

Abstract
The molecular oxygen produced in photosynthesis is generated via water oxidation at a manganese–calcium cluster called the oxygen-evolving complex (OEC). While studies in biophysics, biochemistry, and structural and molecular biology are well known to provide deeper insight into the structure and workings of this system, it is often less appreciated that biomimetic modelling provides the foundation for interpreting photosynthetic reactions. The synthesis and characterization of small model complexes, which either mimic structural features of the OEC or are capable of providing insight into the mechanism of O2 evolution, have become a vital contributor to this scientific field. Our group has contributed to these findings in recent years through synthesis of model complexes, spectroscopic characterization of these systems and probing the reactivity in the context of water oxidation. In this article we describe how models have made significant contributions ranging from understanding the structure of the water-oxidation centre (e.g. contributions to defining a tetrameric Mn3Ca-cluster with a dangler Mn) to the ability to discriminate between different mechanistic proposals (e.g. showing that the Babcock scheme for water oxidation is unlikely).
Keywords: oxygen-evolving complex, water oxidation, manganese, photosynthesis
1. Introduction
Molecular oxygen, which is crucial to life on earth, is produced in cyanobacteria, algae and green plants (Wydrzynski 2005). The enzyme responsible for the conversion of water into dioxygen is the oxygen-evolving complex (OEC), which is located within the larger framework known as photosystem II (PSII). The OEC contains a cluster of manganese (Cheniae & Martin 1970; Sauer 1980; Yocum et al. 1981) and calcium (Ghanotakis et al. 1984; Han & Katoh 1993; Adelroth et al. 1995; van Gorkom & Yocum 2005) ions. The detailed structural aspects of this centre are still to be resolved; however, it is widely accepted that this unit contains four manganese ions, which can change oxidation states, as well as one calcium(II) ion. A chloride ion is known to be necessary for oxygen evolution (Yocum 1992; Lindberg & Andreasson 1996; Homann 2002; van Gorkom & Yocum 2005), but neither its structural location nor its function are fully understood yet. The kinetic profile of the enzyme follows the S-clock, a cycle that is composed of five states given designations Sn, where n=0–4 representing the most reduced to the most oxidized enzyme forms, respectively. Several key issues need to be resolved in order to assess the chemical mechanism of water oxidation. Probably most important are the oxidation states of the Mn ions in each of the S-states, especially S4 and S0. Additionally, the roles of CaII and chloride need to be established. In this article we will address some of these issues using small molecule models as a guide.
2. Structure
The stoichiometric composition of the OEC is widely accepted to be Mn4Ca, whereas the question whether chloride is attached to the cluster, or if it is even a crucial part of photosynthesis, is under debate (Wincencjusz et al. 1999; Yocum & Pecoraro 1999; Popelkova et al. 2006). Researchers agree that the metal ions of the cluster are linked by μ-oxo bridges. Additionally, they are coordinated to amino acids, namely to carboxylate oxygen atoms and a histidine nitrogen. A number of structure suggestions have been put forth by various research groups, mainly derived from electron paramagnetic resonance (EPR), X-ray absorption (XAS) and X-ray diffraction spectroscopies. Early models suggested a Mn cubane structure (Brudvig & Crabtree 1986) which was ruled out by extended X-ray absorption fine structure (EXAFS) data that were inconsistent with such a symmetric structure (Kim et al. 1990; Penner-Hahn et al. 1990). An alternative proposal called the ‘dimer of dimers’ proved very popular for a number of years (Yachandra et al. 1993); however, analysis (Pecoraro & Hsieh 2000; Peloquin et al. 2000) of the EPR spectral features in the S2 state (a g=2 multiline (Dismukes & Siderer 1981) and a broad low-field signal at g∼4 (Casey & Sauer 1984)) suggested that the dimer-of-dimers model was inappropriate, and that a 3+1 formulation was probably more accurate (Pecoraro & Hsieh 2000; Peloquin et al. 2000). Pulsed EPR studies of the enzyme (Peloquin et al. 2000; Britt et al. 2004) and small molecule models suggested an alternative ‘dangler’ model, which was reported in a linear topology but could easily accommodate a more three-dimensional structure. In 2000 the first low-resolution structure appeared which suggested that the Mn formed a 3+1 cluster (Zouni et al. 2000, 2001). Subsequent crystallographic refinement at 3.5 Å resolution done by Barber and co-workers (Ferreira et al. 2004) shows a Mn3Ca–oxo cluster with a dangler Mn ion attached. The manganese and calcium ions are coordinated by glutamic and aspartic acids, a terminal alanine and a histidine. Furthermore, a tyrosine (Yz), which is not part of the metals' first coordination sphere but within a 5 Å distance, is poised to play a vital role in the oxidation mechanism. Most importantly, this structure demonstrated for the first time that calcium was an integral part of the Mn cluster, a point which had been debated for many years based on EXAFS studies (Riggs-Gelasco et al. 1996; Cinco et al. 1998). More recently, a structure from Loll and co-workers (Loll et al. 2005) was obtained at higher resolution (3.0 Å), but unfortunately it fails to locate critical bridging oxygen atoms in the cluster. Also, it differs from the Barber structure (Ferreira et al. 2004) showing a modified spatial distribution of the metal ions, as well as comprising a different ligation of the nearby amino acids. However, available crystal structures of the OEC still lack atomic resolution. More importantly, recent XAS experiments have shown that X-ray structures could be distorted by X-ray-induced photo degradation (Yano et al. 2006). A combination of XAS and crystallography has led to the best available models (Loll et al. 2005; Yano et al. 2006) shown in figure 1. Thus, at present it appears that the best description for the OEC cluster, in the lower oxidation levels, is a tetrameric assembly of Mn (best thought of as a 3+1 Mn–oxo cluster) with a calcium ion integral to the structure.
Figure 1.
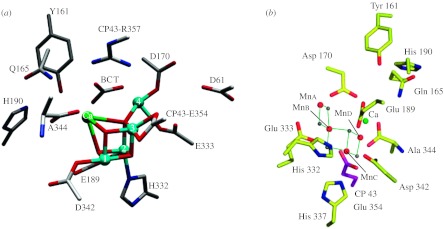
(a) Structure of the OEC based on the X-ray diffraction measurements done by Barber and co-workers (Ferreira et al. 2004) and (b) a recent model (Yano et al. 2006) of the Mn4Ca cluster derived from polarized Mn-EXAFS and the 3.0 Å resolution X-ray structure (Loll et al. 2005). (Reproduced with permission from McEvoy & Brudvig (2006) and Yano et al. (2006).)
Unfortunately, models for such complex structures are difficult to prepare, and the interested reader is referred to numerous reviews on synthetic models for the OEC (Wieghardt 1989; Pecoraro et al. 1994; Law et al. 1998; Mukhopadhyay et al. 2004). Among the best are the various Mn-oxo structures reported by Christou and co-workers (Bashkin et al. 1987; Vincent et al. 1989; Libby et al. 1990, 1991; Bouwman et al. 1992). While most of these structures form butterfly or pseudocubane assemblies that do not match the precise stoichiometry or include calcium, they do provide deeper insight into the system. An alternative formulation has been reported from our group using a class of molecules known as metallacrowns (Gibney et al. 1996; Pecoraro et al. 1997). The 12-MC-4 structure type shown in figure 2 contains four MnIII ions in a ring. A fifth ion is linked to these ring metals through carboxylate bridges and MnIII-O-M linkages (Lah & Pecoraro 1989). Stoichiometrically, these compounds are interesting links to the OEC with a Mn4M composition (Gibney et al. 1996). The structure in figure 2a contains a capping MnII as the fifth ion, whereas figure 2b contains two NaI ions. Of particular interest, the NaI is bridged to the MnIII ions with halides, mimicking a possible role for chloride in the OEC. NaI is a competitive inhibitor (Waggoner et al. 1989) versus calcium of the OEC. Another class of metal inhibitors are lanthanide ions (Bakou et al. 1992). Figure 2c shows the structure with additional carboxylate bridges. The propensity of lanthanides for their higher charge and higher coordination number suggests a mechanism for lanthanide inhibition of the OEC. While we do not yet have crystals of the Mn4Ca or Mn4Sr analogues, we do have mass spectral evidence that such compounds exist. Thus, while these compounds are topologically rather different from the Mn4Ca cluster of the OEC, they are the structures that as a group have most closely modelled the stoichiometric basis for active and related inhibited forms of the enzyme.
Figure 2.

Representation of three 12-MC-4 type manganese(III) metallacrowns with: (a) MnII and acetates as bridges (Lah & Pecoraro 1989), (b) two NaI and two bromide bridges (Gibney et al. 1996) and (c) DyIII and four salicylate bridges. We expect the Ca 12-MC-4 to be isostructural with the structure with MnII (a).
Thinking about the OEC as a 3+1 formulation of Mn ions, it is worth considering trinuclear Mn compounds as models for this centre. A plethora of structure classes is possible in clusters comprising three manganese centres (Mukhopadhyay et al. 2004). May be the simplest structure type is the Mn3O core, in which all three metals are linked by oxygen and further ligated by carboxylates. This core type is usually referred to as the basic carboxylate type. A noteworthy complex was synthesized by Weatherburn and co-workers, in which a peroxo moiety and two carboxylates bridge the Mn centres (Bhula et al. 1988). Despite the poorly resolved crystal structure, this complex provided the first indication of potential dioxygen coordination to a trinuclear manganese cluster. Another structure class of interest is the open structure type, especially, the mixed-valent MnIIIMnIIMnIII clusters. The first compound of this class was published by Pecoraro and co-workers, a tridentate Schiff base complex with acetate and methanol ligands (Kessissoglou et al. 1989). Similar complexes have been synthesized with other Schiff base ligands, all resulting in weakly antiferromagnetically coupled complexes with an EPR signal at approximately g=4, indicating a S=3/2 spin groundstate (Kessissoglou et al. 1992; Tangoulis et al. 1996). Kitajima et al. (1994) have used trispyrazolylborate ligands to synthesize a MnIIIMnIIMnIII complex, in which manganese ions are linked by one μ-hydroxo and two acetato groups. A reversed order of manganese oxidation states, namely MnIIMnIIIMnII was achieved by Tanase and co-workers, who used tris[(N-aldosyl)-aminoethyl]amine as a ligand (Yano et al. 1997). Most trinuclear complexes of this type are low valent. Therefore, higher valent complexes such as MnIIMnIVMnII are very interesting and will be discussed in greater detail in §3.
Trinuclear structures that model the S2 and possibly S3 states include an open trinuclear complex that has been recently reported by Christou and co-workers, comprising a nonlinear Mn3O4 unit (Bhaduri et al. 2002), which can be found in many proposed structures for the OEC. The ligands are bipyridines and acetate. The central metal is solely coordinated by oxygen ions, which seems to increase the stability of this high-valent cluster. In addition, adamantane-like arrangements have been published by Wieghardt and co-workers, who synthesized a series of oxo-bridged complexes using triazacyclononane (Wieghardt et al. 1988). Despite all these advances, true corroborative models of the OEC appear to be far in the future.
3. Oxidation states
While there are five S-states, the two most important are S0, the product state, and S4, the catalytic state that forms and releases dioxygen. In most cases, it is believed that S-state advancement corresponds to manganese oxidation, although there is some controversy as to the S2→S3 (Yachandra et al. 1996; Robblee et al. 2001; Yachandra 2005) transition. But even this ambiguity has as its primary consequence the assignment of the proper manganese oxidation levels in S4. The EPR-active S2 state often serves as the calibration point for the S-clock, being assigned by most workers as the mixed valent oxidation level. There is no disagreement that the S0→S1 and S1→S2 transitions correspond to manganese-centred oxidation. This leads to S1 being assigned as and S0 as Mn4Ca+15. We have chosen this latter formulation because there are two different possible formulations, namely (supported by X-ray absorption near-edge structure spectroscopy—XANES— and EPR; Ahrling et al. 1997; Messinger et al. 1997) and (supported by recent EPR studies). Also, recent 55Mn electron-nuclear double resonance (ENDOR) spectroscopy argues against the presence of MnII in S0 (Kulik Leonid et al. 2005). The arrangement is particularly interesting, as it would be a rare example where manganese ions are found in three different oxidation states.
Model compounds can provide insight into this controversy. There are only two small molecule compounds that have Mn in three different oxidation states (Chan & Armstrong 1990; Mukherjee et al. 2006), while a handful of compounds contain at least two manganese ions separated by two charge units. In the latter case, there are two interesting pairs of compounds that are redox isomers, which serve to evaluate the ability of XANES to discriminate between the two S0-oxidation state formulations (Alexiou et al. 2003; Zaleski et al. submitted).
Comparative spectroscopic and magnetic studies between MnIIMnIVMnII and MnIIIMnIIMnIII compounds suggested that one can properly assign oxidation states to these systems using XANES spectroscopy (Alexiou et al. 2003). The oxidation states in these compounds could be clearly distinguished from each other due to the fact that they display different shapes in the XANES spectra, with MnII-rich compounds exhibiting a more intense X-ray edge absorption. Since XAS spectroscopy is an averaging technique, the ability to resolve oxidation states diminishes as the number of manganese ions increases. Therefore, we have recently investigated a more rigorous test of the XANES technique using tetrameric systems. In particular, we have examined two systems ( (Afrati et al. 2002) versus (Zaleski et al. submitted)). While not as clear cut as with the trinuclear system, we can still resolve the appropriate oxidation states for these compounds. These data suggest that XANES can, in principle, distinguish properly between the and the formulations; however, in practice, this may be very difficult.
The S1 state is almost certainly . Despite being the dark stable state, little attention was focused on this oxidation level, because it was spectroscopically less interesting than the one-electron-oxidized S2 level. Britt's discovery of a parallel mode EPR signal confirmed the non-Kramer's electronic state and strongly supported the accepted manganese oxidation assignment (Campbell et al. 1998). Subsequently, we demonstrated that a tetranuclear manganese complex displays a very similar parallel-mode signal to that of the enzyme (Hsieh et al. 2004). Because this model complex has a oxidation level, the hyperfine shifts are slightly reduced from that of S1. Nonetheless, this is still the only compound known to exhibit a parallel mode multiline signal reminiscent of S1.
The number of model compounds prepared to mimic S2 are too numerous to mention. The interested reader should look at the many reviews on this topic (Mukhopadhyay et al. 2004; Wu et al. 2004). In contrast, models for S3 and S4 are very rare. The highest oxidized S-state is S4, which has a very short lifetime and its individual manganese oxidation states are unclear. The best descriptions are either a or radical (to some extent differentiating these species may be equivalent to asking whether the metal or ligand molecular orbitals have been more depopulated in electron density). Alternatively, one could envision a lower valent S4 state if partial substrate oxidation occurs in S3, prior to the formation of S4. Certainly there are neither reported examples of crystallographically characterized clusters containing MnV nor an equivalent MnIV-oxy radical. There are numerous examples of species (Wieghardt 1989; Pecoraro et al. 1994; Law et al. 1998; Mukhopadhyay et al. 2004), but these complexes are generally inert (non-reactive). The reason for the ambiguity in the S4-oxidation level arises from the conversion of S2→S3. Initially, XANES data were interpreted as corresponding to a non-metal-centred oxidation. Several accounts have suggested that the Mn are oxidized in this transition. Simply put, there are significant amounts of data supporting either metal- or non-metal-centred oxidation, making this point very difficult to resolve at the present time.
4. Mechanisms
Given our present understanding of structure (a Mn4Ca core) and kinetics (five states with S4 either or radical, and S0 either or ), one can begin to evaluate the mechanism for water oxidation (McEvoy & Brudvig 2006). There are several ways to categorize water-oxidation proposals. The most useful starting point is to differentiate models based on whether O–O bond formation begins in S3 or S4. One can then assess whether bridging oxo groups, a combination of bridging oxo and terminal water/hydroxide/oxo functionalities or solely terminal moieties are responsible for the formation of an O–O bond. From there, more subtle points such as the appropriate oxidation states of the manganese ions and the origin of oxo atoms (e.g. bound to Mn versus Ca) can be assessed.
5. O–O bond formation beginning in S3
Proposed as an S3-initiated reaction by Yachandra and co-workers (Yachandra et al. 1996; Robblee et al. 2001; Yachandra 2005), the formation of a Mn-oxyl-radical occurs in the S2→S3 transition, followed by the formation of a second oxygen radical during S3→S4. Both oxo radicals then combine yielding a peroxo species. The μ-oxo bridges of the cluster would be the origin of the oxygen atoms in this model. While this mechanism was originally proposed for a dimer-of-dimers structure, which was ruled out by 55Mn ENDOR spectroscopy (Peloquin et al. 2000) and X-ray crystallography (Zouni et al. 2001; Ferreira et al. 2004; Loll et al. 2005), it can certainly be incorporated into the present 3+1 structural motif. This model would never require the manganese ions of the cluster to exceed a oxidation level. As an advantage, this model accounts for an invariant metal oxidation state from S2→S3 and would be consistent with a structural change in the cluster at higher S-states. One potential drawback of this system is that water exchange between bridging μ-oxo groups would be expected to be much slower than has been reported. Wydrzynski and co-workers have measured the exchange rates of substrate water (Hillier et al. 1998, 2001). They determined that at least two water molecules are bound to the OEC throughout the cycle, one of them by S2. Although μ-oxo exchange could not be measured, the comparison with other metal–oxo complexes leads to the conclusion that the observed rates were too fast for an exchange of a bridging oxygen atom.
Similar to the Yachandra proposal, Messinger and co-workers have invoked oxygen radicals in the S3 and S4 states (Messinger 2004). A major difference to the previous mechanisms, Messinger incorporated a deprotonation step in the S0→S1 transition (Haumann et al. 2005), which rationally explains the Mn–Mn bond-length shortening from 2.85 to 2.70 Å. Such a deprotonation is probably essential in order to maintain a relatively constant potential for the water-oxidation catalyst. This model again suffers from the water-exchange-rate problem described above.
Siegbahn and co-workers proposed an alternative strategy based on density functional theory (Lundberg & Siegbahn 2004; Siegbahn & Blomberg 2005). In their model, oxygen radicals are involved in S3 and S4 and deprotonation leads to a bridging hydroxide. One major difference from the Messinger or Yachandra models is that the oxygen atoms are terminally bound to the Mn ions. These authors propose that terminal MnV-oxo species may be insufficiently reactive for water oxidation, requiring a spin-state alteration to a MnIV-oxyl radical for reactivity. One can think of this model as a homolytic coupling of two MnIV-oxyl radicals.
One of the most influential early proposals for water oxidation was presented by Babcock (Tommos et al. 1995; Hoganson & Babcock 1997). Proton-coupled electron transfer (PCET) and, more specifically, H-atom abstraction significantly influenced the thinking behind this proposal. The mechanism involves the tyrosyl radical Yz not only as an oxidizing agent but also as an H-atom acceptor (the Yz simultaneously accepts protons and electrons). The first critical catalytic step takes place in the transition S2→S3, where a MnIV terminal oxo species in proximity to a hydroxo group (attached to MnIV) is formed. After a final oxidation and proton abstraction in S4, a peroxo bridge linking the two dangler manganese centres is formed. Eventually O2 is released, substrate water is attached and S0 is re-established. While a very appealing process when first published, recent structural and spectroscopic data appear to make this mechanism non-viable. The most recent structures place Yz too far away from the cluster to be involved in direct H-atom abstraction.
Model compounds have served to both support the basic premise of this proposal and refute it. For example, a key requisite for this chemistry (and that proposed by Siegbahn) is the ability to abstract hydrogen atoms from either terminal or bridging oxygen atoms. Several reports on model compounds demonstrated that H-atom abstraction, with homolytic bond dissociation energies (HBDEs) of the order of 87 kcal mol−1, could occur with either terminal or bridging water or hydroxides with biologically relevant manganese oxidation levels. This HBDE is precisely the thermodynamic value necessary for Babcock's H-atom abstraction model (Gardner & Mayer 1995; Caudle & Pecoraro 1997). More recent work on a single set of compounds suggests that going higher than MnIV leads to a large increase in the HBDE (Gupta et al. 2002). Nonetheless, these studies demonstrated that PCET was thermodynamically important in the water-oxidation process.
Model compounds have also served to exclude the Babcock proposal, and any other mechanisms requiring a Mn=O formulation in S3. An important class of mononuclear manganese complexes incorporates Mn terminal oxygen moieties, since many mechanistic proposals are based on manganyl species in the higher S-states. Early synthetic work in this field was done by Collins and co-workers, who published the first structurally characterized MnV-oxo compound, a 1,2-bis(2-hydroxy-2-methylpropanamido)benzene MnV complex, in 1989 (Collins & Gordon-Wylie 1989). Later on, two further MnV complexes, also comprising oxidatively stable and highly negatively charged amidate ligands, were published by the same group (Collins et al. 1990; Miller et al. 1998). XANES spectra of Collins' first (alcoholato) complex, contributed strongly to the understanding of the S3→S4 transition (Weng et al. 2004). An intense XANES pre-edge signal was found in this MnV-oxo compound. The origin of the feature is a consequence of the mixing of metal d- and oxygen p-orbitals, which removes the symmetry forbidden nature of the 1s→3d transition. Furthermore, it was shown that MnIV=O (also having significant metal d- and oxygen p-orbital mixing) should give rise to a similar intense pre-edge transition. In contrast, MnIVOH or other related species do not have such strong pre-edge absorptions. Analysis of the XANES spectra of the OEC demonstrated that a strong pre-edge transition was not present in the native S3. Therefore, this work demonstrated that manganyl species do not exist in the OEC prior to the S4 state (Weng et al. 2004). Dau and co-workers later extended this analysis demonstrating that Mn=O species also do not exist in the region until S4 (Haumann et al. 2005); however, there is no experimental evidence to assess whether such species are viable in the actual water-oxidation step. It should be noted that even though the Babcock model has now been shown not to be the proper description for water oxidation, the components of these model were tremendously inspirational and significantly moved the mechanistic thinking of the field forward.
Meyer and co-workers (Meyer et al. 2007) have proposed a 14-step mechanism that scrupulously accounts for proton movement through the S-cycle. These authors propose the formation of an O–O bond during S3. One oxygen atom of the product is bound to manganese while the second originates on calcium. Subsequent photo-oxidation and proton transfer lead to the release of dioxygen. This is the only model with chemistry beginning in S3 that uses calcium explicitly in the reaction process. In summary, there are several viable water-oxidation mechanisms that require substrate oxidation to begin in S3. The most prominent (Hoganson & Babcock 1997) of these proposals has recently been eliminated. Whether any of these proposals remain viable is dependent on the resolution of the controversy for Mn oxidation in the S2→S3 transition.
6. O–O bond formation beginning in S4
The second major class of mechanisms for water oxidation relies on the entire oxidation process occurring during the S3→S4→S0 transition. An example that invokes oxidation of bridging oxygen atoms was proposed by Dismukes based on a Mn4O4 cuboidal model compound (Ruettinger et al. 2000). Two μ-oxo bridging oxygen atoms combine generating a peroxo bridge. After the liberation of dioxygen, the cluster displays a Mn4O2 butterfly structure. The observation of dioxygen in the gas phase, using mass spectroscopy, has been taken as support of this model. Hückel calculations, which had been carried out for the S4 state of cuboidal Mn–oxo clusters comprising peroxo oxygen atoms coordinated by three Mn ions, have predicted an energetic advantage for this process (Proserpio et al. 1992). It is unclear how this model can be rationalized with the water exchange results of Wydrzynski.
Dau and co-workers suggested that oxygen is formed during a reaction of a terminal oxyl and an exogenous hydroxyl radical in S4 (Dau et al. 2001). Furthermore, they use the concept of proton-assisted oxidation, where the μ-oxide bridges act as bases and abstract H atoms from non-coordinated water molecules. A highly reactive MnV-oxo species that switches to a MnIV-oxyl-radical is proposed in S4. A H atom is abstracted from a coordinated water by an adjacent μ-oxide, providing a poised hydroxyl radical. This OH moiety immediately forms an O–O–H bond, with the hydroperoxo H atom abstracted by another μ-oxide. Model chemistry has been particularly important to assess this mechanistic aspect. It has been shown that μ-OH bridges are generally acidic and this acidity increases as the oxidation level of the cluster increases (Larson et al. 1992; Baldwin et al. 1993; Baldwin et al. 1994). Thus, one would not expect μ-oxo bridges to be sufficiently basic as to act as proton acceptors, especially as the cluster is oxidized.
Pecoraro et al. (1998) proposed a reaction process in 1997 that explicitly accounted for the role of Ca. It was suggested that an electrophilic MnV-oxo is formed in the S4 state. Hydroxide coordinated to a nearby calcium ion could then serve as a nucleophile, attacking this terminal oxo group to directly form dioxygen. In addition to accounting for the need of calcium for the first time, this proposal is consistent with Wydrzynskis experiment (Hillier et al. 1998, 2001), which suggests two water molecules displaying different exchange rates. Most likely, the slower exchanging substrate is bound to manganese and the faster one only to calcium. Subsequently, Brudvig and co-workers expanded on these ideas to provide a detailed step-by-step mechanism that accounts for the chemistry through each S-transition (McEvoy & Brudvig 2006).
Model chemistry has been generally supportive of this proposal although direct proof for the presence of a MnV=O has not been presented. Brudvig and co-workers have produced a MnIIIMnIV terpyridine complex, which served as the first model for catalytic O–O bond formation when sodium hypochlorite or KHSO5 was used as an oxidant (Tagore et al. 2006, 2007). Some of the early experiments have been reinterpreted to release dioxygen through a disproportionation reaction; however, Yagi has indicated that these water-oxidation catalysts are efficient when placed on a clay surface. More recently, McKenzie and co-workers have used a similar complex to probe reactivity towards a mechanistic model, using t-butyl hydroperoxide as an oxidant (Poulsen et al. 2005). It is well known that singlet dioxygen can be liberated from the reaction of Mn dimers and t-butyl hydroxide (Caudle et al. 1996); however, it appears that this may be a legitimate water-oxidation process as 18O-labelled water is found in the product dioxygen. A highly reactive species was suggested to play a crucial role in this process. Further definition of this system is required, with particular attention being focused on the origin of the second oxygen atom.
In summary, the field of photosynthetic oxygen evolution has made tremendous advances in understanding the structure of the cluster responsible for water oxidation. Furthermore, definition of cluster oxidation states has been obtained in several, although not all, of the S-states. Due in part to oxidation state ambiguity, the number of proposed water-oxidation mechanisms seems to be increasing, rather than narrowing. One of the main objectives of the next decade will be to bring a similar level of clarity to the chemical mechanism of this extraordinary process, as is now being revealed for the lower S-state catalyst structure.
Discussion
J. Barber (Imperial College London, UK). In your scheme of O–O bond formation involving a nucleophilic attack of a hydroxyl oxygen on a highly electrophilic oxygen, how important is the directionality of this attack?
V. Pecoraro. The directionality of the attack is important as one would want one of the p orbitals on the hydroxide to be positioned in such a way as to be able to shift electron density into the LUMO of the electron-deficient oxo atom bound to the manganese. This should not be a significant issue, however, because there should be high symmetry for the orbitals on this oxo atom so that the only restriction would really come from the hydroxide group. Again, this is unlikely to be a problem as the only orbitals that are restricted from participating in this attack are the ones engaged in bonds to the Ca and to the proton. Of course, the proton, unless restricted by a strong H-bond, should have rapid movement to all three potential orbitals of the hydroxide oxygen atom.
G. Brudvig (Yale University, USA). What are your thoughts about Ca-hydroxide versus Ca-water as the nucleophile in the O–O bond formation?
V. Pecoraro. I believe that the more proton deficient the oxygen atom becomes, the better base it should be. Therefore, my preference would be to use a hydroxide that had the proton involved in an H bond to another acceptor (making it more oxide-like than even hydroxide-like). I think water is not as good a nucleophile (e.g. it has lower charge) so my preference would be for hydroxide rather than water.
A. Aukauloo (University of Paris-Sud, France). What was the rationale for making your tetranuclear Mn-Ca system and were the Mn atoms of this cluster redox active?
V. Pecoraro. I'll answer the second question first. We have not examined the electrochemistry of this particular compound so I can't give a definitive answer. The corresponding compound with Mn(II) has a one-electron oxidative wave which probably corresponds to the oxidation of Mn(II) to Mn(III). If this new compound parallels the previous molecule, then it is unlikely that the Mn(III) atoms are redox active within a reasonable electrochemical window. As for the rationale, we have long known that the class of molecules we described as metallacrowns leads to controllable assemblies of metal atoms. Given our observations with other related metallacrown systems that bind manganese, lanthanides or calcium, it was rather straightforward to design a synthesis that led to the structures that were described in my lecture. I would refer you to our Progress in Inorganic Chemistry review in 1996 for a general overview of this strategy and some subsequent papers in Inorganic Chemistry and Angewandte Chemie if you'd like more synthetic details.
J. Messinger (Muelheim, Germany). I have a comment I wish to make. On the basis of my first mass spectrometric data on the substrate water exchange in PSII (Messinger et al. 1995) I also proposed a nucleophilic attack mechanism which involves a transient Mn(IV)≡O+ species in the S4 state (this is isoelectronic with Mn(V)=O or Mn(IV)–O·), and a water molecule that was said to be either free, bound to a protein residue or to Ca (symbolized in the corresponding scheme II as X–OH2). At that point in time the exchange rate for the fast exchanging substrate molecule was unresolved due to a mixing time of 30 ms, and therefore appeared to be much faster than the slow exchange. After improving the time-resolution to 8 ms it turned out that the fast exchange rate is only approximately 20 times faster than that of the slowly exchanging substrate molecule (Hillier et al. 1998). On that basis it appears unlikely that the two substrate molecules are bound in completely different environments (i.e. one to Mn, the other being free or ligated only to Ca) and hence I now consider the nucleophilic attack mechanism as not very likely. Furthermore, substrate water exchange measurements on the S2 state show that the slow exchange occurs essentially with the same rate in the S2 and S3 states (Hillier & Wydrzynski 2000). Nucleophilic attack mechanisms during this step often involve the oxidation of the Mn ion to which the slowly exchanging substrate molecule (at this stage usually shown as Mn=O) is bound, which appears to be inconsistent with the above result. Other findings add to this argument: several different experiments and theoretical calculations (summarized in (Messinger 2004; Hillier & Messinger 2005)) indicate the formation of a radical in the S3 state, and consequently argue against a Mn(IV)≡O+ (or Mn(V)=O) formation and, thereby, also against a nucleophilic attack mechanism for O–O bond formation in PSII.
V. Pecoraro. Johannes, you raise a number of interesting points which I will attempt to address in order. With respect to your proposed nucleophilic attack mechanism, there are points about it that never made sense. First, as you point out, Mn(V)=O and Mn(IV)–O· are isoelectronic; however, that does not mean that they react identically or even similarly. In your proposal, an oxygen atom behaves as a non-innocent ligand which is formally oxidized to the radical form by a Mn(V). Such a system would be quite similar to that of Fe(III)catecholate versus Fe(II)(semiquinone) in which there is a non-innocent ligand (catechol) bound to an oxidizing metal. The two compounds do not react the same with the Fe(II)(semiquinone) being air sensitive whereas the Fe(III)catecholate is stable. As Dan Nocera also has pointed out in this symposium session, a reactivity difference is also expected for Mn(V)=O and Mn(IV)–O· The oxygen on a Mn(V)=O is electrophilic, meaning that it is susceptible to attack by a nucleophile. The oxygen of Mn(IV)–O· is non-nucleophilic and reacts as a radical. This is the basis for Per Siegbahn's model from his quantum calculations. Thus, to have invoked a nucleophilic attack on Mn(IV)–O· was simply wrong. Next, I find little insight in describing the nucleophile water as 'either free, bound to a protein residue or to Ca (symbolized in the corresponding scheme II as X-OH2)'. One could even suggest that X could have been manganese. In this case, you would have invoked your non-electrophilic Mn(IV)–O· being attacked by bulk water, water trapped in a protein or water bound to a metal. Said differently, no source of water is excluded. More important, though, is that your proposed nucleophile was water. The reason that we have suggested that water is likely bound to calcium, with its non-redox active Lewis acid properties, is that it makes the water more acidic Thus, calcium helps activate the water to form hydroxide (a much better nucleophile than water) without itself being prone to oxidation. The premise of the idea is that the oxygen atom on manganese must be highly activated (as achieved with an Mn(V)=O) and the nucleophile also must be highly activated (as occurs with a hydroxide bound to calcium). You are correct to question your original proposition as one would never expect a non-electrophilic oxygen (as represented by Mn(IV)–O·) to be attacked by a really poor nucleophile (from bulk water onward to water on X). Thus, I think we are both in agreement that your original proposal is wrong and needs no more discussion. Now let's discuss the water exchange rates. Here one needs to be careful discriminating between experimental results and experimental interpretation. The observation is that the exchange rates for water are within 20 times that of one another and that they do not change going from S2 to S3. Unfortunately, we do not know what process is rate limiting for water exchange in this case. You are assuming that the rate limitation that you and others measure is a consequence of the dissociation rate of water from a metal ion. Gary Brudvig demonstrated in his talk here that the rates of exchange on Mn are kinetically competent with these exchange rates so there would be no problem with our model. In fact, your arguments are of more concern for an oxo bridge (especially for high valent manganese) as water exchange should be very different if one oxygen was in the bridge and the other not. Furthermore, if both were in bridges, one might expect truly slow exchange rates. None-the-less, the main problem with this discussion is that you do not know why you observe the rates that you do. With respect to no change in rate going from S2 to S3, this is also completely consistent with a non-metal dependent rate limitation. While some mechanisms from other groups may invoke a Mn=O in S3, we have said that this is highly unlikely based on our collaborative work with Jim Penner-Hahn (Weng et al. 2004). Thus, there is no inconsistency in our proposal based on the oxidation level of S3. Finally, you are concerned with the appropriate oxidation state description for the Mn ions in higher S states. This is, of course, the controversy to which I referred in my talk. It is wrong to imply that the S3 state is a radical; this has not been proven in my opinion. There are some people, such as yourself, that feel that the manganese ions are not oxidized going from S2 to S3. Others, such as myself and Gary Brudvig, feel the data presented so far is most consistent with a manganese-centered oxidation. There are still others that think that one can not reach Mn(V) because their formulation of the oxidation levels for S2 are Mn(III)3 Mn(IV), not the Mn(III)Mn(IV)3 assignment which I think both you and I prefer. Clearly, reasonable scientists have a disagreement on this point which I do not think has been clarified further at today's meeting. Thus, my conclusion is as I stated in my talk, if one has sequential oxidation of the manganese ions through the entire S cycle, the most reasonable mechanism for water oxidation is by a hydroxide ion bound to calcium attacking an electrophilic oxygen of a manganyl (Mn(V)=O) species.
Acknowledgments
The funding of the NIH (GM39406) is gratefully acknowledged.
Footnotes
One contribution of 20 to a Discussion Meeting Issue ‘Revealing how nature uses sunlight to split water’.
References
- Adelroth P, Lindberg K, Andréasson L.-E. Studies of Ca2+ binding in spinach photosystem II using 45Ca2+ Biochemistry. 1995;34:9021–9027. doi: 10.1021/bi00028a010. doi:10.1021/bi00028a010 [DOI] [PubMed] [Google Scholar]
- Afrati T, Dendrinou-Samara C, Raptopoulou C.P, Terzis A, Tangoulis V, Kessissoglou D.P. A tetranuclear mixed-valence Mn3IIMnIV compound with (μ4-O)Mn4 core. Angew. Chem. 2002;41:2148–2150. doi:10.1002/1521-3773(20020617)41:12<2148::AID-ANIE2148>3.0.CO;2-R [PubMed] [Google Scholar]
- Ahrling K.A, Peterson S, Styring S. An oscillating manganese electron paramagnetic resonance signal from the S0 state of the oxygen evolving complex in photosystem II. Biochemistry. 1997;36:13 148–13 152. doi: 10.1021/bi971815w. doi:10.1021/bi971815w [DOI] [PubMed] [Google Scholar]
- Alexiou M, et al. Models for the lower S states of photosystem II: a trinuclear mixed-valent MnII/MnIV/MnII complex. Inorg. Chem. 2003;42:2185–2187. doi: 10.1021/ic026050h. doi:10.1021/ic026050h [DOI] [PubMed] [Google Scholar]
- Bakou A, Buser C, Dandulakis G, Brudvig G, Ghanotakis D.F. Calcium binding site(s) of photosystem II as probed by lanthanides. Biochim. Biophys. Acta. 1992;1099:131–136. doi:10.1016/0005-2728(92)90209-K [Google Scholar]
- Baldwin M.J, Gelasco A, Pecoraro V.L. The effect of protonation on [Mn(IV)(μ2-O)]2 complexes. Photosynth. Res. 1993;38:303–308. doi: 10.1007/BF00046754. doi:10.1007/BF00046754 [DOI] [PubMed] [Google Scholar]
- Baldwin M.J, Stemmler T.L, Riggs-Gelasco P.J, Kirk M.L, Penner-Hahn J.E, Pecoraro V.L. Structural and magnetic effects of successive protonations of oxo bridges in high-valent manganese dimers. J. Am. Chem. Soc. 1994;116:11 349–11 356. doi:10.1021/ja00104a014 [Google Scholar]
- Bashkin J.S, Chang H.R, Streib W.E, Huffman J.C, Hendrickson D.N, Christou G. Modelling the photosynthetic water oxidation center: preparation and physical properties of a tetranuclear oxide bridged manganese complex corresponding to the native S2 state. J. Am. Chem. Soc. 1987;109:6502–6504. doi:10.1021/ja00255a041 [Google Scholar]
- Bhaduri, S., Pink, M. & Christou, G. 2002 Towards a synthetic model of the photosynthetic water oxidizing complex: [Mn3O4(O2CMe)(4)(bpy)(2)] containing the [Mn-3(IV)(mu-O)(4)](4) core. Chem. Commun. 2352–2353. [DOI] [PubMed]
- Bhula R, Gainsford G.J, Weatherburn D.C. A new model for the oxygen-evolving complex in photosynthesis. A trinuclear μ3-oxomanganese(III) complex which contains a μ-peroxo group. J. Am. Chem. Soc. 1988;110:7550–7552. doi:10.1021/ja00230a053 [Google Scholar]
- Bouwman E, Bolcar M.A, Libby E, Huffman J.C, Folting K, Christou G. Tetranuclear manganese(III)-oxo-carboxylate complexes possessing terminal phenoxide or alkoxide ligands. Inorg. Chem. 1992;31:5185–5192. doi:10.1021/ic00051a008 [Google Scholar]
- Britt R.D, Campbell K.A, Peloquin J.M, Gilchrist M.L, Aznar C.P, Dicus M.M, Robblee J, Messinger J. Recent pulsed EPR studies of the photosystem II oxygen- evolving complex: implications as to water oxidation mechanisms. Biochim. Biophys. Acta. 2004;1655:158–171. doi: 10.1016/j.bbabio.2003.11.009. doi:10.1016/j.bbabio.2003.11.009 [DOI] [PubMed] [Google Scholar]
- Brudvig G.W, Crabtree R.H. Mechanism for photosynthetic O2 evolution. Proc. Natl Acad. Sci. USA. 1986;83:4586–4588. doi: 10.1073/pnas.83.13.4586. doi:10.1073/pnas.83.13.4586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell K.A, Peloquin J.M, Pham D.P, Debus R.J, Britt R.D. Parallel polarization EPR detection of an S1-state ‘Multiline’ EPR signal in photosystem II particles from Synechocystis sp. PCC 6803. J. Am. Chem. Soc. 1998;120:447–448. doi:10.1021/ja972693y [Google Scholar]
- Casey J.L, Sauer K. EPR detection of a cryogenically photogenerated intermediate in photosynthetic oxygen evolution. Biochim. Biophys. Acta. 1984;767:21–28. doi:10.1016/0005-2728(84)90075-6 [Google Scholar]
- Caudle M.T, Pecoraro V.L. Thermodynamic viability of hydrogen atom transfer from water coordinated to the oxygen-evolving complex of photosystem II. J. Am. Chem. Soc. 1997;119:3415–3416. doi:10.1021/ja9641158 [Google Scholar]
- Caudle M.T, Riggs-Gelasco P, Gelasco A.K, Penner-Hahn J.E, Pecoraro V.L. Mechanism for the homolytic cleavage of alkyl hydroperoxides by the manganese(III) dimer MnIII2(2-OHsalpn)2. Inorg. Chem. 1996;35:3577–3584. doi:10.1021/ic951462u [Google Scholar]
- Chan M.K, Armstrong W.H. Tetranuclear manganese-oxo complex with a 2.7 Å Mn–Mn separation and intramolecular H2O–μ2–O hydrogen-bonded contacts: [Mn4O2(TPHPN)2(H2O)2(CF3SO3)2](CF3SO3)3. Possible mode for binding of water at the active site of the oxygen-evolving complex in photosystem II. J. Am. Chem. Soc. 1990;112:4985–4986. doi:10.1021/ja00168a067 [Google Scholar]
- Cheniae G.M, Martin I.F. Sites of function of manganese within photosystem II. Biochim. Biophys. Acta. 1970;197:219–239. doi: 10.1016/0005-2728(70)90033-2. doi:10.1016/0005-2728(70)90033-2 [DOI] [PubMed] [Google Scholar]
- Cinco R.M, Robblee J.H, Rompel A, Fernandez C, Yachandra V.K, Sauer K, Klein M.P. Strontium EXAFS reveals the proximity of calcium to the manganese cluster of oxygen-evolving photosystem II. J. Phys. Chem. B. 1998;102:8248–8256. doi: 10.1021/jp981658q. doi:10.1021/jp981658q [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins T.J, Gordon-Wylie S.W. A manganese(V)-oxo complex. J. Am. Chem. Soc. 1989;111:4511–4513. doi:10.1021/ja00194a063 [Google Scholar]
- Collins T.J, Powell R.D, Slebodnick C, Uffelman E.S. A water-stable manganese(V)-oxo complex: definitive assignment of a ν(Mn–O) triple bond infrared vibration. J. Am. Chem. Soc. 1990;112:899–901. doi:10.1021/ja00158a077 [Google Scholar]
- Dau H, Iuzzolino L, Dittmer J. The tetra-manganese complex during its redox cycle—X-ray absorption results and mechanistic implications. Biochim. Biophys. Acta. 2001;1503:24–39. doi: 10.1016/s0005-2728(00)00230-9. doi:10.1016/S0005-2728(00)00230-9 [DOI] [PubMed] [Google Scholar]
- Dismukes G.C, Siderer Y. Intermediates of a polynuclear manganese center involved in photosynthetic oxidation of water. Proc. Natl Acad. Sci. USA. 1981;78:274–278. doi: 10.1073/pnas.78.1.274. doi:10.1073/pnas.78.1.274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira K.N, Iverson T.M, Maghlaoui K, Barber J, Iwata S. Architecture of the photosynthetic oxygen-evolving center. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. doi:10.1126/science.1093087 [DOI] [PubMed] [Google Scholar]
- Gardner K.A, Mayer J.M. Understanding C–H bond oxidations: H and H− transfer in the oxidation of toluene by permanganate. Science. 1995;269:1849–1851. doi: 10.1126/science.7569922. doi:10.1126/science.7569922 [DOI] [PubMed] [Google Scholar]
- Ghanotakis D.F, Topper J.N, Babcock G.T, Yocum C.F. Water-soluble 17 and 23 kDa polypeptides restore oxygen evolution activity by creating a high-affinity binding site for Ca2+ on the oxidizing side of photosystem-II. FEBS Lett. 1984;170:169–173. doi:10.1016/0014-5793(84)81393-9 [Google Scholar]
- Gibney B.R, Wang H, Kampf J.W, Pecoraro V.L. Structural evaluation and solution integrity of alkali metal salt complexes of the manganese 12-metallacrown-4 (12-MC-4) structural type. Inorg. Chem. 1996;35:6184–6193. doi:10.1021/ic960371+ [Google Scholar]
- Gupta R, MacBeth C.E, Young V.G, Jr, Borovik A.S. Isolation of monomeric MnIII/II–OH and MnIII–O complexes from water: evaluation of O–H bond dissociation energies. J. Am. Chem. Soc. 2002;124:1136–1137. doi: 10.1021/ja016741x. doi:10.1021/ja016741x [DOI] [PubMed] [Google Scholar]
- Han K, Katoh S. Different localization of 2 Ca2+ in spinach oxygen-evolving photosystem II membranes—evidence for involvement of only one Ca2+ in oxygen evolution. Plant Cell Physiol. 1993;34:585. [Google Scholar]
- Haumann M, Liebisch P, Muller C, Barra M, Grabolle M, Dau H. Photosynthetic O2 formation tracked by time-resolved X-ray experiments. Science. 2005;310:1019–1021. doi: 10.1126/science.1117551. doi:10.1126/science.1117551 [DOI] [PubMed] [Google Scholar]
- Hillier W, Messinger J, Wydrzynski T. Kinetic determination of the fast exchanging substrate water molecule in the S3 state of photosystem II. Biochemistry. 1998;37:16 908–16 914. doi: 10.1021/bi980756z. doi:10.1021/bi980756z [DOI] [PubMed] [Google Scholar]
- Hillier W, Hendry G, Burnap R.L, Wydrzynski T. Substrate water exchange in photosystem II depends on the peripheral proteins. J. Biol. Chem. 2001;276:46 917–46 924. doi: 10.1074/jbc.M102954200. doi:10.1074/jbc.M102954200 [DOI] [PubMed] [Google Scholar]
- Hillier W, Wydrzynski T. The affinities for the two substrate water binding sites in the O(2) evolving complex of photosystem II vary independently during S-state turnover. Biochemistry. 2000;39:4399–4405. doi: 10.1021/bi992318d. [DOI] [PubMed] [Google Scholar]
- Hoganson C.W, Babcock G.T. A metalloradical mechanism for the generation of oxygen in photosynthesis. Science. 1997;277:1953–1956. doi: 10.1126/science.277.5334.1953. doi:10.1126/science.277.5334.1953 [DOI] [PubMed] [Google Scholar]
- Homann P.H. Chloride and calcium in photosystem II: from effects to enigma. Invited contribution. Photosynth. Res. 2002;73:169–175. doi: 10.1023/A:1020486729283. doi:10.1023/A:1020486729283 [DOI] [PubMed] [Google Scholar]
- Hsieh W.Y, Campbell K.A, Gregor W, Britt R.D, Yoder D.W, Penner-Hahn J.E, Pecoraro V.L. The first spectroscopic model for the S-1 state multiline signal of the OEC. Biochim. Biophys. Acta. 2004;1655:149–157. doi: 10.1016/j.bbabio.2003.12.001. doi:10.1016/j.bbabio.2003.12.001 [DOI] [PubMed] [Google Scholar]
- Kessissoglou, D. P., Kirk, M. L., Bender, C. A., Lah, M. S. & Pecoraro, V. L. 1989 A bent mixed valence Mn(III/II/III) complex: a new class of trinuclear, acetate bridged Schiff base compounds exhibiting a g=2 multiline ESR sig. J. Chem. Soc. Chem. Commun 84–86.
- Kessissoglou D.P, Kirk M.L, Lah M.S, Li X, Raptopoulou C, Hatfield W.E, Pecoraro V.L. Structural and magnetic characterization of trinuclear, mixed-valence manganese acetates. Inorg. Chem. 1992;31:5424–5432. doi:10.1021/ic00052a018 [Google Scholar]
- Kim D.H, Britt R.D, Klein M.P, Sauer K. The g=4.1 EPR signal of the S2 state of the photosynthetic oxygen-evolving complex arises from a multinuclear manganese cluster. J. Am. Chem. Soc. 1990;112:9389–9391. doi:10.1021/ja00181a049 [Google Scholar]
- Kitajima N, Osawa M, Imai S, Fujisawa K, Morooka Y, Heerwegh K, Reed C.A, Boyd P.D.W. Synthesis, structure and magnetic properties of a linear trimanganese(III,II,III) complex bridged with a (μ-hydroxo)bis(μ-acetato) unit. Inorg. Chem. 1994;33:4613–4614. doi:10.1021/ic00099a004 [Google Scholar]
- Kulik Leonid V, Epel B, Lubitz W, Messinger J. 55Mn pulse ENDOR at 34 GHz of the S0 and S2 states of the oxygen-evolving complex in photosystem II. J. Am. Chem. Soc. 2005;127:2392–2393. doi: 10.1021/ja043012j. doi:10.1021/ja043012j [DOI] [PubMed] [Google Scholar]
- Lah M.S, Pecoraro V.L. Isolation and characterization of {MnII[MnIII(salicylhydroximate)]4(acetate)2(DMF)6}.cntdot.2DMF: an inorganic analog of Mn2+ (12-crown-4) J. Am. Chem. Soc. 1989;111:7258–7259. doi:10.1021/ja00200a054 [Google Scholar]
- Larson, E. J., Riggs, P. J., Penner-Hahn, J. E.& Pecoraro, V. L. 1992 Protonation of (Mniv(Saltn)(Mu-2-O))2 results in significant modification of structure and catalase-like reactivity. J. Chem. Soc. Chem. Commun 102–103.
- Law N.A, Caudle M.T, Pecoraro V.L. Manganese redox enzymes and model systems: properties, structures, and reactivity. Adv. Inorg. Chem. 1998;46:305–440. [Google Scholar]
- Libby E, Folting K, Huffman J.C, Christou G. Feasibility of a ‘building-block’ approach to higher nuclearity manganese/oxygen/RCO2- aggregates: directed conversion of an [Mn4O2] to an [Mn8O4] complex. J. Am. Chem. Soc. 1990;112:5354–5356. doi:10.1021/ja00169a055 [Google Scholar]
- Libby E, McCusker J.K, Schmitt E.A, Folting K, Hendrickson D.N, Christou G. Preparation and properties of models for the photosynthetic water oxidation center: spin frustration in the manganese [Mn4O2(O2CR)7(pic)2]- anion. Inorg. Chem. 1991;30:3486–3495. doi:10.1021/ic00018a019 [Google Scholar]
- Lindberg K, Andreasson L.E. A one-site, two-state model for the binding of anions in photosystem II. Biochemistry. 1996;35:14 259–14 267. doi: 10.1021/bi961244s. doi:10.1021/bi961244s [DOI] [PubMed] [Google Scholar]
- Loll B, Kern J, Saenger W, Zouni A, Biesiadka J. Towards complete cofactor arrangement in the 3.0 Å resolution structure of photosystem II. Nature. 2005;438:1040–1044. doi: 10.1038/nature04224. doi:10.1038/nature04224 [DOI] [PubMed] [Google Scholar]
- Lundberg M, Siegbahn P.E.M. Investigations of structure and mechanism of the oxygen evolving complex in PSII. Phys. Chem. Chem. Phys. 2004;6:4772–4780. doi:10.1039/b406552b [Google Scholar]
- McEvoy J.P, Brudvig G.W. Water-splitting chemistry of photosystem II. Chem. Rev. 2006;106:4455–4483. doi: 10.1021/cr0204294. doi:10.1021/cr0204294 [DOI] [PubMed] [Google Scholar]
- Messinger J, Robblee J, Yu W.O, Sauer K, Yachandra V.K, Klein M.P. The S0 state of the oxygen-evolving complex in photosystem II is paramagnetic: detection of an EPR multiline signal. J. Am. Chem. Soc. 1997;119:11 349–11 350. doi: 10.1021/ja972696a. doi:10.1021/ja972696a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer T.J, Huynh M.H.V, Thorp H.H. The possible role of proton-coupled electron transfer (PCET) in water oxidation by photosystem II. Angew. Chem. Int. Ed. Engl. 2007;46:5284–5304. doi: 10.1002/anie.200600917. doi:10.1002/anie.200600917 [DOI] [PubMed] [Google Scholar]
- Miller C.G, Gordon-Wylie S.W, Horwitz C.P, Strazisar S.A, Peraino D.K, Clark G.R, Weintraub S.T, Collins T.J. A method for driving O-atom transfer: secondary ion binding to a tetraamide macrocyclic ligand. J. Am. Chem. Soc. 1998;120:11 540–11 541. doi:10.1021/ja972922g [Google Scholar]
- Mukherjee, C., Weyhermueller, T., Wieghardt, K. & Chaudhuri, P. 2006 A trinuclear complex containing MnIIMnIIIMnIV, radicals, quinone and chloride ligands potentially relevant to PS II. Dalton Trans 2169–2171. [DOI] [PubMed]
- Mukhopadhyay S, Mandal S.K, Bhaduri S, Armstrong W.H. Manganese clusters with relevance to photosystem II. Chem. Rev. 2004;104:3981–4026. doi: 10.1021/cr0206014. doi:10.1021/cr0206014 [DOI] [PubMed] [Google Scholar]
- Pecoraro, V. L. & Hsieh, W. Y. 2000 The use of model complexes to elucidate the structure and function of manganese redox enzymes. In Metal ions in biological systems, vol. 37 (eds H. Sigel & A. Sigel), pp. 429–504. New York, NY: Marcel-Dekker. [PubMed]
- Pecoraro V.L, Baldwin M.J, Gelasco A. Interaction of manganese with dioxygen and its reduced derivatives. Chem. Rev. 1994;94:807–826. doi:10.1021/cr00027a012 [Google Scholar]
- Pecoraro, V. L., Stemmler, A. J., Gibney, B. R., Bodwin, J. J., Wang, H., Kampf, J. W. & Almut, B. 1997 Progress in inorganic chemistry, vol. 45 (ed. K. D. Karlin), pp. 83–177. New York, NY: Wiley & Sons.
- Pecoraro V.L, Baldwin M.J, Caudle M.T, Hsieh W.-Y, Law N.A. A proposal for water oxidation in photosystem II. Pure Appl. Chem. 1998;70:925–929. [Google Scholar]
- Peloquin J.M, Campbell K.A, Randall D.W, Evanchik M.A, Pecoraro V.L, Armstrong W.H, Britt R.D. 55Mn ENDOR of the S2-state multiline EPR signal of photosystem II: implications on the structure of the tetranuclear Mn cluster. J. Am. Chem. Soc. 2000;122:10 926–10 942. doi:10.1021/ja002104f [Google Scholar]
- Penner-Hahn J.E, Fronko R.M, Pecoraro V.L, Yocum C.F, Betts S.D, Bowlby N.R. Structural characterization of the manganese sites in the photosynthetic oxygen-evolving complex using X-ray absorption spectroscopy. J. Am. Chem. Soc. 1990;112:2549–2557. doi:10.1021/ja00163a011 [Google Scholar]
- Popelkova H, Betts S.D, Lydakis-Symantiris N, Im M.M, Swenson E, Yocum C.F. Mutagenesis of basic residues R151 and R161 in manganese-stabilizing protein of photosystem II causes inefficient binding of chloride to the oxygen-evolving complex. Biochemistry. 2006;45:3107–3115. doi: 10.1021/bi0523759. doi:10.1021/bi0523759 [DOI] [PubMed] [Google Scholar]
- Poulsen A.K, Rompel A, McKenzie C.J. Water oxidation catalysed by a dinuclear Mn complex: a new model for the oxygen evolving center of photosystem II. Angew. Chem. 2005;44:6916–6920. doi: 10.1002/anie.200502114. doi:10.1002/anie.200502114 [DOI] [PubMed] [Google Scholar]
- Proserpio D.M, Hoffmann R, Dismukes G.C. Molecular mechanism of photosynthetic oxygen evolution. A theoretical approach. J. Am. Chem. Soc. 1992;114:4374–4382. doi:10.1021/ja00037a052 [Google Scholar]
- Riggs-Gelasco P.J, Mei R, Ghanotakis D.F, Yocum C.F, Penner-Hahn J.E. X-ray absorption spectroscopy of calcium-substituted derivatives of the oxygen-evolving complex of phostosytem II. J. Am. Chem. Soc. 1996;118:2400–2410. doi:10.1021/ja9504505 [Google Scholar]
- Robblee J.H, Cinco R.M, Yachandra V.K. X-ray spectroscopy-based structure of the Mn cluster and mechanism of photosynthetic oxygen evolution. Biochim. Biophys. Acta. 2001;1503:7–23. doi: 10.1016/s0005-2728(00)00217-6. doi:10.1016/S0005-2728(00)00217-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruettinger W, Yagi M, Wolf K, Bernasek S, Dismukes G.C. O2 evolution from the manganese-oxo cubane core Mn4O46+: a molecular mimic of the photosynthetic water oxidation enzyme? J. Am. Chem. Soc. 2000;122:10 353–10 357. doi:10.1021/ja0005587 [Google Scholar]
- Sauer K. A role for manganese in oxygen evolution in photosynthesis. Acc. Chem. Res. 1980;13:249–256. doi:10.1021/ar50152a001 [Google Scholar]
- Siegbahn P.E.M, Blomberg M.R.A. Methods and models for studying mechanisms of redox-active enzymes. Phil. Trans. R. Soc. A. 2005;363:847–860. doi: 10.1098/rsta.2004.1542. doi:10.1098/rsta.2004.1542 [DOI] [PubMed] [Google Scholar]
- Tagore R, Chen H, Crabtree R.H, Brudvig G.W. Determination of μ-oxo exchange rates in di-μ-oxo dimanganese complexes by electrospray ionization mass spectrometry. J. Am. Chem. Soc. 2006;128:9457–9465. doi: 10.1021/ja061348i. doi:10.1021/ja061348i [DOI] [PubMed] [Google Scholar]
- Tagore R, Crabtree R.H, Brudvig G.W. Distinct mechanisms of bridging-oxo exchange in di-μ-O dimanganese complexes with and without water-binding sites: implications for water binding in the O2-evolving complex of photosystem II. Inorg. Chem. 2007;46:2193–2203. doi: 10.1021/ic061968k. doi:10.1021/ic061968k [DOI] [PubMed] [Google Scholar]
- Tangoulis V, Malamatari D.A, Soulti K, Stergiou V, Raptopoulou C.P, Terzis A, Kabanos T.A, Kessissoglou D.P. Manganese(II/II/II) and manganese(III/II/III) trinuclear compounds, structure and solid and solution behavior. Inorg. Chem. 1996;35:4974–4983. doi: 10.1021/ic960183j. doi:10.1021/ic960183j [DOI] [PubMed] [Google Scholar]
- Tommos C, Tang X.-S, Warncke K, Hoganson C.W, Styring S, McCracken J, Diner B.A, Babcock G.T. Spin-density distribution, conformation, and hydrogen bonding of the redox-active tyrosine YZ in photosystem II from multiple-electron magnetic-resonance spectroscopies: implications for photosynthetic oxygen evolution. J. Am. Chem. Soc. 1995;117:10 325–10 335. doi:10.1021/ja00146a017 [Google Scholar]
- van Gorkom H.J, Yocum C.F. The calcium and chloride cofactors. In: Wydrzynski T.J, Satoh K, editors. Photosystem II: the light-driven water: plastoquinone oxidoreductase. Advances in photosynthesis and respiration. vol. 22. Springer; Dordrecht, The Netherlands: 2005. pp. 307–328. [Google Scholar]
- Vincent J.B, Christmas C, Chang H.R, Li Q, Boyd P.D.W, Huffman J.C, Hendrickson D.N, Christou G. Modeling the photosynthetic water oxidation center preparation and properties of tetranuclear manganese complexes containing [Mn4O2]6+,7+,8+ cores, and the crystal structures of Mn4O2(O2CMe)6(bipy)2 and [Mn4O2(O2CMe)7(bipy)2](ClO4) J. Am. Chem. Soc. 1989;111:2086–2097. doi:10.1021/ja00188a023 [Google Scholar]
- Waggoner C.M, Pecoraro V, Yocum C.F. Mono-valent cations (Na+, K+, Cs+) inhibit calcium activation of photosynthetic oxygen evolution. FEBS Lett. 1989;244:237–240. doi:10.1016/0014-5793(89)81200-1 [Google Scholar]
- Wieghardt K. Die aktiven Zentren in manganhaltigen Metalloproteinen und anorganische Modellkomplexe. Angew. Chem. 1989;101:1179–1198. doi:10.1002/ange.19891010905 [Google Scholar]
- Wieghardt K, Bossek U, Nuber B, Weiss J, Gehring S, Haase W. Synthesis of novel trimeric μ-oxo-bridged manganese (IV) complexes: [L3Mn3IV(μ2-O)3(μ3-XO4)]Br3(X=P, As, or Vv; L=1, 4, 7-triazacyclononane) J. Chem. Soc. Chem. Commun. 1988;17:1145–1146. doi:10.1039/C39880001145 [Google Scholar]
- Wincencjusz H, Yocum C.F, van Gorkom H.J. Activating anions that replace Cl− in the O2-evolving complex of photosystem II slow the kinetics of the terminal step in water oxidation and destabilize the S2 and S3 states. Biochemistry. 1999;38:3719–3725. doi: 10.1021/bi982295n. doi:10.1021/bi982295n [DOI] [PubMed] [Google Scholar]
- Wu A.J, Penner-Hahn J.E, Pecoraro V.L. Structural, spectroscopic, and reactivity models for the manganese catalases. Chem. Rev. 2004;104:903–938. doi: 10.1021/cr020627v. doi:10.1021/cr020627v [DOI] [PubMed] [Google Scholar]
- Wydrzynski T. Springer; Dordrecht, The Netherlands: 2005. Photosystem II: the light-driven water: plastoquinone oxidoreductase. [Google Scholar]
- Yachandra V.K. The catalytic manganese cluster: organization of the metal ions. In: Wydrzynski T.J, Satoh K, editors. Photosystem II: the light-driven water: plastoquinone oxidoreductase. Advances in photosynthesis and respiration. vol. 22. Springer; Dordrecht, The Netherlands: 2005. pp. 235–260. [Google Scholar]
- Yachandra V.K, DeRose V.J, Latimer M.J, Mukerji I, Sauer K, Klein M.P. Where plants make oxygen: a structural model for the photosynthetic oxygen-evolving manganese cluster. Science. 1993;260:675–679. doi: 10.1126/science.8480177. doi:10.1126/science.8480177 [DOI] [PubMed] [Google Scholar]
- Yachandra V.K, Sauer K, Klein M.P. Manganese cluster in photosynthesis: where plants oxidize water to dioxygen. Chem. Rev. 1996;96:2927–2950. doi: 10.1021/cr950052k. doi:10.1021/cr950052k [DOI] [PubMed] [Google Scholar]
- Yano S, Doi M, Tamakoshi S, Mori W, Mikuriya M, Ichimura A, Kinoshita I, Yamamoto Y, Tanase T. Trimanganese complexes with a linear MnIIMnIII MnII assemblage bridged by carbohydrates. Chem. Commun. 1997:997–998. doi:10.1039/a701198i [Google Scholar]
- Yano J, et al. Where water is oxidized to dioxygen: Structure of the photosynthetic Mn4Ca cluster. Science. 2006;314:821–825. doi: 10.1126/science.1128186. doi:10.1126/science.1128186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yocum C.F. In: Manganese redox enzymes. Pecoraro V.L, editor. VCH; New York, NY: 1992. p. 71. [Google Scholar]
- Yocum C.F, Pecoraro V.L. Recent advances in the understanding of the biological chemistry of manganese. Curr. Opin. Chem. Biol. 1999;3:182–187. doi: 10.1016/S1367-5931(99)80031-3. doi:10.1016/S1367-5931(99)80031-3 [DOI] [PubMed] [Google Scholar]
- Yocum C.F, Yerkes C.T, Blankenship R.E, Sharp R.R, Babcock G.T. Stoichiometry, inhibitor sensitivity, and organization of manganese associated with photosynthetic oxygen evolution. Proc. Natl Acad. Sci. USA. 1981;78:7507–7511. doi: 10.1073/pnas.78.12.7507. doi:10.1073/pnas.78.12.7507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaleski, C. M., Dendrinou-Samara, C., Alexiou, M., Kanakaraki, P., Kampf, J., Penner Hahn, J. E., Pecoraro, V. L. & Kessissoglou, D. P. Submitted. Structural and XANES features of tetranuclear [MnSII MnIV] and [Mn2II Mn2III] valence-isomers of manganese clusters.
- Zouni A, Jordan R, Schlodder E, Fromme P, Witt H.T. First photosystem II crystals capable of water-oxidation. Biochim. Biophys. Acta. 2000;1457:103–105. doi: 10.1016/s0005-2728(00)00100-6. doi:10.1016/S0005-2728(00)00100-6 [DOI] [PubMed] [Google Scholar]
- Zouni A, Witt H.-T, Kern J, Fromme P, Krauss N, Saenger W, Orth P. Crystal structure of oxygen evolving photosystem II from Synechococcus elongatus: a 3.8 Å resolution. Nature. 2001;409:739–743. doi: 10.1038/35055589. doi:10.1038/35055589 [DOI] [PubMed] [Google Scholar]
Additional reference
- Hillier, W. & Messinger, J. 2005 Mechanism of photosynthetic oxygen production. In: Photosystem II. The light-driven water: plastoquinone oxidoreductase, advances in Photosynthesis and Respiration, vol. 22 (eds T. Wydrzynski, and K. Satoh), pp. 567–608. Springer: Netherlands.
- Messinger J, Badger M, Wydrzynski T. Detection of one slowly exchanging substrate water molecule in the S3 state of photosystem II. Proc Natl Acad Sci USA. 1995;92:3209–3213. doi: 10.1073/pnas.92.8.3209. doi:10.1073/pnas.92.8.3209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messinger J. Evaluation of different mechanistic proposals for water oxidation in photosynthesis on the basis of Mn4OxCa structures for the catalytic site and spectroscopic data. Phys. Chem. Chem. Phys. 2004;6:4764–4771. doi:10.1039/b406437b [Google Scholar]
- Weng T.C, Hsieh W.Y, Uffelman E.S, Gordon-Wylie S.W, Collins T.J, Pecoraro V.L, Penner-Hahn J.E. XANES evidence against a manganyl species in the S3 state of the oxygen-evolving complex. J. Am. Chem. Soc. 2004;126:8070–8071. doi: 10.1021/ja0494104. doi:10.1021/ja0494104 [DOI] [PubMed] [Google Scholar]