumor progression. It has been hypothesized that c-Myc-triggered apoptotic program must be circumvented in order for Myc to exhibit its full oncogenic potential45. Here, we provide evidence that the major effect of c-Myc and E2F1 interaction in hepatocarcinogenesis is suppression of apoptosis, and describe the molecular mechanisms responsible for E2F1 dependent inhibition of c-Myc-driven cell death. Our findings agree with a previous study showing that c-Myc overexpressing cells require E2F activity for survival18. Although upregulation of E2F1 was also detected in c-Myc single transgenic mice, induction of Akt survival pathway did not occur in this model. This apparent discrepancy might be due to the higher levels of E2F1 upregulation in E2F1 and c-Myc/E2F1 versus c-Myc transgenic mice, resulting in upregulation of Akt upstream inducers GAB2 and PIK3CA only in E2F1-transgenic livers. Nevertheless, we cannot exclude that other tumor modifiers may influence the activation of Akt in E2F1 and c-Myc overexpressing livers.
Overexpression of E2F1 conferred numerous survival advantages to preneoplastic and neoplastic hepatocytes. The major effect was activation of Akt signaling pathway, which partly depended on GAB2 scaffold protein, in accordance with a previous report20. Furthermore, the present findings indicate for the first time that E2F1 activates Akt also via a GAB2-independent mechanism, namely the upregulation of the Akt inducer, PIK3CA. Induction of Akt was associated with inhibition of various pro-apoptotic effectors, including caspase 9, Bad, FOXO1, and suppression of Ask1/p38MAPK/JNK-mediated cell death. In addition, upregulation of Akt and PI3KCA promoted induction of TopBP1, which is able to suppress E2F1-dependent apoptosis but not proliferation40. Accordingly, both mouse and human livers overexpressing E2F1 exhibited downregulation of E2F1 pro-apoptotic targets (AMPK2α and Nr4a3), but not proliferation effectors (CDCA8, TACC3), which were instead induced. Together these data indicate that liver tumor cells with elevated E2F1 levels elaborate a gene expression program leading to the predominance of E2F1 proliferative and anti-apoptotic over pro-apoptotic effects.
Akt induction also triggered activation of mTOR/p70 S6K cascade, a pathway known to contribute to cell growth and survival26,27. The finding that mTOR was activated both via phosphorylation by Akt and through inactivation of the mTOR inhibitor, TSC2, implies a selective pressure toward mTOR activation in E2F1 and c-Myc/E2F1-driven hepatocarcinogenesis. These results underline the importance of aberrant mTOR activity in driving the malignant phenotype of E2F1 overexpressing cells. Furthermore, in vitro experiments aimed at suppressing E2F1 expression showed that activation of the mTOR cascade was dependent on E2F1 activity. To the best of our knowledge, this is the first report indicating that mTOR is an E2F1 target.
We also examined the role of another important anti-apoptotic protein, COX-2. In human HCC, COX-2 activity is presumably mediated by Akt28. However, in c-Myc/E2F1 transgenic lesions, COX-2 was induced via the c-Myb transcription factor, as observed in prostate cancer42. These data highlight the role for c-Myb in the activation of COX-2 and in hepatic malignant transformation/tumor progression in c-Myc/E2F1 and, to a lower extent, in E2F1 transgenic mice. Further studies are needed to unravel the contribution of c-Myb to liver oncogenesis, and to identify its molecular targets. Although the present data underscore a strong anti-apoptotic function of Akt, mTOR, and COX-2 cascades, additional survival pathways may also contribute to c-Myc/E2F1 hepatocarcinogenesis. Both c-Myc and E2F1 protooncogenes are capable of promoting apoptosis via p53-dependent or p53-independent mechanisms14–16, and disruption of these mechanisms cannot be excluded in this mouse model of liver cancer.
Our subsequent studies on human HCC implicated combined upregulation of c-Myc and E2F1 in the development of a more aggressive tumor phenotype. Noticeably, the same downstream effectors, namely activated Akt, mTOR cascade, c-Myb, and COX-2, were activated in human HCC, implying that the molecular pathways triggered by c-Myc and E2F1 are similar across species. Furthermore, these data emphasize the importance of c-Myc, E2F1, and some downstream effectors such as Akt, mTOR cascade, and c-Myb in HCC progression, raising a possibility of using these genes as both prognostic markers and therapeutic targets. In particular, we show that suppression of E2F1-dependent apoptosis is a molecular signature of poor prognosis in human HCC, in agreement with breast and ovarian cancer40. Regarding Akt, our data are consistent with a recent study showing the correlation between levels of phosphorylated Akt and human HCC recurrence and prognosis46. However, it has been reported that activation of mTOR is limited to a small subset of human HCC47. This discrepancy may be due to the differences in etiological factors associated with HCC development or less sensitive methods used for detection previously47.
The present data also demonstrate a strong anti-neoplastic effect of combined treatments with Akt, mTOR and COX-2 inhibitors in vitro suggesting a synergistic activity of the Akt/mTOR and COX-2 pathways in HCC cell growth. The additive anti-growth effect of the combination of the Akt inhibitor LY294002 and the mTOR inhibitor Rapamycin implies that mTOR may be activated independent of Akt in HCC cells. Also, the limited efficacy of Rapamycin on HCC cell growth restraint agrees with the previous observations showing that suppression of mTOR by Rapamycin triggers upregulation of Akt23. Thus, the present study supports the idea of combining Akt and mTOR inhibitors for the treatment of solid tumors23,48. Noticeably, the treatment with the PIK3CA/mTOR dual inhibitor PI-103 was significantly more effective than the combination of LY294002 and Rapamycin in suppressing HCC cell growth. The highest efficacy of PI-103, even at very low doses, might be explained by the strong, combined suppression of PIK3CA and mTOR, which was not achieved by high doses of LY294002 and Rapamycin (Calvisi et al., data not shown), in accordance with previous data in gliomas48. Together with the low toxicity of PI-10348, these data support the potential usefulness of PI-103 in HCC treatment.
In summary, we described the molecular mechanisms by which combined activity of two transcription factors, c-Myc and E2F1, supports the more aggressive tumor phenotype both in mouse models and human liver cancer. In c-Myc transgenic mice, hepatic overexpression of c-Myc alone was associated with activation of ASK1/p38MAPK/JNK and TRAIL cascades, which may be responsible for the high rate of apoptosis and slow development of liver cancer (Figure 7). In contrast, overexpression of E2F1 transgene caused a strong induction of Akt survival pathway, which contributed to the faster tumor growth as compared to c-Myc mice. The molecular signature of c-Myc/E2F1 driven acceleration of hepatocarcinogenesis was preservation of E2F1-mediated Akt activation in combination with universal COX-2 upregulation and inhibition of ASK1/p38MAPK/JNK signaling pathway. The same molecular mechanisms were involved in human hepatocarcinogenesis. These data may advance the prognostic prediction for human HCC and open new perspectives for the development of treatments aimed at suppressing Akt, mTOR, and COX-2 pathways.
Figure 7.
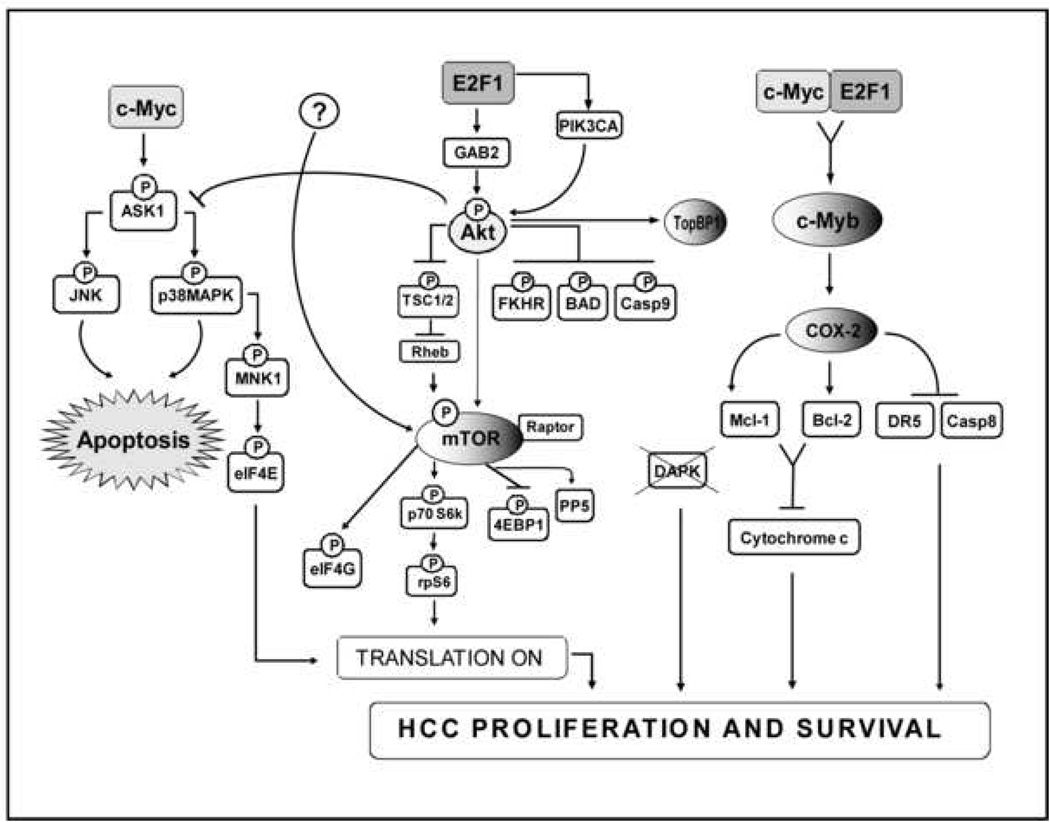
Overview of signal transduction pathways triggered by hepatic co-expression of c-Myc and E2F1 protooncogenes. Overexpression of c-Myc alone results in induction of apoptosis via the ASK1/JNK/p38MAPK cascade and activation of translation via elF4E. Overexpression of E2F1 upregulates Akt and mTOR pathways leading both to inhibition of apoptosis through suppression of the ASK1/JNK/p38MAPK axis and activation of translation via inhibition of 4EBP1 and activation of elF4G. mTOR activation might depend on Akt activation and/or other undefined mechanisms (question mark). Co-expression of c-Myc and E2F1 results in suppression of c-Myc-driven apoptosis and transactivation of the c-Myb protooncogene leading to a strong induction of the COX-2 survival signaling. Once upregulated, COX-2 promotes cell survival via activation of the anti-apoptotic proteins Bcl-2 and MCl-1, and by inhibiting the TRAIL-dependent apoptosis via downregulation of DR5. Resistance to TRAIL-dependent apoptosis is further achieved via promoter methylation of the TRAIL effector DAPK.
Supplementary Material
Acknowledgments
Supported by funds from the Intramural Research Program of National Cancer Institutes of Health; National Cancer Institute; Center for Cancer Research.
Abbreviations used in this paper
- AODN
antisense oligodeoxy nucleotides
- ASK1
apoptosis signal-regulating kinase-1
- DAPK
death-associated protein kinase
- DR5
death receptor 5
- 4E-BP1
eukaryotic initiation factor 4E binding protein 1; FoxO1;
- Gab2
Grb2-associated binder 2
- HCC
hepatocellular carcinoma
- HCCB
hepatocellular carcinoma with better prognosis
- HCCP
hepatocellular carcinoma with poor prognosis
- mTOR
mammalian target of rapamycin
- p70 S6K
p70 ribosomal protein S6 kinase
- PI3K
phosphatidylinositol 3-kinase
- PIK3CA
catalytic subunit p110α of phosphatidylinositol 3-kinase
- Rheb
Ras homologue enriched in brain
- rpS6
ribosomal protein S6
- TopBP1
topoisomerase (DNA) II beta binding protein
- TSC2
tuberin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The Authors state the absence of any conflict of interest to disclose.
References
- 1.Harbour JW, Dean DC. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev. 2000;14:2393–2409. doi: 10.1101/gad.813200. [DOI] [PubMed] [Google Scholar]
- 2.Nevins JR. E2F: a link between the Rb tumor suppressor protein and viral oncoproteins. Science. 1992;258:424–429. doi: 10.1126/science.1411535. [DOI] [PubMed] [Google Scholar]
- 3.Moroy T, Marchio A, Etiemble J, et al. Rearrangement and enhanced expression of c-myc in hepatocellular carcinoma of hepatitis virus infected woodchucks. Nature. 1986;324:276–279. doi: 10.1038/324276a0. [DOI] [PubMed] [Google Scholar]
- 4.Murakami H, Sanderson ND, Nagy P, et al. Transgenic mouse model for synergistic effects of nuclear oncogenes and growth factors in tumorigenesis: interaction of c-myc and transforming growth factor alpha in hepatic oncogenesis. Cancer Res. 1993;53:1719–1723. [PubMed] [Google Scholar]
- 5.Conner EA, Lemmer ER, Omori M, et al. Dual functions of E2F-1 in a transgenic mouse model of liver carcinogenesis. Oncogene. 2000;19:5054–5062. doi: 10.1038/sj.onc.1203885. [DOI] [PubMed] [Google Scholar]
- 6.Blackwell TK, Kretzner L, Blackwood EM, et al. Sequence-specific DNA binding by the c-Myc protein. Science. 1990;250:149–151. doi: 10.1126/science.2251503. [DOI] [PubMed] [Google Scholar]
- 7.Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- 8.Nevins JR, Leone G, DeGregori J, et al. Role of the Rb/E2F pathway in cell growth control. J Cel. Physiol. 1997;173:233–236. doi: 10.1002/(SICI)1097-4652(199711)173:2<233::AID-JCP27>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 9.DeGregori J, Leone G, Miron A, et al. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc Natl Acad Sci USA. 1997;94:7245–7250. doi: 10.1073/pnas.94.14.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson DG, Schwarz JK, Cress WD, et al. Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature. 1993;365:349–352. doi: 10.1038/365349a0. [DOI] [PubMed] [Google Scholar]
- 11.Kaczmarek L, Hyland JK, Watt R, et al. Microinjected c-myc as a competence factor. Science. 1985;228:1313–1315. doi: 10.1126/science.4001943. [DOI] [PubMed] [Google Scholar]
- 12.Hermeking H, Eick D. Mediation of c-Myc-induced apoptosis by p53. Science. 1994;265:2091–2093. doi: 10.1126/science.8091232. [DOI] [PubMed] [Google Scholar]
- 13.Kowalik TF, DeGregori J, Leone G, et al. E2F1-specific induction of apoptosis and p53 accumulation, which is blocked by Mdm2. Cell Growth Differ. 1998;9:113–118. [PubMed] [Google Scholar]
- 14.Wu X, Levine AJ. p53 and E2F-1 cooperate to mediate apoptosis. Proc Natl Acad Sci USA. 1994;91:3602–3606. doi: 10.1073/pnas.91.9.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hiebert SW, Lipp M, Nevins JR. E1A-dependent trans-activation of the human MYC promoter is mediated by the E2F factor. Proc Natl Acad Sci USA. 1989;86:3594–3598. doi: 10.1073/pnas.86.10.3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leone G, Sears R, Huang E, et al. Myc requires distinct E2F activities to induce S phase and apoptosis. Mol Cell. 2001;8:105–113. doi: 10.1016/s1097-2765(01)00275-1. [DOI] [PubMed] [Google Scholar]
- 17.Elliott MJ, Dong YB, Yang H, et al. E2F-1 up-regulates c-Myc and p14 and induces apoptosis in colon cancer cells. Clin Cancer Res. 2001;7:3590–3597. [PubMed] [Google Scholar]
- 18.Santoni-Rugiu E, Duro D, Farkas T, et al. E2F activity is essential for survival of Myc-overexpressing human cancer cells. Oncogene. 2001;21:6498–6509. doi: 10.1038/sj.onc.1205828. [DOI] [PubMed] [Google Scholar]
- 19.Hallstrom TC, Nevins JR. Specificity in the activation and control of transcription factor E2F-dependent apoptosis. Proc. Natl. Acad. Sci USA. 2003;100:10848–10853. doi: 10.1073/pnas.1831408100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chaussepie M, Ginsberg D. Transcriptional regulation of AKT activation by E2F. Mol Cell. 2004;16:831–837. doi: 10.1016/j.molcel.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 21.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 22.Liu K, Paik JC, Wang B, Lin FT, Lin WC. Regulation of TopBP1 oligomerization by Akt/PKB for cell survival. EMBO J. 2006;25:4795–4807. doi: 10.1038/sj.emboj.7601355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hay N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell. 2005;8:179–183. doi: 10.1016/j.ccr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 24.Manning BD, Cantley LC. Rheb fills a GAP between TSC and TOR. Trends Biochem Sci. 2003;28:573–576. doi: 10.1016/j.tibs.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 25.Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 26.Conner EA, Lemmer ER, Sanchez A, et al. E2F1 blocks and c-Myc accelerates hepatic ploidy in transgenic mouse models. Biochem Biophys Res Commun. 2006;302:114–120. doi: 10.1016/s0006-291x(03)00125-6. [DOI] [PubMed] [Google Scholar]
- 27.Calvisi DF, Conner EA, Ladu S, et al. Activation of the canonical Wnt/beta-catenin pathway confers growth advantages in c-Myc/E2F1 transgenic mouse model of liver cancer. J Hepatol. 2005;42:842–849. doi: 10.1016/j.jhep.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 28.Leng J, Han C, Demetris AJ, et al. Cyclooxygenase-2 promotes hepatocellular carcinoma cell growth through Akt activation: evidence for Akt inhibition in celecoxib-induced apoptosis. Hepatology. 2003;38:756–768. doi: 10.1053/jhep.2003.50380. [DOI] [PubMed] [Google Scholar]
- 29.Coulouarn C, Gomez-Quiroz LE, Lee JS, et al. Oncogene-specific gene expression signatures at preneoplastic stage in mice define distinct mechanisms of hepatocarcinogenesis. Hepatology. 2006;44:1003–1011. doi: 10.1002/hep.21293. [DOI] [PubMed] [Google Scholar]
- 30.Frith CH, Ward JM. A morphologic classification of proliferative and neoplastic hepatic lesions in mice. J Environ Pathol Toxicol. 1979;3:329–351. [PubMed] [Google Scholar]
- 31.Lee JS, Chu IS, Heo J, et al. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology. 2004;40:667–676. doi: 10.1002/hep.20375. [DOI] [PubMed] [Google Scholar]
- 32.Laird PW, Zijderveld A, Linders K, et al. Simplified mammalian DNA isolation procedure. Nucleic Acids Res. 1991;19:4293. doi: 10.1093/nar/19.15.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pulling LC, Vuillemenot BR, Hutt JA, et al. Aberrant promoter hypermethylation of the death-associated protein kinase gene is early and frequent in murine lung tumors induced by cigarette smoke and tobacco carcinogens. Cancer Res. 2004;64:3844–3848. doi: 10.1158/0008-5472.CAN-03-2119. [DOI] [PubMed] [Google Scholar]
- 34.Simile MM, De Miglio MR, Muroni MR, et al. Down-regulation of c-myc and Cyclin D1 genes by antisense oligodeoxy nucleotides inhibits the expression of E2F1 and in vitro growth of HepG2 and Morris 5123 liver cancer cells. Carcinogenesis. 2004;25:333–341. doi: 10.1093/carcin/bgh014. [DOI] [PubMed] [Google Scholar]
- 35.Opalinska JB, Machalinski B, Ratajczak J, et al. Multigene targeting with antisense oligodeoxynucleotides: an exploratory study using primary human leukemia cells. Clin Cancer Res. 2005;11:4948–4954. doi: 10.1158/1078-0432.CCR-05-0106. [DOI] [PubMed] [Google Scholar]
- 36.Bader AG, Kang S, Zhao L, et al. Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer. 2005;5:921–929. doi: 10.1038/nrc1753. [DOI] [PubMed] [Google Scholar]
- 37.Sulis ML, Parsons R. PTEN: from pathology to biology. Trends Cell Biol. 2003;13:478–483. doi: 10.1016/s0962-8924(03)00175-2. [DOI] [PubMed] [Google Scholar]
- 38.Kim AH, Khursigara G, Sun X, et al. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol. 2001;21:893–901. doi: 10.1128/MCB.21.3.893-901.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsuzawa A, Ichijo H. Molecular mechanisms of the decision between life and death: regulation of apoptosis by apoptosis signal-regulating kinase 1. J Biochem. 2001;130:1–8. doi: 10.1093/oxfordjournals.jbchem.a002947. [DOI] [PubMed] [Google Scholar]
- 40.Hallstrom TC, Mori S, Nevins JR. An E2F1-dependent gene expression program that determines the balance between proliferation and cell death. Cancer Cell. 2008;13:11–22. doi: 10.1016/j.ccr.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang X, Flynn A, Waskiewicz AJ, et al. Phosphorylation of eukaryotic initiation factor elF4E in response to phorbol esters, cell stresses, and cytokines is mediated by distinct MAP kinase pathways. J Biol Chem. 1998;273:9373–9377. doi: 10.1074/jbc.273.16.9373. [DOI] [PubMed] [Google Scholar]
- 42.Davis JN, McCabe MT, Hayward SW, et al. Disruption of Rb/E2F pathway results in increased cyclooxygenase-2 expression and activity in prostate epithelial cells. Cancer Res. 2005;65:3633–3642. doi: 10.1158/0008-5472.CAN-04-3129. [DOI] [PubMed] [Google Scholar]
- 43.Kern MA, Haugg AM, Koch AF, et al. Cyclooxygenase-2 inhibition induces apoptosis signaling via death receptors and mitochondria in hepatocellular carcinoma. Cancer Res. 2006;66:7059–7066. doi: 10.1158/0008-5472.CAN-06-0325. [DOI] [PubMed] [Google Scholar]
- 44.Tang X, Wu W, Sun SY, et al. Hypermethylation of the death-associated protein kinase promoter attenuates the sensitivity to TRAIL-induced apoptosis in human non-small cell lung cancer cells. Mol Cancer Res. 2004;2:685–691. [PubMed] [Google Scholar]
- 45.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- 47.Nakanishi K, Sakamoto M, Yamasaki S, et al. Akt phosphorylation is a risk factor for early disease recurrence and poor prognosis in hepatocellular carcinoma. Cancer. 2005;103:307–312. doi: 10.1002/cncr.20774. [DOI] [PubMed] [Google Scholar]
- 48.Sahin F, Kannangai R, Adegbola O, et al. mTOR and P70 S6 kinase expression in primary liver neoplasms. Clin Cancer Res. 2004;10:8421–8425. doi: 10.1158/1078-0432.CCR-04-0941. [DOI] [PubMed] [Google Scholar]
- 49.Fan QW, Knight ZA, Goldenberg DD, Yu W, Mostov KE, Stokoe D, Shokat KM, Weiss WA. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell. 2006;9:341–349. doi: 10.1016/j.ccr.2006.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.