

Abstract
Photosystem II (PSII) uses light energy to split water into protons, electrons and O2. In this reaction, nature has solved the difficult chemical problem of efficient four-electron oxidation of water to yield O2 without significant amounts of reactive intermediate species such as superoxide, hydrogen peroxide and hydroxyl radicals. In order to use nature's solution for the design of artificial catalysts that split water, it is important to understand the mechanism of the reaction. The recently published X-ray crystal structures of cyanobacterial PSII complexes provide information on the structure of the Mn and Ca ions, the redox-active tyrosine called YZ and the surrounding amino acids that comprise the O2-evolving complex (OEC). The emerging structure of the OEC provides constraints on the different hypothesized mechanisms for O2 evolution. The water oxidation mechanism of PSII is discussed in the light of biophysical and computational studies, inorganic chemistry and X-ray crystallographic information.
Keywords: calcium, manganese, oxygen-evolving complex, photosystem II, water oxidation
1. Photosystem II and the oxygen-evolving complex
The photosynthetic protein complex photosystem II (PSII) is found in the thylakoid membranes of green plants, algae and oxyphotobacteria, this last group being dominated by the cyanobacteria. PSII is a multi-subunit membrane protein that acts as the first light-transducing complex in the redox pathway of oxygenic photosynthesis. It uses the energy of sunlight to oxidize water, producing dioxygen and protons, and reduces lipid-soluble plastoquinone. The water-derived electrons pass to the cytochrome b6f complex and photosystem I and are eventually used to reduce CO2 in the Calvin cycle. PSII is the source of nearly all of the O2 in the Earth's atmosphere and is, therefore, of considerable importance and practical interest.
Water is oxidized at the metal-containing oxygen-evolving complex (OEC) of PSII, which catalyses the four-electron reaction described in equation (1.1) as follows:
| (1.1) |
where PQ is plastoquinone and PQH2 is plastoquinol.
Protons are liberated from H2O and bound by PQ on opposite sides of the thylakoid membrane; the pH gradient thereby established contributes to the transmembrane free energy gradient used for ATP synthesis. The standard reduction potential of this reaction at pH 5.0, the pH at which the OEC typically operates, is +0.93 V. Because four electrons are transferred, this potential equates to a reaction free energy of 0.93 V×4=3.72 eV, or 359 kJ mol−1. The stringent energetic and catalytic requirements of this reaction are indicated by the fact that the OEC appears to be identical in every type of PSII, while nature is able to use a variety of metal catalysts for many other bioenergetic catalytic reactions (e.g. those performed by hydrogenases, nitrogenases and terminal oxidases).
Kok et al. (1970) established that the OEC accumulates oxidizing equivalents by proceeding through five redox states, commonly called ‘S states’, of which the most reduced during the catalytic cycle is S0 and the most oxidized is the transiently stable S4 (figure 1a). The S4 state oxidizes water and is itself reduced to the S0 state, almost certainly via one or more intermediates (Clausen & Junge 2004; Haumann et al. 2005).
Figure 1.
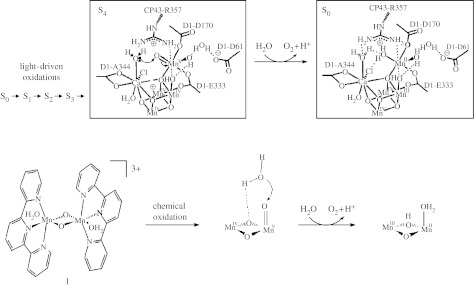
Structure-based mechanisms proposed for the oxidation of water by (a) PSII (McEvoy & Brudvig 2004) and by (b) a functional mixed-valent Mn2III/IV-terpy model complex (complex 1; Limburg et al. 2001).
In the past 6 years, three groups have published X-ray crystal structures of PSII at 3.8 Å (Zouni et al. 2001), 3.7 Å (Kamiya & Shen 2003), 3.5 Å (Ferreira et al. 2004) and 3.0 Å (Loll et al. 2005) resolution. The recent 3.5 Å crystal structure of Barber, Iwata and co-workers (Ferreira et al. 2004) is the first crystal structure of PSII that was refined to atomic resolution. The OEC is modelled as an Mn3CaO4 cuboid, with the fourth manganese ion attached to the exterior of the cuboid via a μ4 oxide ion at one of the cuboid corners (figure 1a). This so-called ‘dangler’ Mn ion (Mn(4)) is positioned so that its coordination sphere is close to that of calcium. It seems probable that the Mn(4)–Ca surface of the metal ion cluster is the area of catalytic activity. The modelled structure of the OEC is generally compatible with a large body of X-ray absorption fine structure (EXAFS) and electron paramagnetic resonance (EPR) data, computational models (Sproviero et al. 2005), and with site-directed mutagenesis studies (Debus 2001; Diner 2001), although there remain many questions of the exact structure of the manganese-oxo cluster and its ligation (McEvoy & Brudvig 2006).
The crystallographic model suggests a well-defined proton exit channel of hydrophilic residues leading from near Mn(4) to the lumenal exterior of PSII. Importantly, the channel is on the opposite side of the Mn4 cluster from YZ (D1-Tyr161) and leads directly away from YZ, which is shown as being hydrogen bonded to D1-His190 alone. This finding casts doubt on YZ's function as the catalytic base in water splitting (Hoganson & Babcock 1997) because the crystallographic evidence suggests that protons would be unable to escape from the YZ/H190 residue pair. Studies of the kinetics of proton release and electron transfer by Junge et al. (2002) also indicate that protons are not released from YZ into the lumen.
We have recently suggested that the arginine residue CP43-Arg357, found to be very close to the OEC, plays the role of the thermodynamically indispensable redox-coupled base (McEvoy & Brudvig 2004). The recent computational study of Knapp and co-workers (Ishikita et al. 2006) has concluded that the pKa of CP43-Arg357 is dramatically lowered by oxidation of the Mn cluster from the S0 state to the higher S states and also that a series of acid/base residues form a pathway for proton transfer leading away from CP43-Arg357 to the lumenal protein surface, in agreement with our proposal that CP43-Arg357 functions as the catalytic base in the OEC.
2. Proposed mechanisms for photosynthetic water oxidation
The mechanism of water oxidation by PSII remains unclear. Mass spectrometry measurements show that none of the lower S states (S0 through S3) contain a non-exchangeable form of water, a result that seems to rule out O–O bond formation before the S4 state is produced (Hillier et al. 1998; Hendry & Wydrzynski 2002; Hillier & Wydrzynski 2004).
There have been a large number of proposals for the mechanism of O–O bond formation since the first modern ‘molecular’ mechanism was proposed by Brudvig & Crabtree (1986); most of the earlier proposals lack molecular detail and are reviewed by Volkov (1989). Proposals for the mechanism of O–O bond formation can be grouped into three major categories based on the nature of the bound substrate water molecules (Hillier & Messinger 2005; McEvoy & Brudvig 2006). The earliest proposals for the mechanism of O–O bond formation involved the coupling of Mn-bridging oxo (μ-oxo) ligands (Brudvig & Crabtree 1986; Vincent & Christou 1987) and some current proposals fall in this category (Ruettinger & Dismukes 2000). Following from evidence that Mn-centred oxidation may not occur in the S2→S3-state transition, mechanisms have been proposed involving coupling reactions of an oxyl radical (Yachandra et al. 1996; Siegbahn 2000; Dau et al. 2001; Messinger 2004). The third category of mechanisms for water oxidation involves nucleophilic attack of a calcium-bound water (or hydroxide) ligand on the electrophilic oxygen atom of an MnV=O intermediate in the O–O bond-forming step (Pecoraro et al. 1998; Szalai et al. 1998). Related proposals involve coupling of an Mn-bound hydroxide and an Mn=O species (Hoganson & Babcock 1997; Hillier & Wydrzynski 2000).
Based on a consideration of both the available biophysical data and the inorganic chemistry of Mn, we proposed a detailed mechanism for the S-state cycle in which O–O bond formation occurs in the S4 state through nucleophilic attack of a calcium-bound water on the electrophilic oxygen atom of an MnV=O species (Limburg et al. 1999a; Vrettos et al. 2001a). Mass spectrometry measurements using H218O corroborate calcium's role in substrate water binding (Hendry & Wydrzynski 2003). The structural model of Ferreira et al. (2004) also fits well with our proposed mechanism. We have extended our mechanistic proposal based on the modelled structure of the OEC; the O–O bond-forming step is shown in figure 1 (McEvoy & Brudvig 2004). In order to test this and other proposed mechanisms, we have investigated the kinetics of ligand exchange in a series of inorganic Mn model complexes, the properties of calcium-substituted PSII, and the mechanism of water oxidation by the mixed-valent Mn2III/IV-terpy complex (complex 1 shown in figure 1; terpy=2,2′: 6,2″-terpyridine). These studies provide insight into the water oxidation chemistry of PSII and are described in §§3–5.
3. Substrate binding to the oxygen-evolving complex
Proposed molecular mechanisms of water oxidation by the OEC must specify the timing and nature of substrate (water) binding to the catalytic centre. The timing of water binding is specified by the S states (S0 through S4) of the OEC at which substrate water binds. The nature of binding is determined by the site and mode of binding; for example, terminal aqua or hydroxo ligands bound to a single Mn or Ca atom (Hoganson & Babcock 1997; Pecoraro et al. 1998; Szalai et al. 1998; Hillier & Wydrzynski 2001; McEvoy & Brudvig 2004), or μ-oxo or μ-hydroxo bridges between metal centres (Brudvig & Crabtree 1986; Vincent & Christou 1987; Pecoraro et al. 1994; Yachandra et al. 1996; Nugent et al. 2001; Dasgupta et al. 2004; Messinger 2004; Isobe et al. 2005; Siegbahn & Lundberg 2005). Important constraints have been provided on the substrate binding to the OEC by isotope exchange experiments between bulk water and evolved oxygen in PSII preparations (Messinger et al. 1995; Hillier et al. 1998; Hillier & Wydrzynski 2000, 2004; Hendry & Wydrzynski 2002, 2003), as well as from studies of proton release from the OEC during the S-state cycle (Junge et al. 2002). However, even mechanisms proposed after the availability of the recent PSII crystal structures disagree on the mode of water binding to the OEC; some authors favour terminal water binding (McEvoy & Brudvig 2004; Isobe et al. 2005), and others invoke conversion of the substrate water into a μ-O bridge (Dasgupta et al. 2004; Messinger 2004; Siegbahn & Lundberg 2005). In order for the μ-O bridges in the OEC to function as binding sites for substrate waters, the substrate water must be able to form a μ-O bridge and the μ-O moiety must exchange with bulk water on a time scale faster than or equal to the time scale of OEC turnover. Measurement of the latter time scale has been provided by the 18O isotope exchange experiments on the OEC referred to above.
Unfortunately, there has been a dearth of information on the rates of ligand exchange in oxo-Mn model complexes to aid in the interpretation of data on substrate exchange in the OEC. In particular, it has not been clear whether or not μ-oxo ligands could exchange with bulk water on a fast enough time scale in the OEC. Using time-resolved electrospray mass spectrometry, we recently determined the rates of isotope exchange between μ-O bridges and 18O-labelled water in a series of di-μ-O dimanganese complexes that are structural models for the OEC (Tagore et al. 2006, 2007).
A comparison of the ligand exchange rates for different Mn model complexes showed that: (i) all MnIV complexes exchange ligands much more slowly than mixed-valence complexes in which the Mn ions can switch between slow-exchanging MnIV and fast-exchanging MnIII states, (ii) terminal water ligands exchange very much faster than μ-O ligands, irrespective of the Mn oxidation states, (iii) the availability of a terminal water-binding site on manganese greatly enhances the μ-O exchange rate, and (iv) the structure, nuclearity and ancillary ligands (other than water) of the Mn complexes do not significantly affect the μ-O exchange rates. These factors place significant constraints on the interpretation of the substrate exchange rates in the OEC in terms of specific modes of water binding to the Mn and Ca ions.
The absolute rates measured in Mn model complexes (Tagore et al. 2006) can be compared with the fast and slow isotope exchange rates measured in PSII samples (Messinger et al. 1995; Hillier et al. 1998; Hillier & Wydrzynski 2000, 2004; Hendry & Wydrzynski 2002, 2003). The S1 and S2 oxidation states of the OEC are widely accepted to be and MnIII, respectively (McEvoy & Brudvig 2006). The exchange rates in mixed-valent complexes are, therefore, considered to be reasonable estimates for the exchange rates in the S1 and S2 states of the OEC. The fast exchange rates in the S1 and S2 states are approximately 105 times greater than the μ-O exchange rate in the mixed-valent -terpy complex 1 (figure 1b). The rate of fast exchange in the OEC is smallest in the S3 state, but is still approximately 103 times greater than in complex 1. Thus, it is very unlikely that the fast isotope exchange rates measured in the OEC are μ-O exchange rates.
The slow isotope exchange rates measured in the S0, S2 and S3 states are approximately 800–4000 times greater than in complex 1 and it is, therefore, also very unlikely that the slow isotope exchange rates measured in the S0, S2 and S3 states of the OEC are μ-O exchange rates. The slow exchange rate in the S1 state of the OEC, however, approaches the μ-O exchange rate for complex 1, being approximately eight times greater. The activation energy for the slow exchange measured in the S1 state is approximately 83 kJ mol−1 (Hillier & Wydrzynski 2004), which is close to the activation energy for μ-O exchange observed for complex 1 (approx. 84 kJ mol−1; Tagore et al. 2007). Thus, the slow-exchanging substrate in the S1 state could be bound as a bridging oxo.
From the perspective of metal oxidation states, the direct comparison of μ-O exchange rates between complex 1 and the OEC is reasonable. However, differences in other factors need to be considered to make a rigorous comparison between the μ-O exchange rates in the OEC and complex 1, such as the dielectric surrounding the OEC, the accessibility of water to the OEC and the functional groups from the protein surrounding the active site, which may be difficult to investigate by a study of model complexes. There is one other system where a comparison can be made between the rates of μ-O ligand exchange in a protein active site with model complexes in solution. The μ-O exchange rate measured for the Fe-(μ-O)-Fe centre of ribonucleotide reductase (Sjoberg et al. 1982) is approximately 8×10−4 s−1. This value is approximately 104 times smaller than the rates of μ-O exchange measured in solution (approx. 1–40 s−1) for inorganic Fe-(μ-O)-Fe porphyrin complexes (Fleischer et al. 1971) and μ-O-bridged Fe(III) dimers coordinated to EDTA and related chelators that are structurally similar to the active site in ribonucleotide reductase (Wilkins & Yelin 1969). The possibility of a similar disparity in the comparison of complex 1 and the OEC should be kept in mind, considering the restricted and rigidly structured nature of the OEC active site, which could inhibit exchange. This would make it even less probable that the substrates are bound to the OEC as μ-O ligands.
The mechanism of μ-O exchange has been studied for di-μ-O dimanganese complexes with and without terminal water-binding sites in order to determine how the rate of μ-O exchange is enhanced in Mn complexes that have terminal water ligands. It was found that the acidic terminal water ligand serves as the proton donor to the μ-O species in the first step of the exchange mechanism (Tagore et al. 2007). The absence of terminal water-binding sites on manganese inhibits μ-O exchange because, in this case, one of the ancillary ligands must dissociate to open up a site for a terminal water ligand to bind (Tagore et al. 2007). Dissociation of ancillary Mn ligands from protein residues seems unlikely in the OEC and, therefore, μ-O exchange would not readily occur in the OEC unless terminally bound waters are present. Thus, if the slow-exchanging substrate in the S1 state of the OEC were to be bound as a μ-O ligand, the OEC would also need to contain terminal water ligands in order to account for the rapid substrate exchange rates.
On the basis of the mechanistic insight gained from studies of Mn model complexes, it can be concluded that the fast-exchanging substrate in the OEC and the slow-exchanging substrate in the S0, S2 and S3 states are terminally bound water or hydroxide ligands. A μ-O-binding mode remains a possibility for the slow-exchanging substrate in the S1 state, with the constraint that the exchange of such a μ-O substrate would need to be facilitated by an adjacent fully protonated water terminally bound to one of the Mn ions in the OEC. Overall, the recent studies of ligand exchange rates in Mn model complexes argue against mechanisms of O2 evolution by the OEC in which the substrate waters are bound as μ-O bridges between Mn atoms.
4. Role of calcium in the oxygen-evolving complex
One prediction of the model shown in figure 1 is that Ca2+ functions as a Lewis acid in the OEC. In order to test this idea, we have studied the binding of a series of cations to the Ca2+ site in PSII (Vrettos et al. 2001b).
Considering the large number of metal ions that will compete for the Ca2+-binding site in PSII, it is surprising that only Ca2+ and Sr2+ support O2 evolution. It would be expected that if the role of Ca2+ in PSII is purely structural, then metal ions of the same size and charge should be functional replacements. For example, Cd2+(0.97 Å) is almost the same size as Ca2+(0.99 Å), carries the same charge, is a closed-shell ion and binds to PSII with an affinity that is comparable to that of Ca2+. Moreover, Cd2+ replaces Ca2+ in other proteins without large structural perturbations and even preserves hydrogen-bonding networks (McPhalen et al. 1991; Bouckaert et al. 2000). If the role of Ca2+ in the OEC is purely structural, then Cd2+ should function too. However, Cd2+-substituted PSII is inactive. What is the distinguishing factor among the metal ions that determines the functional competence of Ca2+ and Sr2+ over other cations that can bind in the Ca2+-binding site in PSII? Values of ionic radii and pKas of the aqua ions are tabulated in table 1; the ions discussed in this section (Ca2+, Sr2+and Cd2+) are highlighted in bold. Owing to its larger size, Sr2+ is not a particularly good match to the size of the Ca2+-binding site in PSII and does not bind to PSII as tightly as Ca2+ (Vrettos et al. 2001b). However, among the cations that can bind with reasonable affinity to the Ca2+-binding site in PSII, only Sr2+ has a pKa of its aqua ion close to that of Ca2+. We postulate that only Sr2+ can functionally substitute for Ca2+ because it is the only cation that can bind with reasonable affinity and whose Lewis acidity is well matched to that of Ca2+. Other metal ions fail to support O2 evolution because either their ionic radius is too large (or too small) to allow good binding, or their Lewis acidity is out of the range required for activity, which makes the coordinated water too strong (or weak) a Brønsted acid. The result of a mismatch in the Lewis acidity is that the H-bonding network of the OEC is disturbed, and the coordinated water either deprotonates to form an unreactive metal cation-bound OH− (too strong a Brønsted acid) or cannot be readily deprotonated in the O–O bond-forming reaction (too weak a Brønsted acid).
Table 1.
Ionic radii and pKas of the aqua ions of metal cations.
| metal ion | ionic radius (Å)a | pKa of aqua ionb |
|---|---|---|
| monovalent cations | ||
| Na+ | 0.97 | 14.77 |
| K+ | 1.33 | 16 |
| Cs+ | 1.67 | >17 |
| divalent cations | ||
| Mg2+ | 0.66 | 11.41 |
| Ni2+ | 0.69 | 9.86 |
| Cu2+ | 0.72 | 8.00 |
| Co2+ | 0.72 | 9.85 |
| Cd2+ | 0.97 | 9.00 |
| Ca2+ | 0.99 | 12.80 |
| Sr2+ | 1.12 | 13.18 |
| Ba2+ | 1.34 | 13.36 |
| trivalent cations | ||
| Lu3+ | 0.85 | 7.94 |
| Dy3+ | 0.91 | 8.10 |
| Gd3+ | 0.92 | 9.78 |
| Pr3+ | 1.01 | 8.91 |
| La3+ | 1.02 | 8.82 |
Data from Weast (1978).
Data from Dean (1985).
There are two possibilities for the protonation state of the Ca2+-bound water, either OH− or H2O. When buried in the hydrophobic interior of a protein, the pKa of water bound to a metal ion is usually lowered several units; nonetheless, the relative ordering of the Lewis acidity of the metal ions in table 1 will remain the same. Taking this into consideration, the Ca2+ aqua ion is still a weak Lewis acid (pKa=12.8) compared with the other metal ions in table 1, and so we expect that H2O, and not OH−, is the form of the Ca2+-bound substrate molecule. Sr2+ is the only other catalytically competent metal ion because its pKa is sufficiently high that it provides H2O and not OH− as a ligand. The conclusion that a Ca2+-OH2 species is present in the OEC, rather than a Ca2+-hydroxide species, fits with the dependence of cation binding on ionic radius (Vrettos et al. 2001b). It was found that both the divalent and trivalent cations bind equivalently for a given ionic radius, which was interpreted to mean that they have the same charge. This can be explained if the trivalent metal ions, which are strong Lewis acids, bind as a metal ion–hydroxide species and that the divalent metal ions bind as a metal ion–water species, both of which have a+2 charge. This has also been proposed to account for structural effects on the tetramanganese cluster induced by substituting Dy3+ for Ca2+ in the OEC (Riggs-Gelasco et al. 1996).
If the role of Ca2+ as a Lewis acid is only required in the O–O bond-forming reaction that occurs during the S3→S0 transition, then it may be expected that other metal cations could functionally substitute for Ca2+ in the earlier S-state transitions. Indeed, in recent work (Lee & Brudvig 2007), it was found that the S2-state multiline EPR signal is formed in Dy3+- and Cd2+-inhibited PSII by 200 K illumination of dark-adapted (S1 state) samples (Brudvig et al. 1983). This observation provides direct support for the proposal that Ca2+ plays a structural role in the early S-state transitions, which can be fulfilled by other cations of similar ionic radius, and that the functional role of Ca2+ to activate water in the O–O bond-forming reaction that occurs in the final step of the S-state cycle can only be fulfilled by Ca2+ and Sr2+, which have similar Lewis acidities.
These results suggest that Ca2+ is not only a structural cofactor in the OEC, but also directly involved in the chemistry of water oxidation as a Lewis acid. They provide good support for the proposed functional role of Ca2+ as a Lewis acid to bind a substrate water molecule and tune its nucleophilic reactivity.
5. Electron transfer between manganese ions in the oxygen-evolving complex
Complex 1 is a water oxidation catalyst in aqueous solution when O-atom transfer reagents such as potassium hydroperoxymonosulphate (KHSO5) or sodium hypochlorite (NaOCl) are used as the primary oxidant (figure 1b) and, thus, provides a functional model system for the oxygen-evolving complex of PSII (Limburg et al. 1999b, 2001). Based on steady-state kinetics and 18O isotope labelling studies, the reaction was shown to exhibit saturation (Michaelis–Menten-like) kinetics and to involve a high-valent Mn species that could exchange with solvent 18OH2 (Limburg et al. 2001). A mechanism was proposed to explain these observations in which an MnV=O intermediate reacts with water to form the O2 product, as shown in figure 1b.
In order to investigate the mechanism of the water oxidation reaction and to characterize the intermediates, we have studied the reaction of complex 1 with HSO5− by using EPR, UV–visible and rapid-mix X-ray spectroscopy (Chen et al. 2007) and by stopped-flow UV–visible spectroscopy (Tagore et al. in press). Conversion of the mixed-valence [MnIII(μ-O)2MnIV]3+ complex (1) to its one-electron oxidized product, [MnIV(μ-O)2MnIV]4+(2) occurs within seconds and in high yield.
With excess HSO5−, the conversion of 1 into 2 is monophasic. The rate of the reaction is first order in [1] and nearly zero order in [HSO5−]. These observations are consistent with a reaction that involves the two-electron oxidation of 1 by HSO5− to form a [MnIV(μ-O)2MnV=O]3+ intermediate, followed by rapid reaction of the two-electron oxidized intermediate with another molecule of 1 to give two molecules of 2 (equations (5.1) and (5.2)).
| (5.1) |
| (5.2) |
However, in the stopped-flow UV–visible spectroscopic measurements, it was found that the reaction is distinctly biphasic for low concentrations of HSO5− and becomes monophasic at higher concentrations of HSO5−. The data are well fit by a model in which redox chemistry proceeds only when HSO5− is bound to MnIII and that interconversion of HSO5− bound to the MnIV and MnIII sites is slow. The kinetic distinctness of the MnIII and MnIV sites allows estimates to be made on the upper limits for the rates of intramolecular electron transfer between MnIV and MnIII sites of an oxo-manganese complex.
It has generally been assumed that the rate of electron transfer between Mn ions in the OEC is much faster than the chemical steps in the water oxidation reaction. However, the water oxidation site in the OEC is probably a manganese ion that is more oxidized than the neighbouring manganese sites. In order to prevent the dissipation of oxidizing power, electron transfer between the water-oxidizing manganese and its neighbours may be required to be slow on the time scale of the water oxidation step. The water oxidation site has been variously proposed to be an MnIV=O adjacent to an MnIII site in the S4 state (Hillier & Wydrzynski 2001), an MnIV=O· adjacent to an MnIII site in the S3 state (Siegbahn & Crabtree 1999), and an MnV=O moiety adjacent to MnIV sites in the S4 state (Pecoraro et al. 1998; McEvoy & Brudvig 2004; Isobe et al. 2005; Sproviero et al. 2005). In the former cases, a high reorganization barrier is expected for electron transfer between the water-oxidizing MnIV site and the Jahn–Teller distorted MnIII site. In the latter case, an octahedral MnV water-oxidizing site would lead to a lower reorganization barrier due to a lower degree of Jahn–Teller distortion in MnV as compared with MnIII, but electron exchange between a square pyramidal MnV (a probable geometry for an MnV=O moiety; Collins & Gordon-Wylie 1989; Collins et al. 1990; MacDonnell et al. 1994) and an adjacent octahedral MnIV would again lead to high reorganization barriers. In all cases, the manganese with the oxo or oxyl radical bound to it would be expected to have a preference for the higher-oxidation state, leading to slow electron exchange. The effects of Jahn–Teller distortion and non-identical ligands on the exchanging partners are, therefore, expected to be operative for electron exchange in the OEC.
Intramolecular electron transfer between MnIII and MnIV is required to be slow in order to account for biphasic kinetics of the reaction of 1 with HSO5−. The maximum electron-transfer rate constant for the interconversion of HSO5− bound to the MnIV and MnIII sites on 1 is 5 s−1 (t1/2≥140 ms; Tagore et al. in press). This can be compared with the measured lifetimes of approximately 200 μs and approximately 1 ms for the S3 and S4 states, respectively, of the OEC (Haumann et al. 2005). Rates for intramolecular electron transfer in oxo-bridged manganese complexes are not available, but an upper limit of 106–108 s−1 has been proposed for the mixed-valent [Mn2III/IV(O)2(bpy)4]3+ complex (bpy=2,2′-bipyridine) on the basis of its EPR and near-IR absorption spectra (Cooper & Calvin 1977; Cooper et al. 1978). Barriers ranging from approximately 13 to approximately 19 kcal mol−1 have been calculated by density functional theory for the same process for MnIII(μ-O)2MnIV complexes with water, hydroxo and formate ligands (Lundberg & Siegbahn 2005). Assuming a pre-exponential factor of kBT/h, these barriers translate to rates of 2400–0.1 s−1. From these observations, it is apparent that slow electron transfer between Mn ions in the OEC could lead to the existence of a localized water oxidation site on a specific manganese in the OEC and this may be important for the mechanism of water oxidation catalysis.
6. Summary and conclusions
As discussed in this article, studies of inorganic Mn model complexes provide important information on the structure and properties of high-valent oxo-manganese complexes, the rates of ligand exchange and the rates of intramolecular electron transfer. This information helps to define the reactions of the OEC leading to water oxidation. There remain many unanswered questions about the structure and function of the Mn4Ca cluster in the OEC. However, a body of evidence provides strong support for binding of the substrate water molecules as terminal ligands to manganese and/or calcium and for a direct role of Ca2+ in the water-oxidation chemistry as a Lewis acid to activate a substrate water molecule as a nucleophile. Mn model chemistry also supports the possibility that water is activated for O–O bond formation in the OEC by binding to a high-valent manganese ion that is more oxidized than its neighbouring manganese sites and that this high-valent Mn does not dissipate its oxidizing power by electron transfer to its neighbouring Mn ions because the inter-Mn electron-transfer reactions are slow on the time scale of the water-oxidation step. Overall, these results are consistent with a mechanism for photosynthetic water oxidation that involves nucleophilic attack of a calcium-bound water ligand on the electrophilic oxygen atom of an MnV=O intermediate in the O–O bond-forming step, as shown in figure 1.
Discussion
V. Pecoraro (University of Michigan, USA). Firstly, I would like to comment that the rate of oxo exchange for di-μ-oxo-MnIII,IV complexes is facilitated by disproportionation. This is unavailable in a protein; therefore, oxo transfer will be even slower. My question is, under what condition were pKa values determined?
G. Brudvig. All of the pKa values I discussed in my talk were determined by pH-dependent measurements in aqueous solutions.
H. Dau (Freie University, Berlin, Germany). You suggested that in an MnIII–MnIV complex the inner-molecule electron transfer may be slow. Why is it so slow? Is the rate limited by ligand movements (or proton movements)?
G. Brudvig. The reaction involves slow conversion of the unreactive H2O-MnIII(O)2MnIV-SO5H-terpy complex into the reactive HSO5-MnIII(O)2MnIV-OH2 complex. Although this reaction could occur by electron transfer from MnIII to MnIV, the electron-transfer reaction will be significantly uphill owing to the asymmetric ligation. I expect that the reaction is limited by ligand movements.
A. Aukauloo (University of Paris-Sud, France). Comparing the mechanisms of formation of the oxygen–oxygen bond, in the natural system you have an Mn-oxyl (terminal oxo) radical that gets attacked by an activated water molecule having some spin density through hydrogen bonding to a μ-oxo fragment. In the synthetic model, you have an Mn(V) oxo low spin or an Mn-oxyl (terminal oxo) type fragment. If in the first case, an activated water molecule comes and attacks the electrophilic oxo fragment, then how do you get triplet dioxygen out? In the second case, the activated water molecule must be a more radical type to do the O–O bond. Can you comment on this?
G. Brudvig. We still do not have any direct information on the electronic state of the Mn-oxo species involved in the O–O bond-forming step for either the OEC or synthetic models. Whether or not it is an Mn-oxyl radical species remains to be determined. Perhaps DFT calculations will clarify this question once a complete atomic-resolution structure of the OEC has been determined. As for formation of triplet oxygen, this is an important question, but I expect that intersystem crossing will be very fast for oxygen bound to paramagnetic manganese complexes.
D. Nocera (MIT, Boston, USA). Do you have an estimate for the reorganization energy of MnIII→MnIV transfers versus electron exchange for a possible MnIV→MnV transfer? How does this play into your ideas of electron localization within the OEC cluster, as it pertains to your model?
G. Brudvig. This will depend greatly on the coordination environments of the Mn ions. For asymmetric ligation, as is probably to be the case in the OEC, the reactions probably will be limited by ligand movements.
P. Siegbalm (Stockholm University, Sweden). I have tested your mechanism with an attack on a terminal Mn=O by a water bound to calcium (Siegbahn 2006). Among the ones I tried, this mechanism has one of the highest barriers (32 kcal mol−1). Actually, an attack by a second-sphere water has a much lower barrier (approx. 20 kcal mol−1). However, by far the lowest barrier is found for an attack by an oxyl radical bound to the dangling manganese on an oxo ligand in the cube. The barrier for this mechanism is as low as 5 kcal mol−1 for the latest Berlin X-ray structure.
J. Messinger (Muelheim, Germany). Your mass spec data on water exchange in model systems are done in acetonitrile with only a few per cent of water. Can you give us an estimate what the actual water exchange rates would be in pure water?
G. Brudvig. There is a first-order dependence of the rate of μ-oxo exchange on the water concentration in acetonitrile solution for concentrations of water up to 0.5 M. We have not measured the rate of exchange in pure water because a rapid mixing experiment cannot be done in this case. However, I do not think an extrapolation of the μ-oxo exchange rates for Mn complexes measured with low water concentrations in acetonitrile solution to pure water will give a valid comparison to the substrate exchange data for PSII because it is very likely that the substrate exchange rates for the protein become saturated with fairly low concentrations of water.
Acknowledgments
Support from the National Institutes of Health (GM32715) is gratefully acknowledged.
Footnotes
One contribution of 20 to a Discussion Meeting Issue ‘Revealing how nature uses sunlight to split water’.
References
- Bouckaert J, Loris R, Wyns L. Zinc/calcium- and cadmium/cadmium-substituted concanavalin A: interplay of metal binding, pH and molecular packing. Acta Cryst. D. 2000;56:1569–1576. doi: 10.1107/s0907444900013342. doi:10.1107/S0907444900013342 [DOI] [PubMed] [Google Scholar]
- Brudvig G.W, Crabtree R.H. Mechanism for photosynthetic O2 evolution. Proc. Natl Acad. Sci. USA. 1986;83:4586–4588. doi: 10.1073/pnas.83.13.4586. doi:10.1073/pnas.83.13.4586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudvig G.W, Casey J.L, Sauer K. The effect of temperature on the formation and decay of the multiline EPR signal species associated with photosynthetic oxygen evolution. Biochim. Biophys. Acta. 1983;723:366–371. doi:10.1016/0005-2728(83)90042-7 [Google Scholar]
- Chen H.Y, Tagore R, Olack G, Vrettos J.S, Weng T.C, Penner-Hahn J.E, Crabtree R.H, Brudvig G.W. Speciation of the catalytic oxygen evolution system: [Mn2III/IV(μ-O)2(terpy)2(H2O)2](NO3)3+ HSO5−. Inorg. Chem. 2007;46:34–43. doi: 10.1021/ic060499j. doi:10.1021/ic060499j [DOI] [PubMed] [Google Scholar]
- Clausen J, Junge W. Detection of an intermediate of photosynthetic water oxidation. Nature. 2004;430:480–483. doi: 10.1038/nature02676. doi:10.1038/nature02676 [DOI] [PubMed] [Google Scholar]
- Collins T.J, Gordon-Wylie S.W. A manganese(V)-oxo complex. J. Am. Chem. Soc. 1989;111:4511–4513. doi:10.1021/ja00194a063 [Google Scholar]
- Collins T.J, Powell R.D, Slebodnick C, Uffelman E.S. A water-stable manganese(V)-oxo complex—definitive assignment of a ν(Mn(V)O) infrared vibration. J. Am. Chem. Soc. 1990;112:899–901. doi:10.1021/ja00158a077 [Google Scholar]
- Cooper S.R, Calvin M. Mixed-valence interactions in di-μ-oxo bridged manganese complexes. J. Am. Chem. Soc. 1977;99:6623–6630. doi:10.1021/ja00462a025 [Google Scholar]
- Cooper S.R, Dismukes G.C, Klein M.P, Calvin M. Mixed valence interactions in di-μ-oxo bridged manganese complexes. Electron paramagnetic resonance and magnetic susceptibility studies. J. Am. Chem. Soc. 1978;100:7248–7252. doi:10.1021/ja00491a021 [Google Scholar]
- Dasgupta J, van Willigen R.T, Dismukes G.C. Consequences of structural and biophysical studies for the molecular mechanism of photosynthetic oxygen evolution: functional roles for calcium and bicarbonate. Phys. Chem. Chem. Phys. 2004;6:4793–4802. doi:10.1039/b408270b [Google Scholar]
- Dau H, Iuzzolino L, Dittmer J. The tetra-manganese complex of photosystem II during its redox cycle—X-ray absorption results and mechanistic implications. Biochim. Biophys. Acta. 2001;1503:24–39. doi: 10.1016/s0005-2728(00)00230-9. doi:10.1016/S0005-2728(00)00230-9 [DOI] [PubMed] [Google Scholar]
- Dean J.A, editor. Lange's handbook of chemistry. McGraw-Hill; New York, NY: 1985. [Google Scholar]
- Debus R.J. Amino acid residues that modulate the properties of tyrosine YZ and the manganese cluster in the water oxidizing complex of photosystem II. Biochim. Biophys. Acta. 2001;1503:164–186. doi: 10.1016/s0005-2728(00)00221-8. doi:10.1016/S0005-2728(00)00221-8 [DOI] [PubMed] [Google Scholar]
- Diner B.A. Amino acid residues involved in the coordination and assembly of the manganese cluster of photosystem II. Proton-coupled electron transport of the redox-active tyrosines and its relationship to water oxidation. Biochim. Biophys. Acta. 2001;1503:147–163. doi: 10.1016/s0005-2728(00)00220-6. doi:10.1016/S0005-2728(00)00220-6 [DOI] [PubMed] [Google Scholar]
- Ferreira K.N, Iverson T.M, Maghlaoui K, Barber J, Iwata S. Architecture of the photosynthetic oxygen-evolving center. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. doi:10.1126/science.1093087 [DOI] [PubMed] [Google Scholar]
- Fleischer E.B, Palmer J.M, Srivastava T.S, Chatterjee A. Thermodyamic and kinetic properties of an iron-porphyrin system. J. Am. Chem. Soc. 1971;93:3162–3167. doi: 10.1021/ja00742a012. doi:10.1021/ja00742a012 [DOI] [PubMed] [Google Scholar]
- Haumann M, Liebisch P, Muller C, Barra M, Grabolle M, Dau H. Photosynthetic O2 formation tracked by time-resolved X-ray experiments. Science. 2005;310:1019–1021. doi: 10.1126/science.1117551. doi:10.1126/science.1117551 [DOI] [PubMed] [Google Scholar]
- Hendry G, Wydrzynski T. The two substrate–water molecules are already bound to the oxygen-evolving complex in the S2 state of photosystem II. Biochemistry. 2002;41:13 328–13 334. doi: 10.1021/bi026246t. doi:10.1021/bi026246t [DOI] [PubMed] [Google Scholar]
- Hendry G, Wydrzynski T. 18O isotope exchange measurements reveal that calcium is involved in the binding of one substrate-water molecule to the oxygen-evolving complex in photosystem II. Biochemistry. 2003;42:6209–6217. doi: 10.1021/bi034279i. doi:10.1021/bi034279i [DOI] [PubMed] [Google Scholar]
- Hillier W, Messinger J. Mechanism of photosynthetic oxygen production. In: Wydrzynski T.J, Satoh K, editors. Photosystem II: the light-driven water : plastoquinone oxidoreductase. Springer; Dordrecht, The Netherlands: 2005. pp. 567–608. [Google Scholar]
- Hillier W, Wydrzynski T. The affinities for the two substrate water binding sites in the O2 evolving complex of photosystem II vary independently during S-state turnover. Biochemistry. 2000;39:4399–4405. doi: 10.1021/bi992318d. doi:10.1021/bi992318d [DOI] [PubMed] [Google Scholar]
- Hillier W, Wydrzynski T. Oxygen ligand exchange at metal sites — implications for the O2 evolving mechanism of photosystem II. Biochim. Biophys. Acta. 2001;1503:197–209. doi: 10.1016/s0005-2728(00)00225-5. doi:10.1016/S0005-2728(00)00225-5 [DOI] [PubMed] [Google Scholar]
- Hillier W, Wydrzynski T. Substrate water interactions within the photosystem II oxygen evolving complex. Phys. Chem. Chem. Phys. 2004;6:4882–4889. doi:10.1039/b407269c [Google Scholar]
- Hillier W, Messinger J, Wydrzynski T. Kinetic determination of the fast exchanging substrate water molecule in the S3 state of photosystem II. Biochemistry. 1998;37:16 908–16 914. doi: 10.1021/bi980756z. doi:10.1021/bi980756z [DOI] [PubMed] [Google Scholar]
- Hoganson C.W, Babcock G.T. A metalloradical mechanism for the generation of oxygen from water in photosynthesis. Science. 1997;277:1953–1956. doi: 10.1126/science.277.5334.1953. doi:10.1126/science.277.5334.1953 [DOI] [PubMed] [Google Scholar]
- Ishikita H, Saenger W, Loll B, Biesiadka J, Knapp E.W. Energetics of a possible proton exit pathway for water oxidation in photosystem II. Biochemistry. 2006;45:2063–2071. doi: 10.1021/bi051615h. doi:10.1021/bi051615h [DOI] [PubMed] [Google Scholar]
- Isobe H, Shoji M, Koizumi K, Kitagawa Y, Yamanaka S, Kuramitsu S, Yamaguchi K. Electronic and spin structures of manganese clusters in the photosynthesis II system. Polyhedron. 2005;24:2767–2777. doi:10.1016/j.poly.2005.08.049 [Google Scholar]
- Junge W, Haumann M, Ahlbrink R, Mulkidjanian A, Clausen J. Electrostatics and proton transfer in photosynthetic water oxidation. Phil. Trans. R. Soc. B. 2002;357:1407–1417. doi: 10.1098/rstb.2002.1137. doi:10.1098/rstb.2002.1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya N, Shen J.R. Crystal structure of oxygen-evolving photosystem II from Thermosynechococcus vulcanus at 3.7 angstrom resolution. Proc. Natl Acad. Sci. USA. 2003;100:98–103. doi: 10.1073/pnas.0135651100. doi:10.1073/pnas.0135651100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok B, Forbush B, McGloin M. Cooperation of charges in photosynthetic O2 evolution—I. A linear four step mechanism. Photochem. Photobiol. 1970;11:457–475. doi: 10.1111/j.1751-1097.1970.tb06017.x. [DOI] [PubMed] [Google Scholar]
- Lee C, Brudvig G.W. Probing the functional role of Ca2+ in the oxygen-evolving complex of photosystem II by metal ion inhibition. Biochemistry. 2007;46:3211–3223. doi: 10.1021/bi062033i. doi:10.1021/bi062033i [DOI] [PubMed] [Google Scholar]
- Limburg J, Szalai V.A, Brudvig G.W. A mechanistic and structural model for the formation and reactivity of a MnV=O species in photosynthetic water oxidation. J. Chem. Soc. Dalton Trans. 1999a:1353–1362. doi:10.1039/a807583b [Google Scholar]
- Limburg J, Vrettos J.S, Liable-Sands L.M, Rheingold A.L, Crabtree R.H, Brudvig G.W. A functional model for O–O bond formation by the O2-evolving complex in photosystem II. Science. 1999b;283:1524–1527. doi: 10.1126/science.283.5407.1524. doi:10.1126/science.283.5407.1524 [DOI] [PubMed] [Google Scholar]
- Limburg J, Vrettos J.S, Chen H, de Paula J.C, Crabtree R.H, Brudvig G.W. Characterization of the O2-evolving reaction catalyzed by [(terpy)(H2O)MnIII(O)2MnIV(OH2)(terpy)](NO3)3 (terpy=2,2′-6,2″-terpyridine) J. Am. Chem. Soc. 2001;123:423–430. doi: 10.1021/ja001090a. doi:10.1021/ja001090a [DOI] [PubMed] [Google Scholar]
- Loll B, Kern J, Saenger W, Zouni A, Biesiadka J. Towards complete cofactor arrangement in the 3.0 Å resolution structure of photosystem II. Nature. 2005;438:1040–1044. doi: 10.1038/nature04224. doi:10.1038/nature04224 [DOI] [PubMed] [Google Scholar]
- Lundberg M, Siegbahn P.E.M. Agreement between experiment and hybrid DFT calculations for O–H bond dissociation enthalpies in manganese complexes. J. Comput. Chem. 2005;26:661–667. doi: 10.1002/jcc.20206. doi:10.1002/jcc.20206 [DOI] [PubMed] [Google Scholar]
- MacDonnell F.M, Fackler N.L.P, Stern C, O'Halloran T.V. Air oxidation of a 5-coordinate Mn(III) dimer to a high-valent oxomanganese(V) complex. J. Am. Chem. Soc. 1994;116:7431–7432. doi:10.1021/ja00095a066 [Google Scholar]
- McEvoy J.P, Brudvig G.W. Structure-based mechanism of photosynthetic water oxidation. Phys. Chem. Chem. Phys. 2004;6:4754–4763. doi:10.1039/b407500e [Google Scholar]
- McEvoy J.P, Brudvig G.W. Water-splitting chemistry of photosystem II. Chem. Rev. 2006;106:4455–4483. doi: 10.1021/cr0204294. doi:10.1021/cr0204294 [DOI] [PubMed] [Google Scholar]
- McPhalen C.A, Strynadka N.C.J, James M.N.G. Calcium-binding sites in proteins: a structural perspective. Adv. Protein Chem. 1991;42:77–144. doi: 10.1016/s0065-3233(08)60535-5. [DOI] [PubMed] [Google Scholar]
- Messinger J. Evaluation of different mechanistic proposals for water oxidation in photosynthesis on the basis of Mn4OxCa structures for the catalytic site and spectroscopic data. Phys. Chem. Chem. Phys. 2004;6:4764–4771. doi:10.1039/b406437b [Google Scholar]
- Messinger J, Badger M, Wydrzynski T. Detection of one slowly exchanging substrate water molecule in the S3 state of photosystem II. Proc. Natl Acad. Sci. USA. 1995;92:3209–3213. doi: 10.1073/pnas.92.8.3209. doi:10.1073/pnas.92.8.3209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent J.H.A, Rich A.M, Evans M.C.W. Photosynthetic water oxidation: towards a mechanism. Biochim. Biophys. Acta. 2001;1503:138–146. doi: 10.1016/s0005-2728(00)00223-1. doi:10.1016/S0005-2728(00)00223-1 [DOI] [PubMed] [Google Scholar]
- Pecoraro V.L, Baldwin M.J, Gelasco A. Interaction of manganese with dioxygen and its reduced derivatives. Chem. Rev. 1994;94:807–826. doi:10.1021/cr00027a012 [Google Scholar]
- Pecoraro V.L, Baldwin M.J, Caudle M.T, Hsieh W.-Y, Law N.A. A proposal for water oxidation in photosystem II. Pure Appl. Chem. 1998;70:925–929. [Google Scholar]
- Riggs-Gelasco P.J, Mei R, Ghanotakis D.F, Yocum C.F, Penner-Hahn J.E. X-ray absorption spectroscopy of calcium-substituted derivatives of the oxygen-evolving complex of phostosystem II. J. Am. Chem. Soc. 1996;118:2400–2410. doi:10.1021/ja9504505 [Google Scholar]
- Ruettinger W.F, Dismukes G.C. Conversion of core oxos to water molecules by 4e−/4H+ reductive dehydration of the Mn4O26+ core in the manganese-oxo cubane complex Mn4O4(Ph2PO2)6: a partial model for photosynthetic water binding and activation. Inorg. Chem. 2000;39:1021–1027. doi: 10.1021/ic9911421. doi:10.1021/ic9911421 [DOI] [PubMed] [Google Scholar]
- Siegbahn P.E.M. Theoretical models for the oxygen radical mechanism of water oxidation and of the water oxidizing complex of photosystem II. Inorg. Chem. 2000;39:2923–2935. doi: 10.1021/ic9911872. doi:10.1021/ic9911872 [DOI] [PubMed] [Google Scholar]
- Siegbahn P.E.M, Crabtree R.H. Manganese oxyl radical intermediates and O–O bond formation in photosynthetic oxygen evolution and a proposed role for the calcium cofactor in photosystem II. J. Am. Chem. Soc. 1999;121:117–127. doi:10.1021/ja982290d [Google Scholar]
- Siegbahn P.E.M, Lundberg M. The mechanism of dioxygen formation in PSII studied by quantum chemical methods. Photochem. Photobiol. Sci. 2005;6:4793–4802. doi: 10.1039/b506746b. [DOI] [PubMed] [Google Scholar]
- Sjoberg B.-M, Loehr T.M, Sanders-Loehr J. Raman spectral evidence for a μ-oxo bridge in the binuclear iron center of ribonucleotide reductase. Biochemistry. 1982;21:96–102. doi: 10.1021/bi00530a017. doi:10.1021/bi00530a017 [DOI] [PubMed] [Google Scholar]
- Sproviero E.M, Gascon J.A, McEvoy J.P, Brudvig G.W, Batista V.S. QM/MM models of the O2-evolving complex of photosystem II. J. Chem. Theory Comput. 2005;2:1119–1134. doi: 10.1021/ct060018l. doi:10.1021/ct060018l [DOI] [PubMed] [Google Scholar]
- Szalai V.A, Stone D.A, Brudvig G.W. A structural and mechanistic model of the O2-evolving complex of photosystem II. In: Garab G, editor. Photosynthesis: mechanisms and effects. Kluwer Academic; Dordrecht, The Netherlands: 1998. pp. 1403–1406. [Google Scholar]
- Tagore R, Chen H, Crabtree R.H, Brudvig G.W. Determination of bridging-oxo exchange in di-μ-oxo dimanganese complexes by electrospray ionization mass spectrometry. J. Am. Chem Soc. 2006;128:9457–9465. doi: 10.1021/ja061348i. doi:10.1021/ja061348i [DOI] [PubMed] [Google Scholar]
- Tagore R, Crabtree R.H, Brudvig G.W. Distinct mechansisms of bridging-oxo exchange in di-μ-oxo di-manganese complexes with and without water binding sites: implications for water binding in the O2-evolving complex of photosystem II. Inorg. Chem. 2007;46:2193–2203. doi: 10.1021/ic061968k. doi:10.1021/ic061968k [DOI] [PubMed] [Google Scholar]
- Tagore, R., Crabtree, R.H. & Brudvig, G.W. In press. Oxygen evolution catalysis by a dimanganese complex and its relation to photosynthetic water oxidation. Inorg. Chem. [DOI] [PubMed]
- Vincent J.B, Christou G. A molecular double-pivot mechanism for water oxidation. Inorg. Chim. Acta. 1987;136:L41–L43. doi:10.1016/S0020-1693(00)81142-1 [PubMed] [Google Scholar]
- Volkov A.G. Oxygen evolution in the course of photosynthesis—molecular mechanisms. Bioelectrochem. Bioenerg. 1989;21:3–24. doi:10.1016/0302-4598(89)87002-3 [Google Scholar]
- Vrettos J.S, Limburg J, Brudvig G.W. Mechanism of photosynthetic water oxidation: combining biophysical studies of photosystem II with inorganic model chemistry. Biochim. Biophys. Acta. 2001a;1503:229–245. doi: 10.1016/s0005-2728(00)00214-0. doi:10.1016/S0005-2728(00)00214-0 [DOI] [PubMed] [Google Scholar]
- Vrettos J.S, Stone D.A, Brudvig G.W. Quantifying the ion selectivity of the Ca2+ site in photosystem II: evidence for direct involvement of Ca2+ in O2 formation. Biochemistry. 2001b;40:7937–7945. doi: 10.1021/bi010679z. doi:10.1021/bi010679z [DOI] [PubMed] [Google Scholar]
- Weast R.C, editor. CRC handbook of chemistry and physics. CRC Press; West Palm Beach, FL: 1978. [Google Scholar]
- Wilkins R.G, Yelin R.E. Kinetics of monomer-dimer interconversion of iron(III) ethylenediaminetetraacetate and related chelates. Inorg. Chem. 1969;8:1470–1473. doi:10.1021/ic50077a020 [Google Scholar]
- Yachandra V.K, Sauer K, Klein M.P. Manganese cluster in photosynthesis: where plants oxidize water to dioxygen. Chem. Rev. 1996;96:2927–2950. doi: 10.1021/cr950052k. doi:10.1021/cr950052k [DOI] [PubMed] [Google Scholar]
- Zouni A, Witt H.T, Kern J, Fromme P, Krauss N, Saenger W, Orth P. Crystal structure of photosystem II from Synechococcus elongatus at 3.8 angstrom resolution. Nature. 2001;409:739–743. doi: 10.1038/35055589. doi:10.1038/35055589 [DOI] [PubMed] [Google Scholar]
Additional reference
- Siegbahn P.E.M. O–O bond formation in the S4 state of the oxygen-evolving complex in photosystem II. Chem. Eur. J. 2006;12:9217–9227. doi: 10.1002/chem.200600774. [DOI] [PubMed] [Google Scholar]