

Abstract
Oxygen–oxygen bond formation and O2 generation occur from the S4 state of the oxygen-evolving complex (OEC). Several mechanistic possibilities have been proposed for water oxidation, depending on the formal oxidation state of the Mn atoms. All fall under two general classifications: the AB mechanism in which nucleophilic oxygen (base, B) attacks electrophilic oxygen (acid, A) of the Mn4Ca cluster or the RC mechanism in which radical-like oxygen species couple within OEC. The critical intermediate in either mechanism involves a metal oxo, though the nature of this oxo for AB and RC mechanisms is disparate. In the case of the AB mechanism, assembly of an even-electron count, high-valent metal-oxo proximate to a hydroxide is needed whereas, in an RC mechanism, two odd-electron count, high-valent metal oxos are required. Thus the two mechanisms give rise to very different design criteria for functional models of the OEC active site. This discussion presents the electron counts and ligand geometries that support metal oxos for AB and RC O–O bond-forming reactions. The construction of architectures that bring two oxygen functionalities together under the purview of the AB and RC scenarios are described.
Keywords: photosynthesis, water oxidation, oxygen-evolving complex, catalysis, solar energy
1. The nature of the Mn–Oxo interaction in OEC
The bond-making and bond-breaking reactions of dioxygen in biology are managed at the metals of cofactors (Holm et al. 1996). Most metallocofactors use oxygen to derive their function; these include the haem iron centres of monooxygenases (Sono et al. 1996; Dunford 1999; Newcomb & Toy 2000; Watanabe 2000; Meunier et al. 2004) and the molybdenum and tungsten of oxotransferases (Kisker et al. 1997; Enemark et al. 2004) to perform substrate oxidation, the Fe–Cu centre of cytochrome c oxidase to drive metabolic respiration (Malmström 1990; Wikström 2004; Bertini et al. 2006; Busenlehner et al. 2006; Hosler et al. 2006) and copper oxidases for oxygen binding, activation and subsequent substrate oxidation (Solomon et al. 1996; Rosenzweig & Sazinsky 2006; Yoon & Solomon 2007). In all cases, metal oxos appear as critical intermediates in the management of oxygen. The oxo may be terminal, bound by a multiple bond to a single metal centre as in the case of haem peroxidases (Watanabe 2000; Newcomb et al. 2006), or the oxo may engage in single bonding to multiple metal centres, as is the case for ribonucleotide reductase (Jordan & Reichard 1998; Stubbe & van der Donk 1998; Stubbe et al. 2003), methane monooxygenase (Feig & Lippard 1994; Hanson & Hanson 1996; Que & Ho 1996; Wallar & Lipscomb 1996; Lieberman & Rosenzweig 2005) and galactose oxidase (Whittaker 2003), to name a few. Both binding modes may be relevant to the most important metallocofactor that is able to produce oxygen—the oxygen-evolving complex (OEC) of photosystem II (PS II). Current structural models of the OEC show oxos within the Mn4Ca cluster singly bonded to two and three Mn centres of the core, and in the dangler model (Peloquin & Britt 2001), an oxo is proposed to be multiply bonded to the dangling Mn.
The appearance of the structures of the photosynthetic membrane (Ferreira et al. 2004; Loll et al. 2005; Yano et al. 2006) together with complementary structural studies of the Kok cycle (Haumann et al. 2005) has led to various proposals for possible mechanisms of O2 generation. Figure 1 lists current proposals for the various electron counts of the S states of the OEC, which were proposed during the Philosophical Discussion at the Royal Society (Dutton et al. 2006). The O–O bond is believed to be formed in the critical S4 state for which the least is known. The precise nature of the active site has profound consequences to the mechanism and consequently the design of catalysts for oxygen generation. If all the oxidizing equivalents are borne by the Mn centres, a [MnIV3MnV] core is obtained. For this model, the oxygen is activated at the ‘dangler’ MnV centre. A tetragonal geometry is effectively assumed for the oxo in the OEC. In this geometry, the oxo donates its orthogonal electron lone pairs to the MnV centre via the e(dxz, dyz) orbital set. The movement of electron pairs from the oxygen to the metal centre makes the oxygen more acidic or electrophilic. Such electrophilic oxos (acids) are susceptible to attack by nucleophiles (bases). For instance, in organic reaction chemistry, this type of oxo is attacked by the two-electron bond of an olefin, thus forming the basis for catalytic oxidations (Zhang et al. 1990; Jacobsen et al. 1999; Yang et al. 2006; Yang & Nocera 2007). In the case of a [MnIV3MnV] centre, a similar mechanism is proposed with the two-electron bond of the olefin replaced by the lone pair of a hydroxide delivered from the calcium of the cofactor (McEvoy & Brudvig 2006). Alternatively, radical character has also been proposed to reside on two oxos of a [MnIIIMnIV3,O·,O·] cofactor or on an oxo and amino acid of a [MnIIIMnIV3,O·,AA·] (AA=amino acid) cofactor. In this case, one may envisage the O–O bond formation to occur by coupling of the biradical pair; this model gives credence to proposals of O2 generation via peroxide-like intermediates (Ruttinger & Dismukes 1997; Siegbahn 2006). A [MnIV4,O·] core places a single-radical spin on oxygen and O2 is generated by an undefined mechanism.
Figure 1.
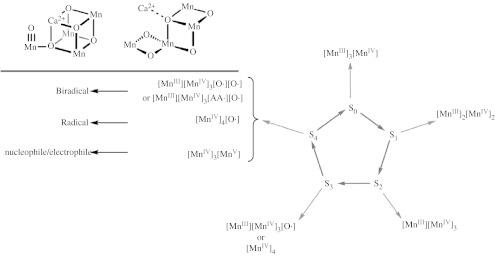
Current proposals for the various electron counts of the S-states of the Kok cycle.
2. Different metal-oxo ligand fields for O–O bond formation
The variegated formalisms of the OEC give rise to fundamentally different mechanisms for the O–O bond formation: acid(electrophilic)–base(nucleophilic) (AB) or radical coupling (RC). These different mechanisms call for different ligand fields in which the metal oxo resides and correspondingly different metal-oxygen coordination environments and electron configurations. Three prevailing ligand fields are shown in figure 2. Discussion of the chemistry derived from these ligand fields is based on orbital energy levels. Differing spin states for an electron count are not treated explicitly, though it is readily acknowledged that the spin state is an important determinant of the metal-oxo bond strength and reaction pathway. Moreover, secondary effects such as Jahn–Teller distortions will also perturb the overall energetics of the system. All such factors must ultimately be considered in the context of the ensuing ligand fields for the proper design of water-splitting catalysts.
Figure 2.
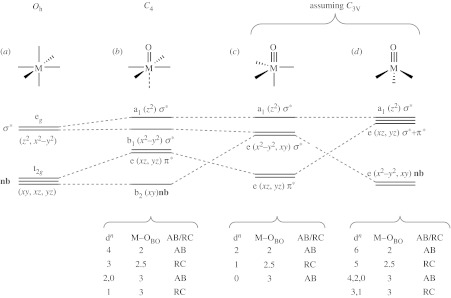
Qualitative frontier molecular orbital splitting diagrams for (a) an octahedral metal complex and a metal oxo residing in a (b) tetragonal field, (c) trigonal bipyramidal and (d) tetrahedral ligand field. The table shows the d-electron count that supports a multiple metal oxo bond and the preferred mechanism for the O–O bond coupling, where AB is acid/base and RC, radical coupling. Only low-spin configurations are considered. The assignment of AB and RC will change with high-spin configurations.
(a) Tetragonal oxo fields
As mentioned in figure 1, a prevailing model for O2 generation at the OEC involves an AB mechanism in which hydroxide attacks an electrophilic oxygen. The origins of the electrophilic oxygen can be found in the molecular orbital diagram shown in figure 2b. The diagram can be obtained by replacing an axial ligand of an ML6 octahedral complex with an oxo. This substitution lowers the t2g orbital symmetry to b2(dxy) and e(dxz, dyz) and the metal-ligand σ antibonding eg orbital symmetry to b1() and a1() levels. Because the metal-oxo bond is short, the orbital is more destabilized than in its ML6 parent. Similarly, the e(dxz, dyz) orbitals are destabilized relative to their octahedral parentage because they are π antibonding with respect to the oxygen 2px and 2py orbitals. Owing to the prevalence of these σ and π interactions of the oxo with the metal, the axial ligand opposite the oxo is significantly weakened or typically absent.
The tetragonal environment (C4 symmetry) described by figure 2b poises a lowest unoccupied molecular orbital (LUMO) directed along the metal-oxo axis for attack by an incoming hydroxide. This situation favours O–O bond formation. From an orbital perspective, an orbital of σ character on the hydroxide (highest occupied molecular orbital) approaches the M–O π* orbital (LUMO). The combination leads to the formation of an O–O σ bond while breaking one of the M–O π bonds, representing a formal 2e− reduction of the metal centre. The AB mechanism effectively requires an even electron system where two electron holes of the (dxz, dyz) LUMO of a d4 metal oxo are filled by the attacking electron pair of the hydroxide. Alternatively, a d2 metal oxo is even more electrophilic as the LUMO is completely unoccupied and hence even more acidic than the partially occupied LUMO of the d4 metal centre. As of now, how the overall spin state of the metal-oxo precursor and its correlation to products determines the reaction kinetics remains undefined. We note that the AB mechanism can be considered the microscopic reverse of the O–O bond heterolysis step in cytochrome P450 to produce H2O and a ferryl (Denisov et al. 2005; Shaik et al. 2005). In this problem, the effect of spin has been accounted for computationally. Similar treatments for the O–O bond-forming reaction are warranted.
The same tetragonal ligand field can support an RC mechanism but for a different electron count. Population of the degenerate e(dxz, dyx) level of the metal oxo with a single electron results in a singly occupied molecular orbital. Thus a d3 metal centre gives rise to the radical character of the metal-oxo moiety. Note, for a d5 centre, most of the metal-oxo π-bonding is lost (formally only 0.5 π bond is preserved) owing to the three-electron occupancy of the e(dxz, dyz) set, which is metal-oxo π* in character. Accordingly, a terminal d5 oxo is difficult to attain in a tetragonal field. The precise location of the radical in the d3 case—metal versus oxygen centred—depends largely on the nature of the metal-oxo interaction. For the most part, the issue of the extent of oxygen radical character at odd-electron metal-oxo centres has remained experimentally unresolved.
Though in the same ligand field, the AB and RC mechanisms give rise to drastically different catalyst design criteria. The O–O bond formation for the AB mechanism requires the assembly of a high-valent oxo in a tetragonal ligand field and a hydroxide, whereas O–O bond formation for the RC mechanism requires the assembly of two high-valent metal oxos in tetragonal fields. This aspect of disparate design for tetragonal metal-oxo moieties will be addressed in §3.
(b) Trigonal oxo fields
The radicals of an RC mechanism are supported by the local C3 symmetry of a metal oxo in a trigonal environment. When an axial ligand is present (the trigonal bipyramidal case shown in figure 2c), the orbital splitting pattern from lowest to highest energy is a degenerate level comprising the dxz and dyz orbitals, another degenerate level formed from the and dxy orbitals and a highest energy orbital. The and dxy orbitals rise above the dxz and dyz orbitals because the former take on substantial M–L σ* character and thus are destabilized. For a d4 case, the LUMO is and dxy and the π bonds are annihilated owing to the 4e− occupancy of the M–O π* e(dxz, dyz) set. Consequently, the terminal metal oxo is inaccessible to d4 metals in a trigonal bipyramidal ligand field. For the d2 electron count, two electrons occupy a degenerate e(dxz, dyz) orbital level. Thus, this system, from an electronic perspective, looks very much like a metal oxo in a tetragonal field with a d4 electron count (vide infra). For the d0 case, the e(dxz, dyz) orbital set is empty and akin to the case of a d2 electron count of a metal oxo in a tetragonal field. As with the d2/d4 even-electron systems of the tetragonal field, a metal oxo with a d0/d2 electron count in a trigonal bipyramidal field lends itself to O–O bond formation by an AB mechanism. More generally, for low electron counts, dn(M–O)tetra∼dn−2(M–O)tbp. This analogy holds for the odd-electron count as well: d1(M–O)tbp and d3(M–O)tbp are similar to d3(M–O)tetra and d5(M–O)tetra, respectively. For the same reasons mentioned for a d5(M–O)tetra centre, the d3(M–O)tbp centre has a 0.5 π bond order, and thus it is fragile and difficult to realize. The d1 system, however, presents a robust metal oxo with putative radical character and accordingly the system lends itself to O–O bond formation by an RC mechanism.
(c) Tetrahedral ligand fields
Metals with higher d-electron counts can exhibit AB and RC oxo reactivity if the apical ligand opposite the metal-oxo vector of the trigonal bipyramid is removed to attain the local coordination environment of a pseudo-tetrahedron. The d-orbital diagram obtained for this metal-oxo ligand coordination geometry is shown in figure 2d. The metal-based orbitals involved in the σ and π M–O interactions are the highest in energy owing to their antibonding character. The ordering of the a1((σ*)) and e(dxz, dyz(π*)) depends on the tridentate ligand's bite angle (Jenkins et al. 2002). The distinguishing trait of the application of this ligand field to the metal oxo is that a degenerate e(, dxy) level is lowest in energy and may accommodate up to four electrons in a low-spin configuration before the metal π* orbitals are populated. Hence, late transition metals may be used to effect AB (metals of even electron counts, d4 and possibly d6) and RC (metals of odd electron count, d5) O–O bond-forming mechanisms. Moreover, high-spin states are likely to be accessible, and thus might be used resourcefully to activate the metal-oxo bond. Targeting this framework, therefore, in principle will allow chemists to design mid-to-late transition metal platforms that are amenable to high oxidation states, but will tolerate higher d-electron counts in a parent complex without destabilization of the M–O linkage.
3. Experimental realization of metal–oxo cofactors in different ligand fields
Approaches to the design of catalysts possessing the metal-oxo cofactor within the ligand fields described by figure 2 are now presented.
(a) Metal-oxo cofactors in tetragonal fields
(i) AB mechanism
Figure 3a shows the structure of the OEC active site that is consistent with an AB mechanism. The key intermediate in the critical bond-forming step of water oxidation is a pre-organized oxo/hydroxyl intermediate. The Ca2+ ion is thought to decrease the pKa of a bound water to produce a nucleophilic hydroxide in the secondary coordination sphere that attacks the electrophilic oxo of a high-valent manganese (Messinger et al. 1995; Pecoraro et al. 1998; Vrettos et al. 2001). The challenges confronting the development of a functional model for O2 generation by the AB mechanism at the OEC are labelled in figure 3:
Active sites must permit control of the secondary coordination environment such that two water molecules may be pre-assembled for the O–O bond coupling.
Water must be activated by coupling proton transfer reactions to electron transfer. The removal of electrons and protons at the OEC active site finds its origins in the Kok cycle (Westphal et al. 2000; Tommos 2002; Haumann et al. 2005). Proton-coupled electron transfer (PCET) activation of the substrate may be managed by a proton exit channel beginning at the D61 residue (Barber 2006).
The high-valent metal oxo must be generated for attack by the water-derived hydroxide.
Figure 3.

(a) The water oxidation centre in photosystem II (Ferreira et al. 2004) and (b) the Hangman porphyrin (I) assemble the oxygen of two waters for coupling, (II) activate the water to oxo by PCET and (III) position a high-valent oxo along the reactive metal hydroxide vector. Though the resting state of the Hangman is a FeIII–OH⋯H2O complex, the Hangman porphyrin is prepared by the introduction of an FeII into the porphyrin core and the assembly of two waters. Production of the Compound I intermediate of the Hangman thus results from an overall (4e−,4H+) process.
We have tackled the challenges presented by (I)–(III) by constructing ‘Hangman’ platforms (Yeh et al. 2001; Chng et al. 2003). The Hangman structure assembles water molecules (challenge I) by using a scaffold to ‘hang’ one water molecule over another that is coordinated to the Fe(III) centre of a haem platform. Figure 3b shows the crystal structure of Hangman porphyrin (Yeh et al. 2001). A carboxylic acid group appended to a xanthene scaffold suspends an exogenous water molecule in the Hangman cleft via hydrogen bonding. The binding energy of the water molecule is 5.8 kcal mol−1 (Chang et al. 2003) and its association within the cleft is reversible. The Hangman's pendant mimics the amino acid residues that orient water in the distal cavities of haem peroxidases by precisely positioning an acid–base functional group over the face of the haem. We have since generalized the Hangman approach to include salen macrocycles (Liu & Nocera 2005). As with haem peroxidases, the Hangman platform supports an electrophilic oxo (challenge III) of the Compound I (Cpd I; De Montellano 1995; Sono et al. 1996; Dunford 1999; Watanabe 2000; Newcomb et al. 2006) type, which is two redox levels above FeIII with a ferryl FeIV=O residing within an oxidized porphyrin π-radical cation, P·+. The electrophilic oxo has been observed by cryogenic stopped-flow spectroscopy (Soper et al. 2007). The production of the oxo is derived from the ability of the Hangman to perform PCET (challenge II; Liu & Nocera 2005; Hodgkiss et al. 2006; Yang et al. 2006; Soper et al. 2007; Yang & Nocera 2007). The importance of the PCET mechanism in the activation of water and other small molecules has been presented in a recent Philosophical Society Discussion (Reece et al. 2006). In short, the pendent H+ donor group in the Hangman system exerts control over the PCET production of the electrophilic oxo from peroxide intermediates by coupling PT to internal 2e− redox events of the redox (salen or porphyrin) cofactor (Yang et al. 2006; Rosenthal et al. 2007; Soper et al. 2007; Yang & Nocera 2007). As with haem peroxidases, the electrophilic oxo, P·+FeIV=O, is susceptible to attack by nucleophiles such as two electrons of the π-bond of olefins (Yang & Nocera 2007). The goal now is to see whether the oxo is sufficiently electrophilic to be attacked by hydroxide. If not, the porphyrin will be modified with electron-withdrawing groups such as pentafluorinated phenyls in the meso positions with the aim of increasing the electrophilicity of the FeIV=O.
The similarities of a potential [MnIV3MnV] S4 state of OEC and the Hangman system are highlighted in figure 3. Both centres assemble two water molecules within a cleft. An electrophilic oxo in a tetragonal ligand may be produced at either centre by PCET. For the case of the proposed model for the S4 state of OEC, the oxo is bonded to a d2 centre of Mn(V), whereas for the Hangman cofactor, the oxo is bonded to a d4 metal centre of Fe(IV). Both OEC and the Hangman porphyrins possess a proximate site to deliver hydroxide (from the Ca2+ in OEC and from the hanging group in Hangman) to the oxo, which is susceptible to nucleophilic attack. But in the case of the Hangman cofactor, the use of hydroxide as the nucleophile remains to be demonstrated.
(ii) RC mechanism
Meyer's (Gersten et al. 1982) blue dimer complex [(bpy)2(OH2)RuIII(μ-O)RuIII(OH2)(bpy)2]4+(1 in figure 4) catalyses the oxidation of water to molecular oxygen at 1.4 V versus normal hydrogen electrode (NHE; pH 1) with 10–25 turnovers. This species is composed of a dimeric ruthenium core connected by a nearly linear μ-oxo bridge. Kinetic analysis reveals that the initial [(OH2)RuIII(μ-O)RuIII(OH2)] core undergoes a series of coupled proton and electron transfers to produce a [(O)RuV(μ-O)RuV(O)] species that precedes O–O bond formation (Binstead et al. 2000). Although there remains much mechanistic uncertainty over the nature of the discrete mechanism by which the O–O bond formation occurs, the precursor to oxygen formation is two d3 metal oxos in tetragonal ligand fields disposed in a side-by-side orientation. From the perspective of figure 2b, the odd-electron system may be formulated as side-by-side oxo radical centres, though their direct coupling has not been implicated as a dominant reaction pathway. Structural and electronic features unique to the blue dimer and related analogues (Sens et al. 2004; Zong & Thummel 2005) indeed inhibit a straightforward RC reaction path. Geometric constraints imposed by the ancillary ligands and the μ-oxo bridge prevent oxygen atoms from attaining the correct geometry to form an O–O bond. It has been estimated that rotation about the μ-O bond to yield an eclipsed geometry of the M–O vectors still places the O atoms 3.2 Å apart (Gilbert et al. 1985). Coupling at this distance would require a significant distortion or the participation of water to mediate the radical coupling. In addition, putative radical character may be diminished by antiferromagnetic coupling through the μ-O bridge (Yang & Baik 2004, 2006).
Figure 4.

Bimetallic ruthenium compounds capable of water splitting: Meyer's blue oxo dimer complex [(bpy)2(OH2)RuIII(μ-O)RuIII(OH2)(bpy)2]4+(1) and cofacial diruthenium platforms on the scaffolds of dimethylxanthene (2), dibenzofuran (3) and anthracene (4,5).
In the light of these barriers to an RC mechanism, we have designed the Pacman systems of figure 4 (2 and 3) to remove impediments to RC while largely retaining the d3 oxo character of the blue dimer system. The Pacman complexes use dimethylxanthene (DTX; 2) and dibenzofuran (DPD; 3) organic spacers to connect two tridentate terpyridine ligands. These organic scaffolds have been previously applied to create cofacially oriented Pacman porphyrins (Chang et al. 2000a,b, 2002a,b; Deng et al. 2000). In the Pacman approach, the two Ru oxos are oriented towards each other to enable their direct interaction. Moreover, the oxo centres are electronically isolated from each other by imposing the long covalent pathway between them via the scaffold. We have shown that the Pacman scaffolds are flexible enough to accommodate over a four-angstrom range in metal–metal distance (Deng et al. 2000). We envision that this flexibility would allow the metal centres to clamp down, facilitating O–O coupling, while retaining the ability to spring open, release O2 and bind water. Electrochemical studies indicate that the Pacman complexes are stable through the range of oxidation states of RuII to RuIV(O), in contrast to the analogous blue dimer fragment that is unstable towards disproportionation. Further oxidation of the RuIV(O) at 1.4 V versus Ag/AgCl (pH=7) for both the DTX (2) and DTD (3) systems leads to oxygen production, which has been verified by a standard pyrogallol analysis. The mechanism of oxygen production is currently under study. It is noteworthy that the monomeric Ru centres are inert for water oxidation to O2 (McHatton & Anson 1984; Takeuchi et al. 1984). Our observations of the DTX and DTD Pacman complexes agree with those obtained for Pacman complexes employing an anthracene spacer between the Ru terpyridine centres (Wada et al. 2001a,b). These coordination spheres are completed by either a bipyridine (4, figure 4) or a 3,6-di-tert-butyl-1,2-benzoquinone (5, figure 4) ligand. Interestingly, the bipyridine complex is reported to exhibit modest activity for O2 production at potentials beyond 1.31 V. Conversely, the introduction of the redox-active benzoquinone ligand in 5 (figure 4) leads to a pronounced increase in O2 production. The reasons for this enhanced activity of Ru Pacman systems possessing redox-active, ancillary ligands are unknown.
(b) Metal-oxo cofactors in trigonal bipyramidal fields
As shown in figure 2c, the lowest lying metal-based orbital for a metal oxo in a trigonal bipyramidal field is a degenerate level composed primarily of the dxz and dyz orbitals. This limits the stabilization of the metal-oxo multiple bond to metals with a d0 or d1 configuration. To test the limit of oxo stability in this geometry, we have created the modified tris(pyrrolyl-α-methyl)amine framework (Betley & Nocera submitted a) shown in figure 5 to survey group V oxo chemistry. The ligand design is based on Odom's parent scaffold (Shi et al. 2004). Substitution of 2-arylpyrroles (Rieth et al. 2004) for pyrrole in Odom's reported synthesis produces the target tris(pyrrolyl-α-methyl)amine ligand in high yields (aryl=mesityl). A family of VIV species and VV oxo complexes have been synthesized that uses this ligand framework. The trigonally coordinated V(mesityl)3 readily condenses with single O-atom bridges to form VIV species [(mesityl)3VIV]2(μ-O) (Odom & Cummins 1995). The large aryl substituents of the ligand framework block the μ-oxo dinuclear structure from forming. Currently, the terminal oxo of VIV (d1) has been targeted to probe where the radical character largely resides, i.e. on the metal or the oxygen.
Figure 5.

The trigonal bipyramidal field offered by the tris(pyrrolide)amine framework. The complex is stabilized for the oxo complexes of VIV and VV formal oxidation states.
Appearance of radical character at the oxygen of the (M–O)tbp centre is critical to advancing the RC mechanism for O–O bond coupling. However, this criterion alone is insufficient. The trigonal bipyramidal scaffold needs to be elaborated so that two oxos may be brought together for coupling. We have set out on a course of study to address this challenge with the synthesis of the cryptands shown in figure 6. The active site combines the features of the TREN-based (TREN=tris(2-aminoethyl)amine) metal-binding site (Schrock 1997) and the neutral hexacarboxamide cryptand (Kang et al. 2003). These ligands proffer a trigonal monopyramidal coordination site for the metal ions in high oxidation states, engendered by the trianionic charge of the deprotonated TREN ligand. The ligands are diametrically positioned by attaching three carboxamide meta-phenyl spacers between the TREN metal-binding sites. The design results in the formation of a cavity that can accommodate two high-valent metal centres in a cofacial geometry. Manganese and cobalt have been introduced into the cavity; the molecules shown in figure 6 have been structurally characterized (Alliger et al. in preparation). A comparison of the two structures shed light on the nature of the cavity created by the hexacarboxamido ligand. The Mn complex (figure 6, 6) possesses a hydroxide ligand that imposes a Mn–Mn distance of 4.058 Å. With the absence of a bridging ligand, the metals relax to a Co–Co distance of 6.073 Å (figure 6, 7). The flex in the cavity results from rotation about the metal–amide bond, which allows the ligand to bring the metal centres together in an accordion-like motion. The new ligand thus has several attributes pertaining to an RC mechanism: it (i) presents binding sites that can accommodate high-valent metal centres, a necessity for a trigonal bipyramidal ligand geometry; (ii) permits the assembly of two metal oxos within the cavity in a convergent orientation; and (iii) possesses a cavity able to flex over 2 Å, thus allowing oxos to be brought together for their subsequent RC. Moreover, the use of carboxamide linkers should render these ligands resistant to oxidation, thus avoiding ligand degradation by reactive oxygen intermediates. Current efforts are indeed focused at generating high-valent oxos within the cavity of the new ligand.
Figure 6.
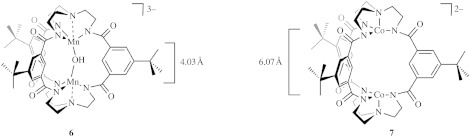
Hexaanionic cryptands to support the coupling of two high-valent oxos by an RC mechanism.
(c) Metal-oxo cofactors in tetrahedral fields
The cryptands of §4 sterically shield the metal-oxo radical species from deleterious reactivity and proximally orient the two metal-oxo vectors. The trigonal bipyramidal approach, however, does not favour metal-oxo species with d-electron counts exceeding d2. Thus, targeting pseudo-tetrahedral frameworks will allow us, in principle, to stabilize and probe both AB and RC mechanistic possibilities with high-valent, mid-to-late transition metal ions with up to six d electrons (figure 2d).
The ligand scaffold for a tetrahedral field should be (i) C3-symmetric and trianionic to achieve the desired oxidation states for the metal-oxo complexes, (ii) modular such that the steric protection about the oxo moiety can be tempered for isolation or promotion of bimolecular AB and RC reactions, (iii) oxidatively robust such that the redox events are localized within the metal-oxo framework without deleterious ligand oxidation events, and (iv) ultimately protolytically insensitive, as the desired reaction medium involves water as a substrate. We have synthesized a library of tridentate scaffolds (Betley & Nocera submitted b); figure 7 illustrates select platforms. They include the known tris(3-methylindolyl)methane ligand (8), which has been suitably employed to stabilize main group elements in the +3 oxidation state (Müller & Pindur 1984; Barnard & Mason 2001), a tris(tert-butylurea)methane species (9) evocative of platforms synthesized by Borovik (MacBeth et al. 2000; MacBeth et al. 2004), and a tris(2-mesitylpyrrolyl)methane ligand (10) that presents a sterically shielded trigonal environment for the metal ions. Because the target metal ion species involve mid-to-late transition elements, the trianionic ligand platforms are chosen because the N-donor basicity is attenuated by their pendent organic moiety, that is the indole, urea and pyrrole frameworks. With this library of ligand platforms in hand, investigations of the transition metal-oxo chemistry engendered by this ligand field may now be explored. These studies are currently underway.
Figure 7.
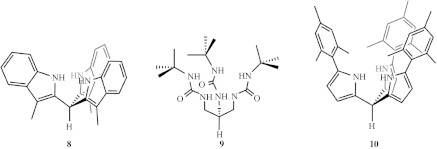
Trianionic platforms to support high-valent, late transition metal-oxo cores in a tetrahedral ligand field.
4. Summary and outlook
The realization of a water-splitting catalyst addresses one of the greatest challenges facing our planet in the coming century—the development of a clean and renewable fuel source (Eisenberg & Nocera 2005; Lewis & Nocera 2006; Nocera 2006). The combination of water and light from the Sun can be used to produce hydrogen and oxygen. The blueprint for accomplishing this solar energy conversion is provided by photosynthesis, which converts sunlight into a wireless current which in turn is captured cathodically by photosystem I to reduce the protons to ‘hydrogen’ (i.e. NADPH) and anodically by OEC to produce oxygen. It is this latter transformation that poses the greatest difficulty to duplicating photosynthesis outside of the leaf.
The various proposals for the redox equivalency of the S4 state of the Mn4Ca cluster lead to different mechanisms for O–O bond coupling and O2 generation. Current models of the OEC suggest the prevalence of either acid–base or radical-coupling pathways. The AB mechanism requires an electrophilic oxo for bond formation with an exogenous oxygen nucleophile. To attain such an acidic oxygen, the lone pairs of the oxo donate into empty d-orbitals of a high-valent metal to form a double or triple metal-oxo bond. We note, however, that the formation of such strong bonds imposes kinetic barriers, since the multiple metal-oxo bond must ultimately be broken for O2 release. To this end, the AB mechanism hinges on the balance between a strong metal-oxo bond to ensure electrophilicity but it must be weak enough to allow it to be broken with facility. The participation of high-spin states in the OEC complex may provide a resolution to this quandary. Alternatively, the coupling of oxygen radicals may circumvent the requirements for a strong metal-oxo bond. Oxygen radicals may be supported by single or multiple bonds to metals in the mid-to-late transition series. The open questions surrounding the mechanism for O2 production by the OEC provides an imperative for the design of metal oxos in coordination environments that permit the full range of stereoelectronic factors associated with O–O bond coupling to be explored. Thus it is a task for the synthetic chemist to design new ligand environments that capitalize on the discovery of OEC structure and function. Whereas an alternative approach might be pursued with solid-state materials and surfaces, a molecular approach lends itself better to mechanistic analysis. We report here progress towards this end.
Discussion
C. Dismukes (Princeton University, USA). What type of ligand field is needed at the OEC Mn4 site to create the MnV=O species that you say could form?
D. Nocera. We have addressed this issue in detail in the paper accompanying the Discussion (§§2a and 3a). In short, an oxo of a MnV oxidation state would be supported by a tetragonal field and a d2 electron count. For this situation, the Mn oxo triple bond would be of order three. For a MnIV oxo, our inclination is to expect radical character involving the oxo.
A. Aulauloo (University of Paris-Sud, France). Concerning the ligand design for the biscompartmental cryptand, once you generate Mn=O inside the cavities, don't you think it will attack the aromatic ring?
D. Nocera. Any practical catalysts for water oxidation will have to be stable and stand up to a highly active oxo core. It seems almost a paradox—the presence of a high-valent and reactive oxo in an environment rife with reducible moieties—i.e. the C–C and C–H bonds of a ligand framework. But these are the same moieties in proteins and enzymes, and many of these are oxidases whose function is derived from a reactive metal oxo. Biology overcomes this paradox in many ways. A metallocofactor will be fixed with its surrounding reactive bonds directed away from the metal oxo. Alternatively, the metal-oxo cofactor is sometimes insulated in a non-reactive shell (except for the site of substrate binding). And if this is still not enough, then as the participants of this Discussion certainly are familiar with, biology incorporates repair mechanisms. It may come as no surprise that the S4 state of OEC is particularly damaging; thus the D1 protein is replaced every 30–60 min. Such repair mechanisms do not exist for synthetic catalytic active sites—but they will have to be ‘designed’ to ensure that catalysts can withstand the harsh oxidizing conditions of oxygen generation from water. This is a frontier area of research (along with the design of working catalysts!). Now to the specifics of the question….the cryptate (compound 5) in my manuscript is designed with the idea of robustness in mind. The use of the carboxamide linker should render these ligands resistant to oxidation, thus avoiding ligand degradation by reactive oxygen intermediates. Of course, we may also need to worry about the CH bonds on the phenyl spacer. If this is a problem, we will have to teflonize the spacer and replace the reactive CH bonds with CF bonds.
Acknowledgements
The National Science Foundation supported this work with a Chemical Bonding Center grant CHE-0533150. We also acknowledge support from the Department of Energy, DOE DE-FG02-05ER15745. Y.S. thanks the DoD for a NDSEG pre-doctoral fellowship and T.A.B. thanks the NIH for a post-doctoral research fellowship.
Footnotes
One contribution of 20 to a Discussion Meeting Issue ‘Revealing how nature uses sunlight to split water’.
References
- Alliger, G. E., Breiner, B., Fu, R., Cummins, C. C. & Nocera, D. G. In preparation. Dimanganese (II) and dicobalt (II) cryptates supported by a hexaanionic cryptand ligands.
- Barber J. Photosystem II: an enzyme of global significance. Biochem. Soc. Trans. 2006;34:619–631. doi: 10.1042/BST0340619. doi:10.1042/BST0340619 [DOI] [PubMed] [Google Scholar]
- Barnard T.S, Mason M.R. Synthesis, structure, and coordination chemistry of the bicyclic π-acid phosphatri(3-methylinolyl)methane. Organometallics. 2001;20:206–214. doi:10.1021/om000793y [Google Scholar]
- Bertini I, Cavallaro G, Rosato A. Cytochrome c: occurence and functions. Chem. Rev. 2006;106:90–115. doi: 10.1021/cr050241v. doi:10.1021/cr050241v [DOI] [PubMed] [Google Scholar]
- Betley, T. A. & Nocera, D. G. Submitted a Metal complexes supported by the tris(2-mesityl-pyrrolyl-α-methyl)amine ligand.
- Betley, T. A. & Nocera, D. G. Submitted b Design criteria for O–O bond formation via metal oxo complexes. [DOI] [PubMed]
- Binstead R.A, Chronister C.W, Ni J, Hartshorn C.M, Meyer T.J. Mechanism of water oxidation by the μ-oxo dimer [(bpy)2(H2O)RuIIIORuIII(OH2)(bpy)2]4+ J. Am. Chem. Soc. 2000;122:8464–8473. doi:10.1021/ja993235n [Google Scholar]
- Busenlehner L.S, Salomonsson L, Brzezinski P, Armstrong R.N. Mapping protein dynamics in catalytic intermediates of the redox-driven proton pump cytochrome c oxidase. Proc. Natl Acad. Sci. USA. 2006;103:15 398–15 403. doi: 10.1073/pnas.0601451103. doi:10.1073/pnas.0601451103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.J, Deng Y, Heyduk A.F, Chang C.K, Nocera D.G. Xanthene-bridged cofacial bisporphyrins. Inorg. Chem. 2000a;39:959–966. doi: 10.1021/ic990987+. doi:10.1021/ic990987+ [DOI] [PubMed] [Google Scholar]
- Chang C.J, Deng Y, Shi C, Chang C.K, Anson F.C, Nocera D.G. Electrocatalytic four-electron reduction of oxygen to water by a highly flexible cofacial cobalt bisporphyrin. Chem. Commun. 2000b:1355–1356. doi:10.1039/b001620i [Google Scholar]
- Chang C.J, Baker E.A, Pistorio B.J, Deng Y, Loh Z.-H, Miller S.E, Carpenter S.D, Nocera D.G. Structural, spectroscopic, and reactivity comparison of xanthene- and dibenzofuran-bridged cofacial bisporphyrins. Inorg. Chem. 2002a;41:3102–3109. doi: 10.1021/ic0111029. doi:10.1021/ic0111029 [DOI] [PubMed] [Google Scholar]
- Chang C.J, Yeh C.Y, Nocera D.G. Porphyrin architectures bearing functionalized xanthene spacers. J. Org. Chem. 2002b;67:1403–1406. doi: 10.1021/jo016095k. doi:10.1021/jo016095k [DOI] [PubMed] [Google Scholar]
- Chang C.J, Chng L.L, Nocera D.G. Proton-coupled O–O activation on a redox platform bearing a hydrogen-bonding scaffold. J. Am. Chem. Soc. 2003;125:1866–1876. doi: 10.1021/ja028548o. doi:10.1021/ja028548o [DOI] [PubMed] [Google Scholar]
- Chng L.L, Chang C.J, Nocera D.G. Catalytic O–O activation chemistry mediated by iron Hangman porphyrins with a wide range of proton-donating abilities. Org. Lett. 2003;5:2421–2424. doi: 10.1021/ol034581j. doi:10.1021/ol034581j [DOI] [PubMed] [Google Scholar]
- De Montellano P.R.O, editor. Cytochrome P-450 structure, mechanism and biochemistry. 2nd edn. Plenum Press; New York, NY: 1995. [Google Scholar]
- Deng Y, Chang C.J, Nocera D.G. Direct observation of the ‘pac-man’ effect from dibenzofuran-bridged cofacial bisporphyrins. J. Am. Chem. Soc. 2000;122:410–411. doi:10.1021/ja992955r [Google Scholar]
- Denisov I.G, Makris T.M, Sligar S.G, Schlichting I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. doi:10.1021/cr0307143 [DOI] [PubMed] [Google Scholar]
- Dunford H.B. Wiley; New York, NY: 1999. Heme peroxidases. [Google Scholar]
- Dutton P.L, Munro A.W, Scrutton N.S, Sutcliffe M.J. Quantum catalysis in enzymes: beyond the transition state theory paradigm. Phil. Trans. R. Soc. B. 2006;361:1293–1455. doi: 10.1098/rsif.2006.0122. doi:10.1098/rstb.2006.1879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg R, Nocera D.G. Overview of the forum on solar and renewable energy. Inorg. Chem. 2005;44:6799–6801. doi: 10.1021/ic058006i. doi:10.1021/ic058006i [DOI] [PubMed] [Google Scholar]
- Enemark J.H, Cooney J.J.A, Wang J.-J, Holm R.H. Synthetic analogues and reaction systems relevant to the molybdenum and tungsten oxotransferases. Chem. Rev. 2004;104:1175–1200. doi: 10.1021/cr020609d. doi:10.1021/cr020609d [DOI] [PubMed] [Google Scholar]
- Feig A.L, Lippard S.J. Reactions of non-heme iron(II) centers with dioxygen in biology and chemistry. Chem. Rev. 1994;94:759–805. doi:10.1021/cr00027a011 [Google Scholar]
- Ferreira K.N, Iverson T.M, Maghlaoui K, Barber J, Iwata S. Architecture of the photosynthetic oxygen-evolving center. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. doi:10.1126/science.1093087 [DOI] [PubMed] [Google Scholar]
- Gersten S.W, Samuels G.J, Meyer T.J. Catalytic oxidation of water by an oxo-bridged ruthenium dimer. J. Am. Chem. Soc. 1982;104:4029–4030. doi:10.1021/ja00378a053 [Google Scholar]
- Gilbert J.A, Eggleston D.S, Murphy W.R, Jr, Geselowitz D.A, Gersten S.W, Hodgson D.J, Meyer T.J. Structure and redox properties of the water-oxidation catalyst [(bpy)2(OH2)RuORu(OH2)(bpy)2]4+ J. Am. Chem. Soc. 1985;107:3855–3864. doi:10.1021/ja00299a017 [Google Scholar]
- Hanson R.S, Hanson T.E. Methanotrophic bacteria. Microbiol. Rev. 1996;60:439–471. doi: 10.1128/mr.60.2.439-471.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haumann M, Liebisch P, Muller C, Barra M, Grabolle M, Dau H. Photosynthetic O2 formation tracked by time-resolved X-ray experiments. Science. 2005;310:1019–1021. doi: 10.1126/science.1117551. doi:10.1126/science.1117551 [DOI] [PubMed] [Google Scholar]
- Hodgkiss J.M, Damrauer N.H, Pressé S, Rosenthal J, Nocera D.G. Electron transfer driven by proton fluctuations in a hydrogen-bonded donor–acceptor assembly. J. Phys. Chem. A. 2006;110:18 853–18 858. doi: 10.1021/jp056703q. [DOI] [PubMed] [Google Scholar]
- Holm R.H, Kennepohl P, Solomon E.I. Structural and functional aspects of metal sites in biology. Chem. Rev. 1996;96:2239–2314. doi: 10.1021/cr9500390. doi:10.1021/cr9500390 [DOI] [PubMed] [Google Scholar]
- Hosler J.P, Ferguson-Miller S, Mills D.A. Energy transduction: proton transfer through the respiratory complexes. Annu. Rev. Biochem. 2006;75:165–187. doi: 10.1146/annurev.biochem.75.062003.101730. doi:10.1146/annurev.biochem.75.062003.101730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen E.N, Pfaltz A, Yamamoto H, editors. Comprehensive asymmetric catalysis. vol. I–III. Springer; New York, NY: 1999. [Google Scholar]
- Jenkins D.M, Di Bilio A.J, Allen M.J, Betley T.A, Peters J.C. Elucidation of a low spin cobalt(II) system in a distorted tetrahedral geometry. J. Am. Chem. Soc. 2002;124:15 336–15 350. doi: 10.1021/ja026433e. doi:10.1021/ja026433e [DOI] [PubMed] [Google Scholar]
- Jordan A, Reichard P. Ribonucleotide reductases. Annu. Rev. Biochem. 1998;67:71–98. doi: 10.1146/annurev.biochem.67.1.71. doi:10.1146/annurev.biochem.67.1.71 [DOI] [PubMed] [Google Scholar]
- Kang S.O, Llinares J.M, Powell D, VanderVelde D, Bowman-James K. New polyamide cryptand for anion binding. J. Am. Chem. Soc. 2003;125:10 152–10 153. doi: 10.1021/ja034969+. doi:10.1021/ja034969+ [DOI] [PubMed] [Google Scholar]
- Kisker C, Schindelin H, Rees D.C. Molybdenum-cofactor-containing enzymes: structure and mechanism. Annu. Rev. Biochem. 1997;66:233–267. doi: 10.1146/annurev.biochem.66.1.233. doi:10.1146/annurev.biochem.66.1.233 [DOI] [PubMed] [Google Scholar]
- Lewis N.S, Nocera D.G. Powering the planet: chemical challenges in solar energy utilization. Proc. Natl Acad. Sci. USA. 2006;103:15 729–15 735. doi: 10.1073/pnas.0603395103. doi:10.1073/pnas.0603395103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman R.L, Rosenzweig A.C. Crystal structure of membrane-bound metalloenzymes that catalyses the biological oxidation of methane. Nature. 2005;434:177–182. doi: 10.1038/nature03311. doi:10.1038/nature03311 [DOI] [PubMed] [Google Scholar]
- Liu S.-Y, Nocera D.G. Hangman salophens. J. Am. Chem. Soc. 2005;127:5278–5279. doi: 10.1021/ja042849b. doi:10.1021/ja042849b [DOI] [PubMed] [Google Scholar]
- Loll B, Kern J, Saenger W, Zouni A, Biesiadka J. Towards complete cofactor arrangement in the 3.0 Å resolution of photosystem II. Nature. 2005;438:1040–1044. doi: 10.1038/nature04224. doi:10.1038/nature04224 [DOI] [PubMed] [Google Scholar]
- MacBeth C.E, Golombek A.P, Young V.G, Jr, Yang C, Kuczera K, Hendrich M.P, Borovik A.S. O2 activation by nonheme iron complexes: a monomeric Fe(III)-oxo complex derived from O2. Science. 2000;289:938–941. doi: 10.1126/science.289.5481.938. doi:10.1126/science.289.5481.938 [DOI] [PubMed] [Google Scholar]
- MacBeth C.E, Gupta R, Mitchell-Koch K.R, Young V.G, Jr, Lushington G.H, Thompson W.H, Hendrich M.P, Borovik A.S. Utilization of hydrogen bonds to stabilize M–O(H) units: synthesis and properties of monomeric iron and manganese complexes with terminal oxo and hydroxo ligands. J. Am. Chem. Soc. 2004;126:2556–2567. doi: 10.1021/ja0305151. doi:10.1021/ja0305151 [DOI] [PubMed] [Google Scholar]
- Malmström B.G. Cytochrome c oxidase as a redox-linked proton pump. Chem. Rev. 1990;90:1247–1260. doi:10.1021/cr00105a008 [PubMed] [Google Scholar]
- McEvoy J.P, Brudvig G.W. Water-splitting chemistry of photosystem II. Chem. Rev. 2006;106:4455–4483. doi: 10.1021/cr0204294. doi:10.1021/cr0204294 [DOI] [PubMed] [Google Scholar]
- McHatton R.C, Anson F.C. Electrochemical behavior of Ru(trpy)(bpy)(OH2)3+ in aqueous solution and when incorporated in nafion coatings. Inorg. Chem. 1984;23:3935–3942. doi:10.1021/ic00192a020 [Google Scholar]
- Messinger J, Badger M, Wydrzynski T. Detection of one slowly exchanging substrate water molecule in the S3 state of photosystem II. Proc. Natl Acad. Sci. USA. 1995;92:3209–3213. doi: 10.1073/pnas.92.8.3209. doi:10.1073/pnas.92.8.3209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier B, de Visser S.P, Shaik S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. doi:10.1021/cr020443g [DOI] [PubMed] [Google Scholar]
- Müller J, Pindur U. Reactions of electron-rich heterocycles with derivatives of carboxylic ortho acids, II: acid catalyzed reactions of 3-substituted indoles with ethyl orthoformates. Arch. Pharm. 1984;317:555–561. doi:10.1002/ardp.19843170612 [Google Scholar]
- Newcomb M, Toy P.H. Hypersensitive radical probes and the mechanisms of cytochrome P450-catalyzed hydroxylation reactions. Acc. Chem. Res. 2000;33:449–455. doi: 10.1021/ar960058b. doi:10.1021/ar960058b [DOI] [PubMed] [Google Scholar]
- Newcomb M, Zhang R, Chandrasena R.E.P, Halgrimson J.A, Horner J.H, Makris T.M, Sligar S.G. Cytochrome P450 compound I. J. Am. Chem. Soc. 2006;128:4580–4581. doi: 10.1021/ja060048y. doi:10.1021/ja060048y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nocera D.G. On the future of global energy. Daedalus. 2006;135:112–115. doi:10.1162/daed.2006.135.4.112 [Google Scholar]
- Odom A.L, Cummins C.C. Nitric oxide cleavage: synthesis of terminal chromium(VI) nitride complexes via nitrosyl deoxygenation. J. Am. Chem. Soc. 1995;117:6613–6614. doi:10.1021/ja00129a034 [Google Scholar]
- Pecoraro V.L, Baldwin M.J, Caudle M.T, Hsieh W.Y, Law N.A. A proposal for water oxidation in photosystem II. Pure Appl. Chem. 1998;70:925–929. [Google Scholar]
- Peloquin J.M, Britt R.D. EPR/ENDOR characterization of the physical and electronic structure of the OEC Mn cluster. Biochim. Biophys. Acta Bioenerg. 2001;1503:96–111. doi: 10.1016/s0005-2728(00)00219-x. doi:10.1016/S0005-2728(00)00219-X [DOI] [PubMed] [Google Scholar]
- Que L, Jr, Ho R.Y.N. Dioxygen activation by enzymes with mononuclear non-heme iron active sites. Chem. Rev. 1996;96:2607–2624. doi: 10.1021/cr960039f. doi:10.1021/cr960039f [DOI] [PubMed] [Google Scholar]
- Reece S.Y, Hodgkiss J.M, Stubbe J, Nocera D.G. Proton-coupled electron transfer: the mechanistic upderpinning for radical transport and catalysis in biology. Phil. Trans. R. Soc. B. 2006;361:1351–1364. doi: 10.1098/rstb.2006.1874. doi:10.1098/rstb.2006.1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieth R.D, Mankad N.P, Calimano E, Sadighi J.P. Palladium-catalyzed cross-coupling of pyrrole anions with aryl chlorides, bromides, and iodides. Org. Lett. 2004;6:3981–3983. doi: 10.1021/ol048367m. doi:10.1021/ol048367m [DOI] [PubMed] [Google Scholar]
- Rosenthal J, Chng L.L, Fried S.D, Nocera D.G. Stereochemical control of H2O2 dismutation by Hangman porphyrins. Chem. Commun. 2007:2642–2644. doi: 10.1039/b616884a. doi:10.1039/b616884a [DOI] [PubMed] [Google Scholar]
- Rosenzweig A.C, Sazinsky M.H. Structural insights into dioxygen-activating copper enzymes. Curr. Opin. Struct. Biol. 2006;16:729–735. doi: 10.1016/j.sbi.2006.09.005. doi:10.1016/j.sbi.2006.09.005 [DOI] [PubMed] [Google Scholar]
- Ruttinger W, Dismukes G.C. Synthetic water-oxidation catalysts for artificial photosynthetic water oxidation. Chem. Rev. 1997;97:1–24. doi: 10.1021/cr950201z. doi:10.1021/cr950201z [DOI] [PubMed] [Google Scholar]
- Schrock R.R. Transition metal complexes that contain a triamidoamine ligand. Acc. Chem. Res. 1997;30:9–16. doi:10.1021/ar950195t [Google Scholar]
- Sens C, Romero I, Rodriguez M, Llobet A, Parella T, Benet-Buchholz J. A new Ru complex capable of catalytically oxidizing water to molecular dioxygen. J. Am. Chem. Soc. 2004;126:7798–7799. doi: 10.1021/ja0486824. doi:10.1021/ja0486824 [DOI] [PubMed] [Google Scholar]
- Shaik S, Kumar D, De Visser S.P, Altun A, Thiel W. Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes. Chem. Rev. 2005;105:2279–2328. doi: 10.1021/cr030722j. doi:10.1021/cr030722j [DOI] [PubMed] [Google Scholar]
- Shi Y, Cao C, Odom A.L. Synthesis and group 4 complexes of tris(pyrrolyl-α-methyl)amine. Inorg. Chem. 2004;43:275–281. doi: 10.1021/ic035049v. doi:10.1021/ic035049v [DOI] [PubMed] [Google Scholar]
- Siegbahn P.E.M. O–O bond formation in the S4 state of the oxygen-evolving complex in photosystem II. Chem. Eur. J. 2006;12:9217–9227. doi: 10.1002/chem.200600774. doi:10.1002/chem.200600774 [DOI] [PubMed] [Google Scholar]
- Solomon E.I, Sundaram U.M, Machonkin T.E. Multicopper oxidases and oxygenases. Chem. Rev. 1996;96:2563–2605. doi: 10.1021/cr950046o. doi:10.1021/cr950046o [DOI] [PubMed] [Google Scholar]
- Sono M, Roach M.P, Coulter E.D, Dawson J.H. Heme-containing oxygenases. Chem. Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. doi:10.1021/cr9500500 [DOI] [PubMed] [Google Scholar]
- Soper J.D, Kryatov S.V, Rybak-Akimova E.V, Nocera D.G. Proton-directed redox control of O–O bond activation by heme hydroperoxidase models. J. Am. Chem. Soc. 2007;129:5069–5075. doi: 10.1021/ja0683032. doi:10.1021/ja0683032 [DOI] [PubMed] [Google Scholar]
- Stubbe J, van der Donk W.A. Protein radicals in enzyme catalysis. Chem. Rev. 1998;98:705–762. doi: 10.1021/cr9400875. doi:10.1021/cr9400875 [DOI] [PubMed] [Google Scholar]
- Stubbe J, Nocera D.G, Yee C.S, Chang M.C.Y. Radical initiation in the class I ribonucleotide reductase: long-range proton-coupled electron transfer? Chem. Rev. 2003;103:2167–2201. doi: 10.1021/cr020421u. doi:10.1021/cr020421u [DOI] [PubMed] [Google Scholar]
- Takeuchi K.J, Thompson M.S, Pipes D.W, Meyer T.J. Redox and spectral properties of monooxo polypyridyl complexes of ruthenium and osmium in aqueous media. Inorg. Chem. 1984;23:1845–1851. doi:10.1021/ic00181a014 [Google Scholar]
- Tommos C. Electron, proton and hydrogen-atom transfers in photosynthetic water oxidation. Phil. Trans. R. Soc. B. 2002;357:1383–1394. doi: 10.1098/rstb.2002.1135. doi:10.1098/rstb.2002.1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrettos J.S, Limburg J, Brudvig G.W. Mechanism of photosynthetic water oxidation: combining biophysical studies of photosystem II with inorganic model chemistry. Biochim. Biophys. Acta Bioenerg. 2001;1503:229–245. doi: 10.1016/s0005-2728(00)00214-0. doi:10.1016/S0005-2728(00)00214-0 [DOI] [PubMed] [Google Scholar]
- Wada T, Tsuge K, Tanaka K. Electrochemical oxidation of water to dioxygen catalyzed by the oxidized form of the bis(ruthenium-hydroxo) complex in H2O. Angew. Chem. Int. Ed. 2001a;39:1479–1482. doi: 10.1002/(sici)1521-3773(20000417)39:8<1479::aid-anie1479>3.0.co;2-4. doi:10.1002/(SICI)1521-3773(20000417)39:8<1479::AID-ANIE1479>3.0.CO;2-4 [DOI] [PubMed] [Google Scholar]
- Wada T, Tsuge K, Tanaka K. Syntheses and redox properties of bis(hydroxoruthenium) complexes with quinone and bipyridine ligands: water-oxidation catalysis. Inorg. Chem. 2001b;40:329–337. doi: 10.1021/ic000552i. doi:10.1021/ic000552i [DOI] [PubMed] [Google Scholar]
- Wallar B.J, Lipscomb J.D. Dioxygen activation by enzymes containing binuclear non-heme iron clusters. Chem. Rev. 1996;96:2625–2658. doi: 10.1021/cr9500489. doi:10.1021/cr9500489 [DOI] [PubMed] [Google Scholar]
- Watanabe Y. High-valent intermediates. In: Kadish K.M, Smith K.M, Guilard R, editors. The porphyrin handbook. vol. 4. Academic Press; San Diego, CA: 2000. pp. 97–117. [Google Scholar]
- Westphal K.L, Tommos C, Cukier R.I, Babcock G.T. Concerted hydrogen-atom abstraction in photosynthetic water oxidation. Curr. Opin. Plant Biol. 2000;3:236–242. [PubMed] [Google Scholar]
- Whittaker J.W. Free radical catalysis by galactose oxidase. Chem. Rev. 2003;103:2347–2363. doi: 10.1021/cr020425z. doi:10.1021/cr020425z [DOI] [PubMed] [Google Scholar]
- Wikström M. Cytochrome c oxidase: 25 years of the elusive proton pump. Biochim. Biophys. Acta. 2004;1655:241–247. doi: 10.1016/j.bbabio.2003.07.013. doi:10.1016/j.bbabio.2003.07.013 [DOI] [PubMed] [Google Scholar]
- Yang X, Baik M.-H. Electronic structure of the water-oxidation catalyst [(bpy)2(OHx)RuORu(OHy)(bpy)2]z+: weak coupling between the metal centers is preferred over strong coupling. J. Am. Chem. Soc. 2004;126:13 222–13 223. doi: 10.1021/ja0462427. doi:10.1021/ja0462427 [DOI] [PubMed] [Google Scholar]
- Yang X, Baik M.-H. cis,cis-[(bpy)2RuVO]2O4+ catalyzes water oxidation formally via in situ generation of radicaloid RuIV–O·. J. Am. Chem. Soc. 2006;128:7476–7485. doi: 10.1021/ja053710j. doi:10.1021/ja053710j [DOI] [PubMed] [Google Scholar]
- Yang J.Y, Nocera D.G. Catalase and epoxidation activity of manganese salen complexes bearing two xanthene scaffolds. J. Am. Chem. Soc. 2007;129:8192–8198. doi: 10.1021/ja070358w. doi:10.1021/ja070358w ASAP article. [DOI] [PubMed] [Google Scholar]
- Yang J.Y, Bachmann J, Nocera D.G. Hangman salen platforms containing two xanthene scaffolds. J. Org. Chem. 2006;71:8706–8714. doi: 10.1021/jo0613075. doi:10.1021/jo0613075 [DOI] [PubMed] [Google Scholar]
- Yano J, et al. Where water is oxidized to dioxygen: structure of the photosynthetic Mn4Ca cluster. Science. 2006;314:821–825. doi: 10.1126/science.1128186. doi:10.1126/science.1128186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh C.-Y, Chang C.J, Nocera D.G. Hangman porphyrins for the assembly of a model heme water channel. J. Am. Chem. Soc. 2001;123:1513–1514. doi: 10.1021/ja003245k. doi:10.1021/ja003245k [DOI] [PubMed] [Google Scholar]
- Yoon J, Solomon E.I. Electronic structures of exchange coupled trigonal trimeric Cu(II) complexes: spin frustration, antisymmetric exchange, pseudo-A terms, and their relation to O2 activation in the multicopper oxidases. Coord. Chem. Rev. 2007;251:379–400. doi:10.1016/j.ccr.2006.04.012 [Google Scholar]
- Zhang W, Loebach J.L, Wilson S.R, Jacobsen E.N. Enantioselective epoxidation of unfunctionalized olefins catalyzed by (salen)manganese complexes. J. Am. Chem. Soc. 1990;112:2801–2803. doi:10.1021/ja00163a052 [Google Scholar]
- Zong R, Thummel R.P. A new family of Ru complexes for water oxidation. J. Am. Chem. Soc. 2005;127:12 802–12 803. doi: 10.1021/ja054791m. doi:10.1021/ja054791m [DOI] [PubMed] [Google Scholar]