

Abstract
Heat shock protein 27 (Hsp27), a recently discovered member of the heat shock protein family, is markedly induced in the brain after cerebral ischemia and other injury states. In non-neuronal systems, Hsp27 has potent cell death-suppressing functions. However, the mechanism of Hsp27-mediated neuroprotection has not yet been elucidated. Using transgenic and viral overexpression of Hsp27, we investigated the molecular mechanism by which Hsp27 exerts its neuroprotective effect. Overexpression of Hsp27 conferred long-lasting tissue preservation and neurobehavioral recovery, as measured by infarct volume, sensorimotor function, and cognitive tasks up to 3 weeks following focal cerebral ischemia. Examination of signaling pathways critical to neuronal death demonstrated that Hsp27 overexpression led to the suppression of the MKK4/JNK kinase cascade. While Hsp27 overexpression did not suppress activation of an upstream regulatory kinase of the MKK/JNK cascade, ASK1, Hsp27 effectively inhibited ASK1 activity via a physical association through its N-terminal domain and the kinase domain of ASK1. The N-terminal region of Hsp27 was required for neuroprotective function against in vitro ischemia. Moreover, knockdown of ASK1 or inhibition of the ASK1/MKK4 cascade effectively inhibited cell death following neuronal ischemia. This underscores the importance of this kinase cascade in the progression of ischemic neuronal death. Inhibition of PI3K had no effect on Hsp27-mediated neuroprotection, suggesting that Hsp27 does not promote cell survival via activation of PI3K/Akt. Based on these findings, we conclude that overexpression of Hsp27 confers long-lasting neuroprotection against ischemic brain injury via a previously unexplored association and inhibition of ASK1 kinase signaling.
Keywords: cerebral ischemia, neuroprotection, heat shock proteins, apoptosis signaling kinase-1, JNK, mitochondria
Introduction
Neuronal injury following cerebral ischemia initiates a complex series of signaling cascades that lead to cell death (Lipton, 1999; Graham and Chen, 2001; Lo et al., 2003). Many of these pathways ultimately converge on the mitochondria, resulting in the release of mitochondrial proteins such as cytochrome c and Smac into the cytosol and subsequent activation of cell death execution molecules (Fujimura et al., 1998; Sugawara et al., 1999). Neuroprotective interventions targeted beyond the level of mitochondrial damage at times serve only to delay rather than prevent cell death, with limited effects on long-term functional outcomes (Loetscher et al., 2001; Bilsland and Harper, 2002). Accordingly, defining critical upstream prodeath pathways and identifying neuroprotective agents targeting these pathways have become important strategies in developing novel neuroprotective therapies for stroke.
The heat shock protein (Hsp) family consists of members within a wide molecular-size range. Heat shock proteins perform cell survival-promoting functions, ranging from protein chaperoning (exemplified by Hsp70) to more recently discovered pathway-specific cell death suppressive functions (Garrido et al., 2006; Arya et al., 2007). In non-neuronal systems, Hsp27 in particular has been found to possess potent anti-apoptotic functions. The majority of studies have focused on the effects of Hsp27 on downstream death-execution pathways; Hsp27 may directly inactivate caspase-3 and caspase-9, or indirectly suppress caspase activation via inhibition of apoptosome formation (Bruey et al., 2000; Concannon et al., 2001, 2003; Voss et al., 2007). Recently, several reports indicate that independent of its downstream effects, Hsp27 may affect upstream signaling cascades, including MK2 and Akt (Konishi et al., 1997; Mearow et al., 2002; Rane et al., 2003; Zheng et al., 2006; Wu et al., 2007; Havasi et al., 2008).
In neuronal systems, Hsp27 can exert neuroprotective effects against neuronal injury, including cerebral ischemia (Badin et al., 2006; An et al., 2008). Endogenous induction of Hsp27 has been observed in cells surviving ischemic insults (Kato et al., 1995) and in ischemic preconditioning models (Currie et al., 2000; Dhodda et al., 2004), suggesting that Hsp27 is associated with cellular survival against cerebral ischemia. However, the precise mechanism underlying this protection is currently unknown. Oxidative stress, including cerebral ischemia, triggers neuronal cell death via activation of the mitochondrial prodeath pathway (Christophe and Nicolas, 2006). The initiation of this pathway leads to release of prodeath proteins, resulting in activation of both caspase-dependent and caspase-independent mechanisms and neuronal death (Graham and Chen, 2001). Therefore, in the present study we seek to determine whether Hsp27-mediated neuroprotection is due to a direct or indirect suppression of the mitochondrial cell death pathway in ischemic neurons.
In the context of cerebral ischemia, several upstream cell death regulatory signaling pathways have been identified. Activation of the c-Jun N-terminal kinase (JNK) has been observed in many neuronal injury models, and appears to be critical in mediating neuronal cell death (Gao et al., 2005; Philpott and Facci, 2008). The upstream kinase apoptosis signal-regulating kinase (ASK1) can lead to the activation of JNK via MKK4 and MKK7 in response to oxidative stress, endoplasmic reticular stress, DNA damage, or inflammation (Nagai et al., 2007). All of these stressors also contribute to cerebral ischemic pathology (Paschen, 2004; Martin, 2008), thus supporting the hypothesis that activation of ASK1 may be an important converging point of multiple prodeath signals that participate in ischemic neuronal death (Nagai et al., 2007). In the present study, we present new evidence that Hsp27 overexpression leads to robust and prolonged neuroprotection against stroke in mice. Furthermore, we have identified ASK1 as a novel target for Hsp27, and show that Hsp27 directly inhibits ASK1-dependent MKK4/JNK activation and, subsequently, mitochondrial prodeath signaling.
Materials and Methods
Creation of the Hsp27 transgenic mice.
The chimeric transgene that we used to create the transgenic mice contains human Hsp27 cDNA under the control of cytomegalovirus enhancer and a chicken β-actin promoter with the first intron. An HA tag was added before the Hsp27 coding region. The microinjection and ES screening procedures were done at the University of Pittsburgh Transgenic Core. Founders were used to establish independent transgenic lines by breeding to wild-type F1 hybrid mice. In all experiments, wild-type mice backbred from the same transgenic colonies were used as controls. All lines of mice were backbred to the C57/B6 background for at least eight generations before use to minimize the potential influence of genetic heterogeneity on the accessibility to stroke. Only male adult mice (25–30 g) were used in the study.
Murine model of transient focal ischemia.
All animal experiments were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Male 2- to 3-month-old C57/B6 mice were anesthetized with 1.5% isoflurane in a 30% O2/68.5% N2O mixture under spontaneous breathing. Focal cerebral ischemia was produced by intraluminal occlusion of the left middle cerebral artery (MCAO) as described previously (Cao et al., 2002). Rectal temperature was controlled at ∼37.0°C throughout experiment via a temperature-regulated heating pad. Arterial blood pressure was monitored through a tail cuff (XBP1000 Systems, Kent Scientific Corporation), and arterial blood gas was analyzed 15 min after induction of ischemia and 15 min after reperfusion. The animals underwent MCA occlusion for 60 min and then reperfusion for the indicated duration. After recovering from anesthesia, the animals were maintained in an air-conditioned room at 20°C. In all experiments, the surgeon was blinded to the genotypes of mice.
To confirm the induction of ischemia and successful reperfusion, changes in regional cerebral blood flow (rCBF) before, during and after MCA occlusion were evaluated in animals using laser-Doppler flowmetry (Cao et al., 2002). In two groups of mice (wild-type or Tg-Hsp27H), quantitative rCBF measurement was performed at 30 min after the onset of MCA occlusion using [14]-iodoantipyrine autoradiography as described previously (Sawada et al., 2000).
Neurobehavioral tests.
Behavioral tests were performed to assess sensorimotor deficits (1–7 d after ischemia) and long-term cognitive deficits (15–21 d after ischemia) in Hsp27 transgenic mice (Tg-Hsp27H) and wild-type littermates. All behavioral assessments were made during the light phase of the circadian cycle starting ∼4 h after the light phase onset. The individual conducting the behavioral assessments was not informed of experimental group assignment. The assessment for sensorimotor deficits consisted of three different tests. (1) The corner test was performed as described previously (Zhang et al., 2002; Gao et al., 2005), in which the ischemic mouse turns preferentially toward the nonimpaired (left) side. The direction of each turn was recorded from 10 trials for each test. (2) The rotarod test which began 2 d before surgery consisted of 5 trials each day. On the day of surgery the mice underwent 5 trials, the mean of which was used as the presurgery baseline value for each animal. After surgery, each animal was tested for 5 trials on a daily basis for 7 d (Gibson et al., 2005). The data are expressed as the percentage of mean duration per day compared with the presurgery baseline value. (3) The fault test was performed daily for 7 d after surgery as described (Gibson et al., 2005). In brief, mice were placed on an elevated grid surface (30 L × 35 W × 31 H cm) with a grid opening of 2.5 cm2. Each animal was tested for 3 trials lasting 1 min each, with an interval of 1 min between trials. The data are expressed as the number of errors made by the contralateral limbs as a percentage of the total faults.
For detecting cognitive deficits in stroke mice, the Morris water maze was performed to assess spatial reference memory using a modified protocol (Hattori et al., 2000; Gibson et al., 2005) from the original method (Morris, 1984). The Morris water maze paradigm is an open-field procedure in which mice learn to find a hidden platform to escape from a forced swimming task. Mice were placed in the maze on day 14 after surgery for two acclimatization trials without platform. Thereafter, the mice were tested for 4 trials each day for 7 d. To avoid confounding effects from motor deficits, mice were tested on days 15–21 after surgery, when motor deficits have recovered in this model. Data are expressed as the time (in seconds) that was required for the animal to reach the submerged platform on each day (latency). In the last session of 2 trials, the mice underwent the “probe trial,” in which the platform was removed and the animal was allowed to swim freely for 30 s. The time spent in the goal quadrant (where the platform was previously located) and the swim speed were calculated.
Measurement of infarct volume.
At 48 h after MCA occlusion, animals were killed, brains were removed, and the forebrain was sliced into 7 coronal sections each 1 mm thick. Sections were stained with 3% 2,3,5-triphenyltetrazolium in saline for 20 min, and then fixed in 4% paraformaldehyde in PBS, pH 7.4. Infarct volume was determined using MCID image analysis (Cao et al., 2002). For long-term assessment of infarct size, animals were killed at 21 d after surgery. Frozen coronal sections were prepared for cresyl violet staining (Cao et al., 2002), and infarct volume measured as described above by an observer who was blinded to the experimental group assignment.
Immunohistochemistry.
Animals were killed in a carbon dioxide chamber at the indicated time points after induction of ischemia, and the brains were prepared for freshly frozen coronal sectioning (15 μm thick). For immunohistochemical staining of phospho-c-Jun (p-c-Jun) or active caspase-3 (cleaved form), the rabbit polyclonal anti-p-c-Jun (Ser63) antibody (dilution 1:200) and the rabbit monoclonal anti-caspase-3 antibody (dilution 1:250) were used, respectively. For double-label immunofluorescence staining, sections were first incubated with anti-p-c-Jun antibody at 4°C for 48 h followed by incubation for 2 h at room temperature with goat anti-rabbit Cy3.18 immunoconjugate (Jackson ImmunoResearch) at 1:2500 dilutions. Sections were then subjected to incubation for 24 h in mouse anti-NeuN antibody (dilution at 1:500, Chemicon). This was followed by incubation with biotin-conjugated anti-mouse antibody (dilution 1:3000) and then fluorescein-avidin D (Vector Laboratories) at 8 μg/ml. For the assessment of nonspecific staining, alternating sections from each experimental condition were incubated without the primary antibody.
To quantify caspase-3-positive cells after ischemia, immunohistochemistry was performed using the above procedures except that the secondary antibodies were detected using the Elite Vectastain ABC kit and the diaminobenzidine substrate kit (Vector). Cell counting was done with stereology using the Bioquant Image Analysis program (Bioquant) as detailed previously (Cao et al., 2007).
cDNA isolation, site-directed mutagenesis, and generation of recombinant proteins.
The human full-length Hsp27 was obtained using PCR from a human kidney cDNA library. The human full-length ASK1 cDNA (ASK1 h) was from Dr. Yong Lee (University of Pittsburgh, Pittsburgh, PA). Various truncated Hsp27 or ASK1 cDNAs were derived from the full-length sequences using PCR. The kinase-inactive ASK1 dominant negative (containing the K709R mutation) was generated by site-directed mutagenesis using the Gene Editor system (Promega). The sequences of all cDNAs were verified at the University of Pittsburgh Gene Service Facility by sequencing reactions on both strands.
Recombinant proteins of full-length or truncated Hsp27 were generated using the standard method (Cao et al., 2001b). In brief, the cDNA was fused into the glutathione S-transferase (GST) gene in PGEX-2T vector (GE Healthcare). The GST fusion protein was expressed in Escherichia coli BL21 cells, absorbed to a glutathione-Sepharose 4B column, and then cleaved by thrombin to remove GST.
Construction of viral vectors.
Adeno-associated virus (AAV) vectors carrying either the human full-length (AAV-Hsp27) or truncated (AAV-Hsp27ΔN, AAV-Hsp27ΔC) Hsp27 cDNA were constructed using the expressing vector plasmid described previously (Cao et al., 2004). Large-scale production of the AAV vector was performed using the adenovirus-free triple-plasmid cotransfection method (Xiao et al., 1998). A total of 50 μg of plasmid mixture was cotransfected into human HEK293 cells with the assistance of 0.25 m CaCl2, and the cells were grown in DMEM containing 10% fetal bovine serum for 48 h and then harvested. AAV vectors were purified using a fast protein liquid chromatography system in conjunction with HiTrap Heparin columns (GE Healthcare).
To construct lentiviral vectors overexpressing human full-length ASK1 (Lenti-ASK1 h) or its dominant negative form (Lenti-ASK1dn), the cDNA (HA tagged) was inserted into the lentiviral transfer vector FSW under the control of neuron-specific Synapsin I promoter. To construct lentiviral vectors expressing short hairpin interfering RNA (shRNA) against murine ASK1 or MLK3, or rat Hsp27, the gene-specific targeting sequence (ASK1t: 5′-CCAACAACATCATCCTCTA-3′ and 5′-TGAACACCATTACCGAAGA-3′; MLK3t: 5′-GTGTAACAGCTGAGAGTGT-3′ and 5′-GTCCAACAACATTCTGTTG-3′; Hsp27, 5′-GCTACATCTCTCGGTGCTTCA-3′ and 5′-GCCCAAAGCAGTCACACAATC-3′) or its counterpart scramble sequence was inserted into the transfer vector FSW under the control of U6 promoter. The constructed transfer vectors were transformed into Stbl3 E. coli, and then isolated using the EndoFree Plasmid Maxi Kit (Qiagen). For large-scale production of the virus, a slightly modified protocol (Ramezani and Hawley, 2002) was used. In brief, a plasmid mixture containing 435 μg of pCMVΔR8.9 (packaging construct), 237 μg of pVSVG (envelope plasmid) and 675 μg of FSW (transfer vector) was suspended in 34.2 ml of CaCl2 (250 mm) and then added volume to volume into 2× BES buffer (pH 6.95). The DNA-CaCl2 precipitate was added into human kidney 293 FT cells (on 15 cm plates at the density of 1.1 × 107/plate) drop by drop (1.125 ml each plate) and allowed to incubate for 12 h before switching to fresh culture medium. The supernatant was collected 72 h after transfection, filtered through the 0.45 μm filter flask and centrifuged at 21,000 rpm for 2 h using the SW28 rotor (Beckman Coulter). Viruses were further purified by sucrose gradient ultracentrifuge. The pellet was suspended in 3 ml of PBS, loaded on the top of 2 ml of 20% sucrose solution, and centrifuged at 22,000 rpm for 2 h using the SW50.1 rotor (Beckman Coulter). The resulting pellet was resuspended in 200 μl of DMEM, aliquoted, and stored at −70°C. The titer of vector stock was determined using ELISA.
Primary neuronal cultures and in vitro model of ischemia.
Cortical neurons with two different genetic phenotypes, including Tg-Hsp27H and wild type, were derived from embryos of mice and maintained 10–12 d in vitro (DIV) before experiments, when cultures consisted primarily of neurons (∼97%) as determined using cell phenotype-specific immunocytochemistry (Cao et al., 2001a). In additional experiments, primary cultures of cortical neurons were prepared from 17-d-old Sprague Dawley rat embryos as described previously (Cao et al., 2001b), and experiments were conducted at 10–12 DIV.
To model ischemia-like conditions in vitro, primary cultures were exposed to transient oxygen and glucose deprivation (OGD) for 45 or 60 min as described previously (Cao et al., 2001b, 2007). Control glucose-containing cultures were incubated for the same periods of time in humidified 95% air and 5% CO2. Fluorescence of Alamar blue (Accumed International), an indicator that changes from blue to red and fluoresces when reduced by cellular metabolic activity, was used to measure the viability of the cultured neuron at 24–48 h after OGD (Cao et al., 2007). One-half of the culture medium was replaced with MEM-Pak containing 10% (v/v) Alamar blue, and cultures were incubated for 1.5 h at 37°C in humidified 95% air and 5% CO2. Fluorescence was determined in a Millipore CytoFluor 2300 automated plate-reading fluorimeter, with excitation at 530 nm and emission at 590 nm.
OGD-induced cell death was quantified by measuring lactate dehydrogenase (LDH) release from damaged cells into the culture medium (Cao et al., 2003). In brief, 10 μl aliquots of medium taken from the cell culture wells were added to 200 μl of LDH reagent (Sigma). Using a spectrophotometer plate reader (Molecular Devices), the emission was measured at 340 nm, which is proportional to the amount of LDH in the medium. The percentage of cell death was calculated as described previously (Cao et al., 2003).
In selective experiments, cell death was evaluated after OGD using Hoechst 33258 nuclear staining or Klenow-mediated DNA nick-end labeling. The percentages of cells showing chromatin condensation or DNA damage were quantified by counting at least 3000 cells under each experimental condition (three randomly selected fields per well, four to six wells per condition per experiment, and three independent experiments).
Gene transfection in primary neurons by viral vectors and in HEK293 cells using Lipofectamine.
The neuronal cultures were infected with the AAV-Hsp27, AAV-Hsp27ΔN, AAV-Hsp27ΔC, or the control vector (AAV-GFP or empty AAV) for 6 h as previously described (Cao et al., 2004) and then incubated in vector-free normal media for 72 h. The overexpression of Hsp27 in neurons was confirmed by immunocytochemistry and Western blot, respectively, using the anti-hemagglutinin (HA) antibody. For gene transfection using lentivirus vectors, the neuronal cultures were infected for 3 d with the Lenti-ASK1t, Lenti-ASK1 s, Lenti-Hsp27t, Lenti-Hsp27 s, Lenti-MLK3t, Lenti-MLK3 s, Lenti-ASK1 h, or Lenti-ASK1dn or the control vector (Lenti-GFP). Knockdown of ASK1, MLK3, or Hsp27 in neurons by lentivirus vectors was assessed using Western blot.
To study protein interactions between Hsp27 and ASK1 in cells, expression plasmids containing cDNA for the full-length or truncated human ASK1 (all Flag-tagged) or Hsp27 (all HA-tagged) were cotransfected into human HEK293 cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol.
Western blot.
Western blot was performed using the standard method and enhanced chemiluminescence detection reagents (GE Healthcare). The following antibodies were used: rabbit monoclonal antibodies against JNK3 (clone 55A8, 1:1000), active caspase-3 (1:1000), p-JNK (Thr183/Tyr185) (clone 81E11, 1:1000), p-ERK (clone 137F5, 1:2000), p-p38 (Thr180/Tyr182) (clone 3D7, 1:1000), p-c-Jun (Ser63) (clone 54B3,1:1000), p-Akt (Ser473) (clone 193H12, 1:1000), purchased from Cell Signaling; rabbit polyclonal antibodies against p-MKK4 (Ser257/Thr261) (1:1000), p-MKK7 (Ser271/Thr275) (1:1000), p-ASK1 (Thr845) (1:1000), MLK3 (1:1000), from Cell Signaling; rabbit polyclonal antibodies against Bax (1:1000), Bim (1:500), β-actin (1:2000), α-tubulin (1:2000), cytochrome c (1:1000), Smac (1:1000), HA (1:1000), Flag (1:1000), from Santa Cruz Biotechnology; mouse monoclonal anti-cytochrome c oxidase IV antibody (1:1000), from Invitrogen; and rabbit polyclonal anti-PUMA antibody, a gift from Dr. Jian Yu (Pittsburgh Cancer Institute, Pittsburgh, PA).
Protein kinase assays.
Cell lysates were prepared under nondenaturing conditions as described (Gao et al., 2005), and 150 μg of protein was used for each kinase assay. To assay for JNK activity (JNK 1 or JNK3), a capture JNK assay was performed using a nonradioactive kinase assay kit according to the manufacturer's instructions (Cell Signaling). Briefly, the cell lysates were first subjected to JNK1 capture using the specific anti-JNK1 antibody (clone F-3, Santa Cruz), and then the immunoprecipitates were incubated with recombinant GST-c-Jun (1–79) in the presence of ATP and subsequently immunoblotted using the anti-phospho-c-Jun (Ser-63) antibody. To assay for the JNK3-specific kinase activity, cell lysates were immunoprecipitated with a mixture of a monoclonal antibody that recognizes JNK1/2 (clone G151–666, BD PharMingen) and a monoclonal antibody that recognizes JNK1 (clone F-3, Santa Cruz) to remove both JNK1 and JNK2 from the lysates (Gao et al., 2005). The remaining kinase activity (JNK3) in the supernatant was assayed by the capture JNK assay as described above. To assay for ASK1 kinase activity, the kinase in cell lysates was captured using the anti-ASK1 antibody (clone s.c.-7931, Santa Cruz) and then incubated with recombinant myelin basic protein (MBP) in the presence of [γ-32P]ATP, and MBP phosphorylation was detected using autoradiogram. To determine the direct effects of recombinant Hsp27 on ASK1-related kinase activities, in vitro cell-free kinase assays were performed using purified active ASK1, MKK4, MKK7, JNK1, and JNK3 proteins according to the specific protocols provided by the manufacturer (Upstate).
Microinjection and cell death assessment.
Microinjection was performed in murine cortical neuron cultures using the procedure described by others (Deshmukh and Johnson, 1998; Cregan et al., 2002). Microinjection solution containing rhodamine dextran (5 mg/ml) alone or the dye plus bovine cytochrome c (1–20 μm) or the synthesized BH3-domain peptide (0.1–3 nm) was injected into the cytoplasm of neurons in 35 mm dishes using Femtotips needles (Eppendorf). At various times after injections, neurons were fixed in 4% paraformaldehyde, washed in PBS, and stained with Hoechst 33258 (1 μg/ml). Cell death was determined by counting the fraction of rhodamine dextran positive neurons exhibiting condensed and/or fragmented nuclei. A minimum of 150 neurons was analyzed for each treatment and data were derived from three independent experiments.
Statistical analysis.
Results are reported as mean ± SEM. The difference between means was assessed by the Student's t test (single comparisons) or by ANOVA and post hoc Bonferroni/Dunn tests (for multiple comparisons), with p < 0.05 considered statistically significant.
Results
Hsp27 overexpression confers long-term neuroprotection against cerebral ischemia and improves postischemic neurofunctional recovery
We have successfully generated several lines of Hsp27 transgenic mice overexpressing human Hsp27. As shown (supplemental Fig. 1A–D, available at www.jneurosci.org as supplemental material), the low-expressing Hsp27 transgenic line (Tg-Hsp27L) demonstrated twofold to threefold increases in Hsp27 protein expression (2.6 ± 0.33, mean ± SEM, n = 6) in the forebrain, whereas the high-expressing Hsp27 transgenic line (Tg-Hsp27H) yielded fivefold to sevenfold increases (5.8 ± 0.45, mean ± SEM, n = 7) in the brain. We confirmed the increased Hsp27 protein expression at the cellular levels using immunohistochemistry. Robust increases in Hsp27 were found in various brain regions, including the cortex and striatum (supplemental Fig. 1E, available at www.jneurosci.org as supplemental material). Double-label immunofluorescence revealed that Hsp27 transgenic expression is localized primarily in neurons and, to a lesser extent, in astrocytes and microvessels. The intracellular localization of the overexpressed Hsp27 protein was predominantly cytosolic (supplemental Fig. 1E, available at www.jneurosci.org as supplemental material).
We determined the effects of Hsp27 transgenic expression on infarct volume following focal cerebral ischemia. Using either transient focal ischemia (60 min of MCA occlusion followed by 48 h of reperfusion) or permanent MCA occlusion (48 h) experimental paradigms, Hsp27 overexpression decreased infarct volume in a dose-dependent manner (Fig. 1 A–C). In particular, Tg-Hsp27H mice showed greater neuroprotection against both transient and permanent ischemic conditions compared with Tg-Hsp27L mice. To determine the long-term effect of Hsp27 overexpression against focal cerebral ischemia, infarct volume was also measured in Tg-Hsp27H and wild-type mice at 21 d after induction of ischemia. Tg-Hsp27H mice showed significantly smaller infarct volume than the wild-type mice (Fig. 1 D). The results suggest that Hsp27 transgenic expression conferred permanent neuroprotection against ischemic infarct induced by transient focal cerebral ischemia.
Figure 1.

Neuroprotective effects of HSP27 transgenic overexpression in mice against focal ischemia. A, Representative photographs of TTC-stained brain coronal sections recovered from 60 min of MCAO and 48 h reperfusion. Data in B represents the total infarct volume determined in wild-type (n = 7) or HSP27 transgenic mice (Tg-HSP27L, low-overexpression mice, n = 8; Tg-HSP27H, high-overexpression mice, n = 8) at 48 h after transient MCAO (tMCAO) or permanent MCAO (pMCAO), whereas data in C illustrates the infarct areas at 48 h after tMCAO taken from consecutive coronal sections beginning +3.0 mm from the bregma and progressing every 1 mm through the entire MCA territory. D, Infarct volume determined at 21 d after 60 min of MCAO in cresyl violet-stained brain sections (n = 9 per group). All data are mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001 versus wild-type mice. E, F, The neuroprotective effect of HSP27 overexpression is independent of rCBF changes after focal ischemia. Representative [14]-iodoantipyrine autoradiographs obtained at 30 min after the onset of MCAO show similar distribution of low-flow regions between Tg-HSP27H and wild-type brains (E). Data are expressed as low perfusion (<50, 40, 30, or 20% levels of contralateral nonischemic hemisphere) volumes in the ischemic hemisphere (F), n = 7 mice per group.
Brain surface vascular anatomy appeared unaffected by Hsp27 transgenic overexpression (Table 1). Physiological parameters, including blood pressure, blood gases, and glucose levels, did not differ significantly between the transgenic or wild-type groups (Table 2). Furthermore, regional blood flow in ischemic brains, as determined using laser Doppler flowmetry and [14]-iodoantipyrine autoradiography quantitative analysis, was also not significantly different between groups (Fig. 1 E–F; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). These results suggest that the reduced brain injury observed in Hsp27 transgenic mice was not the result of a diminished ischemic insult.
Table 1.
Evaluation of surface cerebral blood vessels in MCA territory and PcomA plasticity in wild-type and Tg-HSP27 mice
| MCA distance from the midline
a
at the specified coronal plane of:
b
|
Number of hemispheres with PcomA scores of:
c
|
||||||
|---|---|---|---|---|---|---|---|
| 2 mm | 4 mm | 6 mm | 0 | 1 | 2 | 3 | |
| Wild type | 2.32 (0.05) | 2.13 (0.06) | 2.52 (0.06) | 2 | 6 | 4 | 0 |
| Tg-HSP27L | 2.29 (0.06) | 2.32 (0.04) | 2.42 (0.11) | 1 | 7 | 3 | 1 |
| Tg-HSP27H | 2.36 (0.08) | 2.10 (0.07) | 2.49 (0.08) | 2 | 6 | 4 | 0 |
Comparison of surface cerebral blood vessels in MCA territory and PcomA plasticity in wild-type and Tg-HSP27 mice.
aValues shown are mean (SE) in millimeters (n = 6/group).
bA coronal plane is specified based on its distance from the frontal pole in millimeters.
cPcomA (posterior communicating artery) plasticity scoring criteria: 0, no anastomoses between PCA and SCA; 1, anastomoses between PCA and SCA in capillary phase; 2, small truncal anastomoses between PCA and SCA; 3, truncal anastomoses between PCA and SCA.
Table 2.
Hemodynamics during and after transient MCAO
| Time a | BP | pH | pO2 | pCO2 | Glucose | |
|---|---|---|---|---|---|---|
| Wild type | During MCAO | 88.3 (4.1) | 7.33 (0.02) | 124.3 (7.60) | 40.1 (1.5) | 128.5 (5.8) |
| After MCAO | 85.6 (3.8) | 7.35 (0.01) | 120.4 (6.88) | 41.8 (2.3) | 137.3 (7.4) | |
| Tg-HSP27L | During MCAO | 92.0 (4.6) | 7.32 (0.03) | 127.4 (7.11) | 41.4 (2.3) | 144.1 (4.2) |
| After MCAO | 87.4 (5.0) | 7.31 (0.02) | 120.5 (5.62) | 42.4 (2.1) | 139.5 (5.5) | |
| Tg-HSP27H | During MCAO | 87.7 (3.5) | 7.34 (0.03) | 130.2 (6.54) | 39.7 (3.4) | 134.5 (6.1) |
| After MCAO | 85.5 (3.6) | 7.35 (0.03) | 124.6 (5.22) | 40.8 (2.7) | 140.6 (7.3) |
Comparison of hemodynamics during and after transient MCAO between wild-type and Tg-HSP27 mice. Hemodynamics parameters measured are: BP (mean blood pressure; mmHg); pO2 (artery O2 pressure; mmHg); pCO2 (artery CO2 pressure; mmHg); glucose (blood glucose level, mg/dl). Data are mean (SE).
aMeasurements were taken 15 min after the onset of MCAO (during) and 15 min after reperfusion (after) on wild-type (n = 7), 7 Tg-HSP27L (n = 8), and Tg-HSP27H (n = 8) mice.
To determine the impact of Hsp27 transgenic expression on neurofunctional outcomes after stroke, a series of neurobehavioral assays were administered during both the acute (1–7 d) and long-term (14–21 d) recovery stages following transient focal ischemia. Sham control mice showed no significant difference in neurobehavioral performance regardless of their transgenic phenotypes (wild type or Tg-Hsp27H). In contrast, postischemic behavior significantly improved in Tg-Hsp27H mice after transient focal ischemia compared with their wild-type littermates. Acute sensorimotor dysfunction was significantly improved in Tg-Hsp27H mice as assessed by rotarod performance, gridwalking, and the corner test (Fig. 2 A–C). Poststroke cognitive function was also significantly improved in Tg-Hsp27H, determined using the Morris water maze task (Fig. 2 D,E). High-level Hsp27 transgenic overexpression improved long-term cognitive performance to the extent that there was no significant difference between sham-operated and Tg-Hsp27H animals following transient focal ischemia.
Figure 2.

HSP27 transgenic overexpression improves motor function and cognitive performance after focal ischemia and reperfusion. A–C, Assessment of sensorimotor function in sham control mice and ischemic mice. Tg-HSP27H or wild-type mice were subjected to rotarod (A), foot fault (B), and corner tests (C) over 7 d following 60 min of MCAO. D, E, Assessment of cognitive function in mice using Morris water maze 2–3 weeks following MCAO both in the presence of the platform to assess the ability of task acquiring and learning (D) and with the subsequent removal of the platform to assess memory on the position of platform (time spent in goal quadrant, E). All three groups showed similar swim speed during the probe trial (F). At the end of the testing, mice were killed for infarct size measurement (results presented in Fig. 1). All data are presented as mean ± SEM, where *p < 0.05, **p < 0.01, ***p < 0.001, n = 9 mice per group.
Hsp27 attenuates mitochondrial release of cytochrome c and Smac following cerebral ischemia
Figure 3 A illustrates the protective effects of Hsp27 against ischemic cell death, determined by assessing active caspase-3 and apoptotic DNA fragmentation at 24 h after transient focal ischemia. Stereology-assisted cell counting for the number of DNA-damaged or caspase-3-immunoreactive cells was performed in brain sections. Cell death and caspase-3-positive cells were significantly decreased in both ischemic cortex and striatum of Tg-Hsp27H mice compared with their wild-type littermates (Fig. 3 B,C).
Figure 3.
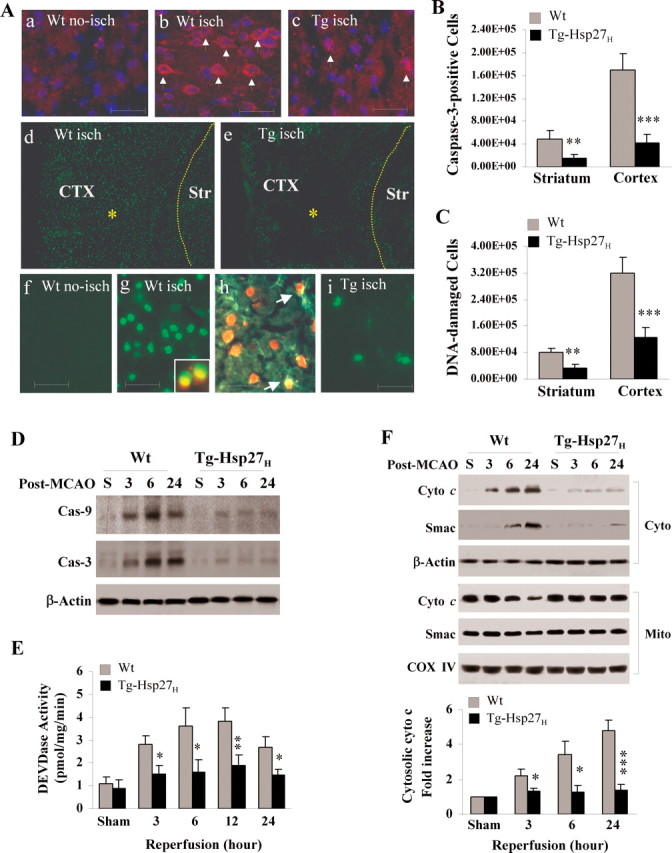
Transgenic overexpression of HSP27 attenuates cell death and mitochondrial prodeath signaling after ischemia in vivo. A–C, Tg-HSP27H or wild-type mice were subjected to 60 min of MCAO and brain sections were processed for in situ detection of DNA fragmentation at 24 h of reperfusion and immunohistochemistry for cleaved caspase-3. Representative fluorescence images (Aa–c) demonstrate the reduced number of caspase-3-positive cell in Hsp27 transgenic brain than in the wild-type brain after ischemia (a, wild-type nonischemic control; b, wild-type ischemic brain; c, transgenic ischemic brain). Images in Ad–e show the reduced density of DNA damage in cortex (CTX) and striatum (Str) in Hsp27 transgenic brain than in the wild-type brain after ischemia; high power images are presented in panels Af–i, showing that DNA damage occurred mainly in neurons (insert in g, costained with NeuN) and astrocytes (GFAP-positive, arrows in h). The numbers of caspase-3 immunoreactive cells (B) and DNA-damaged (C) were quantified using stereology principles at the coronal levels between −1.5 and 1.5 mm from the bregma. Data are mean ± SEM, **p < 0.01, ***p < 0.001 versus wild-type mice, n = 7 per group. D, E, Activation of caspases 3–24 h following 60 min of MCAO. Cortical cell lysates were immunoblotted against active caspase-9 and active caspase-3 (D) or assessed for DEVDase activity using the DEVD.afc substrate-based enzymatic assay (E). F, Mitochondrial release of cytochrome c (Cyto c) and Smac was detected by Western blot following subcellular fractionation (cyto, cytosol; mito, mitochondria) in ischemic cortex 3–24 h following MCAO. The graph in the lower panel illustrates the semiquantitative results from four independent sets of data. Data are presented as mean ± SEM, where *p < 0.05, **p < 0.01, ***p < 0.001 versus wild-type mice.
It has been shown in non-neuronal systems that Hsp27 can interact with prodeath signaling cascades via direct inactivation of executioner caspases (e.g., caspase-3 inhibition) or inhibition of apoptosome formation downstream of mitochondrial cytochrome c release (Bruey et al., 2000; Concannon et al., 2001, 2003; Voss et al., 2007). In the present study, we detected marked inhibitory effects of Hsp27 on caspase-9 and caspase-3 activation 3–24 h after transient focal ischemia (Fig. 3 D,E), suggesting that Hsp27 interfered with the activation of the intrinsic prodeath signaling pathway. We next determined whether inhibition of caspase activation by Hsp27 in ischemic brain was due to reduced mitochondrial release of cytochrome c or Smac. Using subcellular fractionation and immunoblots, we found that ischemia-induced mitochondrial release of both cytochrome c and Smac were attenuated in Tg-Hsp27H mice (Fig. 3 F). These results suggest that Hsp27 might prevent caspase activation by targeting an upstream prodeath signaling pathway.
To further elucidate the cellular mechanisms underlying the neuroprotective effects of Hsp27, we used an in vitro model of neuronal ischemia, oxygen-glucose deprivation (OGD), using nearly pure neuronal primary cultures (>97% neurons). Cortical cultures derived from Tg-Hsp27H mice or wild-type mice were maintained in vitro for 12 d before 45 or 60 min of OGD. Hsp27-overexpressing cultures were significantly more resistant to OGD-induced cell death than cultures from wild-type mice (Fig. 4 A,B). To further confirm the neuroprotective effects of Hsp27 in cultured neurons, we transfected human Hsp27 into wild-type cortical neuronal cultures using adeno-associated virus (AAV) vectors. AAV vectors consistently achieved transfection efficiency of >90% and increased Hsp27 expression levels five to eightfold as determined by Western blot (data not shown). Using the AAV transfection approach, we were able to confirm the robust neuroprotective effect of Hsp27 against OGD (Fig. 4 C,D). Consistent with the in vivo data, AAV-mediated Hsp27 overexpression in neurons attenuated caspase-3 and caspase-9 activation and cytochrome c release following OGD (Fig. 4 E–I).
Figure 4.

Overexpression of HSP27 prevents mitochondrial prodeath signaling following in vitro ischemia. A, B, Cortical neuronal cultures derived from either wild-type or Tg-HSP27H mice were challenged at 12 DIV with OGD for 45 or 60 min, then returned to normal culture conditions for 24 h. Cell viability was assessed using Alamar blue (A), while cell death was quantified by measuring LDH release from neurons (B, 100% is based on total cell lysis by Triton X-100). C, D, Cortical neurons from rat embryos were infected with AAV-GFP or AAV-HSP27 for 3 d and then challenged at 12 DIV with 60 min of OGD. Cell viability (C) and cell death (D) were determined at 24 h after OGD using Alamar blue and Hoescht staining (percentages of condensed/fragmented nuclei), respectively. E, Representative triple-label fluorescence images from AAV-infected cortical cultures 3 d after infection either without OGD (top) or subjected to 60 min of OGD followed by 24 h of reperfusion (middle and bottom). Neurons were transfected with GFP alone (top, middle) or coinfected with GFP and HSP27 (bottom), and labeled (GFP, green; Hoechst, blue; active caspase-3, red; arrows indicate dying neurons). F–I, AAV-infected cortical cultures were subjected to 60 min of OGD. At 6 and 16 h after OGD, neurons were assessed for active caspase-9 and -3 (F) or fluorogenic substrate-based DEVDase activity assay (G), release of cytochrome c by Western blot following subcellular fractionation (H) or immunocytochemistry with Hoescht counterstaining (Ia–d: empty AAV, e–h: AAV-HSP27; arrows in c, d indicate neurons that lost cytochrome c immunoreactivity and contained apoptotic nuclei at 16 h after OGD). All data are presented as mean ± SEM; for cell viability or cell death assessment, each data point was from at least 12 culture wells out of 3 independent experiments, where **p < 0.01, ***p < 0.001 versus wild-type neurons; for DEVDase activity and cytosolic cytochrome c analysis, data were derived from 4 experiments, *p < 0.05 versus empty AAV- or AAV-GFP infected cultures.
Hsp27 does not directly protect against targeted mitochondrial prodeath insults
Recent reports suggest that, under certain circumstances, activation of death-execution caspases such as caspase-3 can trigger cytochrome c release from mitochondria (Lakhani et al., 2006). Thus, while the above data suggest that Hsp27 may prevent caspase-9 and -3 activation via attenuating cytochrome c release, it is plausible that Hsp27 could directly inhibit caspase activity and thereby prevent the positive feedback loop to the mitochondria. To assess this possibility, we induced caspase-9- and caspase-3-dependent cell death by microinjection of recombinant cytochrome c into cultured cortical neurons (Deshmukh and Johnson, 1998). As expected, microinjection of bovine cytochrome c, but not the inactive yeast cytochrome c, resulted in apoptosis in a protein concentration- (Fig. 5 B) and time-dependent (supplemental Fig. 3, available at www.jneurosci.org as supplemental material) manner. Neurons derived from animals overexpressing Hsp27, however, failed to protect against cell death induced by cytochrome c microinjection (Fig. 5 A,B). These results suggest that the cytochrome c/caspase-3-dependent cell death cascade is not a direct target for Hsp27.
Figure 5.

Hsp27 is neuroprotective upstream of mitochondrial cell death signaling. A–D, Apoptosis induced by microinjection of cytochrome c or BH3 peptide is not protected by Hsp27 overexpression. Cortical neurons derived from Hsp27 transgenic mice or their wild-type littermates were microinjected with either cytochrome c or the BH3 peptide at the indicated concentrations combined with the tracer rhodamine dextran. BH3 microinjection was also performed using neurons from Bcl-xL transgenic mice. Neurons were counterstained with Hoescht at 6 h after cytochrome c injection or 2 h after BH3 peptide injection to visualize apoptosis. Representative images (A, C) show cytochrome c- or BH3-induced apoptosis (arrows) in neurons; quantitative analysis of apoptosis under various conditions are presented as graphs (B and D; in B, the caspase inhibitor z-DEVD.fmk was added at 100 μm to wild-type cultures 30 min before cytochrome c injection). All data are mean ± SEM, where *p < 0.05, **p < 0.01, ***p < 0.001 versus wild-type cultures; from five independent experiments, each data point was derived from at least 150 neurons. E, Hsp27 or Bcl-x-L transgenic cultures were infected with adenovirus carrying Bax, Puma, or BimEL and assessed for cell survival at 16 h after infection. Data were analyzed based on three experiments, ***p < 0.001 versus wild-type cultures. F, G, Hsp27 overexpression inhibits bleomycin- or OGD-induced translocation of proapoptotic proteins from cytosol to mitochondria and release of cytochrome c from mitochondria. Cortical cultures were infected with AAV-Hsp27 or AAV-GFP for 3 d, exposed to bleomycin (10 μm) or 60 min of OGD followed by reperfusion, then subjected to cellular fractionation (cyto, cytosol; mito, mitochondria) and Western blotting. H, Bleomycin-induced cytochrome c release is attenuated in neurons derived from Bax or Puma knock-out mice, compared with the wild-type cultures. Blots are representatives of 3 sets of independent experiments. I, Hsp27 overexpression inhibits bleomycin-induced apoptosis. Cortical cultures were infected with AAV-Hsp27 or AAV-GFP for 3 d, exposed to bleomycin (10 μm) for 16 h, and then assessed for apoptosis using Hoescht nuclear staining. Data are presented as mean ± SEM, where ***p < 0.001 versus noninfected or AAV-GFP-infected cultures.
We next examined the effect of Hsp27 overexpression on BH3 peptide-induced cell death, another mitochondrial-targeted prodeath cascade. Synthetic BH3 peptide recapitulates the essential prodeath domain of BH3-containing proteins, including Bax and BimEL, and can potently induce cell death by directly targeting mitochondria (Moreau et al., 2003). Following microinjection, BH3 peptide rapidly induced apoptosis in cortical neurons. BH3-mediated cell death was partially prevented by Bcl-xL overexpression. In contrast, overexpression of Hsp27 could not diminish BH3-induced cell death (Fig. 5 C,D). Likewise, Hsp27 overexpression could not inhibit cell death induced by adenovirus-mediated overexpression of proapoptotic molecules Bax, Puma, or BimEL in cultured cortical neurons (Fig. 5 E). Conversely, transgenic overexpression of Bcl-xL robustly inhibited cell death initiated by adenovirus-delivered prodeath stimuli (Fig. 5 E). The inability of Hsp27 to protect neurons against direct activation of several mitochondrial prodeath signaling pathways strongly suggests that Hsp27 targets a mechanism upstream of the mitochondrial damage pathways.
To determine whether Hsp27 inhibited ischemia-induced cytochrome c release by preventing the translocation of proapoptotic proteins to mitochondria, we infected cultured neurons with AAV-Hsp27 or AAV-GFP (control) for 3 d and then exposed the cultures to OGD or the DNA-damaging agent bleomycin. Bleomycin is a well characterized agent that induces apoptosis by the JNK-mediated, Bax-dependent mitochondrial prodeath signaling pathway (Lee et al., 2005; Souhir et al., 2007). Both OGD and bleomycin resulted in robust translocation of Bax, PUMA, and BimEL to the mitochondria in cortical neurons, and this was significantly attenuated by overexpression of Hsp27 (Fig. 5 F). Similar to OGD, bleomycin markedly induced cytochrome c release into the cytosol (Fig. 5 G). The mitochondrial release of cytochrome c was dependent, at least in part, on the presence of Bax or PUMA, as cultures derived from Bax- or PUMA-knock-out mice had attenuated cytochrome c release (Fig. 5 H). Hsp27 overexpression also inhibited cytochrome c release from the mitochondria as well as apoptotic cell death following bleomycin exposure (Fig. 5 G,I). Together, these data confirm that Hsp27 confers neuroprotection by blunting the initiation of the Bax- and PUMA-mediated mitochondrial prodeath signaling pathway.
Hsp27 inhibits the JNK signaling pathway
Two important signaling cascades upstream of the mitochondrial prodeath signaling pathway, the PI3K/Akt and the JNK/c-Jun pathways, play integral and typically opposing roles in the pathogenesis of cell death following ischemic insults (Zhang et al., 2007). The ability of Hsp27 overexpression to inhibit bleomycin-induced cell death (Fig. 5 I) raised the possibility that Hsp27 could inhibit or counteract JNK/c-Jun signaling. We therefore sought to determine whether the neuroprotective effects of Hsp27 overexpression could be attributed to either the activation of the prosurvival PI3K/Akt pathway, or to the suppression of cell death pathways, such as JNK/c-Jun. Following OGD, robust phosphorylation of both c-Jun and JNK was observed in cortical cultures, and this was significantly suppressed by overexpression of Hsp27 (Fig. 6 A). Pharmacological inhibition of JNK signaling using SP600125 mimicked the protective effects of Hsp27, including inhibition of cytochrome c release and increased cell survival against both OGD and bleomycin toxicity (Fig. 6 B–D).
Figure 6.

Alteration in JNK, but not Akt/PKB, signaling contributes to Hsp27 neuroprotection. A, Overexpression of HSP27 suppresses OGD-induced JNK and c-Jun phosphorylation. Cortical neurons were infected with AAV-HSP27 or AAV-GFP for 3 d and then exposed to 60 min of OGD. Cell lysates obtained at 1, 2, and 6 h following OGD were subjected to Western blot using phospho-specific antibodies. Optical density was quantified by averaging four independent experiments. B–D, JNK activity plays a critical role in OGD- or DNA damage-induced cytochrome c release and cell death. Cortical neurons were treated with the JNK inhibitor SP600125 (3 μm) for 30 min and then exposed to either 60 min of OGD (B) or bleomycin (10 μm) (C). Cytochrome c was assessed by Western blot in cytosolic (cyto) and mitochondrial (mito) fractions, respectively, at 6 and 16 h after OGD (B) and at 2 and 6 h after bleomycin exposure (C); cell viability was determined 24 h after OGD or bleomycin by Alamar blue (D). E, HSP27 overexpression leads to sustained expression of phospho-Akt following OGD or bleomycin exposure. Neurons were infected with AAV vectors for 3 d before OGD or bleomycin exposure. The blots are representatives of four sets. F, Inhibition of Akt activation does not affect HSP27-mediated neuroprotection against OGD or bleomycin toxicity. The PI3K/Akt inhibitor LY294002 (10 μm) was added to cortical cultures 30 min before bleomycin or OGD challenge, and assessed for cell survival 24 h later using Alamar blue. All data are presented as mean ± SEM, where *p < 0.05, **p < 0.01 versus vehicle controls.
The inhibition of JNK activation and the suppression of cytochrome c release and cell death by Hsp27 overexpression suggest that Hsp27 neuroprotection occurs via suppression of this kinase arm. However, cross talk between PI3K/Akt and JNK pathways has been reported previously and could occur in our neuronal injury models. For example, activation of PI3K/Akt may directly suppress JNK signaling (Zhang et al., 2007). As shown by diminished phosphorylation of Ser473, OGD or bleomycin exposure resulted in sustained suppression of Akt activity in cortical neuronal cultures (Fig. 6 E). Overexpression of Hsp27 restored the levels of phospho-Akt following OGD or bleomycin (Fig. 6 E). However, pharmacological inhibition of the upstream kinase, PI3K, failed to affect Hsp27-induced neuroprotection against OGD or bleomycin (Fig. 6 F). Thus, the PI3K/Akt prosurvival pathway does not appear to be essential for Hsp27 inhibition of mitochondrial cell death signaling.
Hsp27 inhibits upstream JNK signaling pathways by directly interacting with and suppressing ASK1 activity
Since Hsp27-mediated neuroprotection appears to depend on the inhibition of JNK and not the activation of Akt (Fig. 6), we further investigated components of the JNK kinase signaling pathway targeted by Hsp27. In CNS neurons, the JNK1 and JNK3 isoforms of JNK are activated by a series of multipotent upstream kinases, including MKK4/7, ASK1, and MLK3 (Kuan et al., 2003). Using phospho-specific antibodies, we found that MKK4 and ASK1 were robustly phosphorylated following OGD in cortical cultures, while MKK7 showed only modest activation. Overexpression of Hsp27 nearly abolished the activation of MKK4 (Fig. 7 A), but did not affect the phosphorylation state of ASK1, suggesting that Hsp27 might target downstream of ASK1 activation but upstream of MKK4 activation, or by an indirect process that does not alter ASK1 phosphorylation status.
Figure 7.

Hsp27 inhibits and forms a physical association with ASK1. A, Hsp27 overexpression decreases the activation of MKK4 but does not affect the phosphorylation of ASK1 following OGD. Cortical cultures were infected with AAV-Hsp27 or AAV-GFP for 3 d and then challenged with 60 min of OGD and immunoblotted using phospho-specific antibodies at 1, 2, and 6 h after OGD. The graph in the bottom panel illustrates the semiquantitative results for p-MKK4 and p-MKK7, based on 3 independent experiments. Data are presented as mean ± SEM, *p < 0.05, **p < 0.01 versus AAV-GFP-infected cultures. B, Hsp27 directly inhibits in vitro ASK1, but not MKK4/7 or JNK1/3, activity. Recombinant active kinases were incubated with recombinant Hsp27 protein at the indicated concentrations and then assessed for the incorporation of radiolabeled phosphate onto the substrate protein (MBP) as evidence of kinase activity. Data are presented as mean ± SEM, ***p < 0.001 versus reactions without adding Hsp27 protein. C, Hsp27 and ASK1 form a physical association. Cortical neurons were infected with HSP27 or GFP (both HA-tagged) for 3 d and exposed to 60 min of OGD, followed by 2 h of reperfusion. Lysates were collected for coimmunoprecipitation using anti-HA (left) or anti-ASK1 (right) and immunoblotted for HA, ASK1, or MLK3. Blots are representatives of 3 sets.
The interference of kinase signaling pathways can occur via direct or indirect mechanisms. In some non-neuronal systems, Hsp27 can form a complex with MK2 or Akt, altering kinase activity of the latter (Mearow et al., 2002; Rane et al., 2003; Zheng et al., 2006; Wu et al., 2007). We speculated that Hsp27 could inhibit ASK1 activity via a direct association with components of the ASK1/MKK/JNK pathway. Using an in vitro kinase activity assay with purified recombinant proteins, we assessed the ability of active kinases (ASK1, MKK4, MKK7, JNK1, JNK3) to incorporate a radiolabeled phosphate into their corresponding substrates in the presence of Hsp27 (Fig. 7 B). The kinase activities of JNK1 and JNK3, and the immediate upstream kinases MKK4 and MKK7 were unaffected by the presence of recombinant Hsp27; however, ASK1 kinase activity was significantly decreased in the presence of recombinant Hsp27 in the assay (Fig. 7 B). Addition of a truncated Hsp27 lacking the N-terminal domain (1–120 aa) failed to inhibit ASK1 activity (data not shown).
To determine whether Hsp27 physically interacts with ASK1, we infected neurons with either AAV-Hsp27 or AAV-GFP (control) for 3 d and then, at 2 h after OGD, performed coimmunoprecipitation with antibodies against HA (to pull down HA-tagged Hsp27 or GFP) and ASK1, respectively. We found that Hsp27 coimmunoprecipitated with ASK1 (Fig. 7 C), but not MLK3, another kinase that could activate JNK via MKK4/7 (Pan et al., 2007).
To determine which protein domains are critical for the interaction between Hsp27 and ASK1, we constructed a series of Flag-tagged ASK1 truncates (Fig. 8 A). Various ASK1 constructs were then cotransfected with either HA-tagged full-length or truncated Hsp27 into HEK293 cells, followed by coimmunoprecipitation. Interestingly, all ASK1 constructs that contained the kinase domain retained the ability to coimmunoprecipitate with Hsp27, including the kinase-only construct (Fig. 8 B). However, any constructs lacking the kinase domain failed to interact with Hsp27 (Fig. 8 B). This suggests that the kinase domain of ASK1 alone was capable of and required for interaction with Hsp27.
Figure 8.

Characterization of functional domains required for Hsp27 and ASK1 interaction and suppression of ASK1 activity. A, Schematic representation of various ASK1 constructs. All ASK1 constructs were Flag-tagged. B, Interaction with Hsp27 requires the kinase domain of ASK1. Human HEK293 cells were cotransfected with Flag-tagged ASK1 constructs and HA-tagged full-length Hsp27 for 16 h. Cell lysates were immunoprecipitated with either anti-HA or anti-Flag antibody and immunoblotted against HA or Flag. C, Interaction with ASK1 requires the N-terminal domain of Hsp27. HSP27 deletion mutants lacking either the C-terminal (ΔC) or the N-terminal (ΔN) domain were HA-tagged and cotransfected with full-length Flag-tagged ASK1 into HEK293 cells for 16 h. Cell lysates were immunoprecipitated with either anti-HA or anti-Flag antibody and immunoblotted against HA or Flag. D, E, The N-terminal domain of Hsp27 efficiently suppresses ASK1 activity. HEK293 cells were cotransfected with Flag-tagged ASK1 and either HA-tagged full-length Hsp27 (D) or HA-tagged ΔC truncated Hsp27 (E) at the indicated plasmid quantity (μg) for 16 h. Cell lysates were immunoprecipitated with anti-Flag and assessed for ASK1 activity present in cell lysates. All blots presented in B–E are representatives of at least 3 independent experiments. F, The N-terminal domain is required for Hsp27-mediated neuroprotection against OGD. Cortical cultures were infected with AAV carrying full-length Hsp27 or truncated Hsp27 lacking either the C-terminal (ΔC) or the N-terminal (ΔN) domain. Three days after infection, cultures were exposed to 60 min of OGD and assessed for viability using Alamar blue. All data are presented as mean ± SEM, *p < 0.05, ***p < 0.001, and each data point is derived from at least 12 culture wells based on 3 independent experiments.
Hsp27 is comprised of two dominant molecular domains: the N-terminal region, which contains critical phosphorylation sites necessary for functional regulation; and the α-crystallin-like C-terminal, necessary for protein oligomerization. Using C-terminal and N-terminal deletion truncates, we found that the N-terminal region of Hsp27 is necessary for the physical association between Hsp27 and ASK1 (Fig. 8 C). This is consistent with our results demonstrating that truncated Hsp27 lacking the N-terminal domain failed to inhibit ASK1 activity (data not shown). Furthermore, transfection of the N-terminal Hsp27 construct (ΔC) was able to inhibit ASK1 activity in a plasmid concentration-dependent manner similar to the full-length Hsp27 (Fig. 8 D,E), suggesting that the presence of the N-terminal region alone is sufficient to inhibit ASK1 activity. Consistent with its role in interacting with and inhibiting ASK1 activity, AAV-mediated overexpression of the N terminus of Hsp27 (ΔC), but not the C terminus, significantly protected neurons against OGD-induced cell death (Fig. 8 F).
ASK1 is a critical mediator for ischemic neuronal death
Activation of ASK1 has been implicated in various cell toxicity models; however, its role in neuronal ischemia is not well defined. ASK1 is an important activator of the MKK4/JNK signaling pathway under conditions such as oxidative stress and DNA damage, although MKK4/JNK may also be activated via other MKKKs (Pan et al., 2007). Thus, we sought to determine whether ASK1 has a major role in the activation of MKK4/JNK in the OGD model of ischemic injury. Primary cortical neurons were transduced for 3 d with lentiviral vectors expressing either an ASK1-targeting shRNA (Lenti-ASK1t) or a scrambled control sequence (Lenti-ASK1 s). As determined using Western blot, transfection of Lenti-ASK1t but not Lenti-ASK1 s resulted in reduction of ASK1 expression by >90% (Fig. 9 A). Targeted ASK1 knockdown abolished OGD-induced MKK4 and JNK phosphorylation (Fig. 9 B) and significantly increased cell survival following OGD (Fig. 9 C). In neuronal cultures derived from Hsp27 transgenic mice, which are resistant to OGD injury, transfection of Lenti-ASK1t failed to confer additional neuroprotection compared with Lenti-ASK1 s-transfected or empty vector-transfected cultures (Fig. 9 C). These results support the hypothesis that ASK1 and Hsp27 may interact with the same prodeath signaling pathway in OGD injury.
Figure 9.

ASK1 plays a critical role in OGD-induced neuronal death. A–E, Knockdown of ASK1 expression is neuroprotective against OGD. Murine cortical cultures were infected for 3 d with lentivirus-shRNA targeting the ASK1 mouse sequence (Lnt-ASK1t) or a scrambled sequence (Lnt-ASK1 s), or the empty lentivirus (Lnt). Lnt-ASK1t effectively suppressed ASK1 protein expression for >90% (A) and inhibited activation of the downstream kinase MKK4 and JNK at 2 and 6 h after 60 min of OGD (B). Knockdown of ASK1 promoted neuron survival in wild-type mice-derived cultures at 24 h after 60 min of OGD, but failed to confer additional neuroprotection against OGD in Tg-Hsp27H mice-derived cultures (C). Coinfection of HA-tagged human ASK1, which is resistant to Len-ASK1t knockdown (D), restored sensitivity of wild-type cortical cultures to 60 min of OGD (E), confirming that the effects observed from ASK1 knockdown were specific. F, G, Infection of lentivirus encoding an HA-tagged dominant-negative form of ASK1 (F) promoted cell survival assessed by Alamar blue at 24 h after 60 min of OGD (G). H–J, Knockdown of endogenous Hsp27 promotes ASK1 activity and cell death after OGD. Cortical cultures were infected for 3 d with lentivirus-shRNA targeting Hsp27 (Lnt-Hsp27t) or a scrambled sequence (Lnt-Hsp27 s) and then challenged with OGD (60 min) and reperfusion. Cell extracts were subjected to Western blot for Hsp27 (H) or ASK1-pull down for ASK1 activity assay (I). Cell viability was measured at 24 h after OGD (J). All data are presented as mean ± SEM, where *p < 0.05, ***p < 0.001, and each data point was derived from at least 12 culture wells out of 3 independent experiments.
Using the above approach, we determined the role of MLK3 in OGD-induced activation of MKK4 and JNK and cell death. The results revealed that MLK3 knockdown has no neuroprotective effects in the neuronal OGD model (data not shown).
We confirmed the neuroprotective effects of Lenti-ASK1t-mediated ASK1 knockdown in neurons by coinfection with human ASK1 (Lenti-ASK1 h) which is resistant to shRNA targeting the mouse ASK1 (Fig. 9 D). Overexpression of ASK1 h restored the vulnerability of ASK1-deficient neurons to OGD injury (Fig. 9 E). Finally, lentiviral vector-mediated overexpression of a dominant-negative form of ASK1 (ASK1dn) also conferred significant neuroprotection against OGD injury in cultures (Fig. 9 F,G).
To determine whether endogenous Hsp27 has a role in neuroprotection and suppressing ASK1 activity, cortical cultures were subjected to Hsp27 knockdown using Lenti-Hsp27t (Fig. 9 H). In control neurons (noninfected or Lenti-Hsp27 s-infected), the basal level of Hsp27 expression was low but readily detectable; in response to 60 min of OGD, Hsp27 expression was transiently increased at 2 h after OGD by approximately twofold and then subsided. In Lenti-Hsp27t-infected neurons, Hsp27 was not detectable either before or after OGD. Targeted Hsp27 knockdown promoted ASK1 activity (Fig. 9 I) and significantly decreased cell survival following OGD (Fig. 9 J).
Hsp27 protects against ischemic brain injury via inhibition of the ASK1-JNK signaling cascade
Having determined that the ASK1/JNK pathway is integral to both ischemic cell death and Hsp27 neuroprotection in vitro, we wished to determine whether this is also the underlying mechanism in the in vivo model of cerebral ischemia. We performed kinase activity assays on cerebral lysates obtained at 4 and 8 h after 60 min of transient focal ischemia from wild-type and Tg-Hsp27H mice. Compared with sham-surgery control animals, ASK1 as well as JNK1 and JNK3 activities (but not JNK2 activity, data not shown) were increased in wild-type mice, and these activities were significantly attenuated in Tg-Hsp27H mice (Fig. 10 A,B). The inhibitory effect of Hsp27 overexpression on JNK activation was also demonstrated at the cellular level by p-c-Jun immunohistochemistry on brain sections (Fig. 10 C). While p-c-Jun immunoreactivity was robustly increased in both cerebral cortex and striatum of wild-type mice at 8 h after 60 min of transient focal ischemia, the increases in p-c-Jun immunoreactivity were only modest in the brains of the Tg-Hsp27H mice.
Figure 10.

Hsp27 transgenic overexpression inhibits ischemia-induced ASK1/JNK signaling pathways in vivo. A–C, Hsp27 transgenic overexpression suppresses ASK1 and JNK1 and JNK3 activities following transient ischemia. Tg-Hsp27H or wild-type mice were subjected to 60 min of MCAO followed by reperfusion. Whole-cell protein extracts prepared from the cortical MCA territory at 4 or 8 h after ischemia or sham operation (S) were assessed for kinase activities (A, ASK1 activity; B, JNK1, JNK2, and JNK3 activities). The graphs in the right panel of A and B illustrate the quantitative results from four independent sets of samples. Data are presented as mean ± SEM, *p < 0.05, **p < 0.01 versus wild-type mice. Brain sections obtained at 4 h of postischemic reperfusion were processed for immunohistochemistry for phospho-c-Jun (C), and wild-type sections were counterstained with the neuronal markers NeuN (C, bottom). The majority of p-c-Jun positive cells are also NeuN positive. Scale bars, 50 μm. D, Hsp27 overexpression inhibits JNK activation but has no effect on the status of p38 or ERK1/2 activation at 4 or 8 h after 60 min of MCAO. Blots are representatives of four sets. E, HSP27 overexpression inhibits mitochondrial translocation of the prodeath proteins Bax and BimEL. Lysates were collected 4 and 8 h after 60 min of MCAO, fractionated to separate mitochondria from cytosol, and immunoblotted. Blots are representatives of four sets. F, Pharmacological inhibition of JNK decreases infarct volume in wild-type mice, but has no additional effect in Tg-Hsp27H mice. Mice were injected (i.p.) with the JNK inhibitor SP6000125 (10 mg/kg) or with vehicle at the onset of postischemic reperfusion. Infarct volume was determined using TTC staining at 48 h after 60 min of MCAO. Data are mean ± SEM, **p < 0.01, ***p < 0.001, n = 8 per group.
Using Western blot, the phosphorylation states of JNK and two other MAP kinases, p38 MAPK and ERK1/2, were next examined at 4 and 8 h after 60 min of transient focal ischemia. The levels of p-JNK (Fig. 10 D) and p-ERK1/2 (data not shown) were remarkably increased at both time points after ischemia in wild-type mice, while no increase in p-38 MAPK was detected. In contrast, ischemia-induced increases in p-JNK were abolished in the Tg-Hsp27H mice (Fig. 10 D), whereas the increases in p-ERK1/2 were not attenuated (data not shown), suggesting that Hsp27 selectively targets the JNK pathway rather than attenuating the ischemic stress response in general. Transgenic overexpression of Hsp27 also reduced the translocation of Bax and BimEL to mitochondria at 4 and 8 h after focal ischemia (Fig. 10 E), consistent with the critical role JNK plays in contributing to Bax/BimEL-dependent mitochondrial prodeath signaling after ischemia (Gao et al., 2005).
Finally, to determine whether JNK and Hsp27 interact with different prodeath signaling pathways after ischemia, wild-type or Tg-Hsp27H mice received the JNK inhibitor SP600125 (10 mg/kg) before 60 min of transient focal ischemia. SP600125 treatment significantly decreased infarct volume measured 48 h after ischemia in the wild-type mice, but failed to further reduce infarct volume in the Tg-Hsp27H mice (Fig. 10 F). These results support the hypothesis that inhibition of JNK signaling may be an integral component of the mechanism underlying the neuroprotective effect of Hsp27 against ischemic brain injury.
Discussion
The complex and intricate signaling pathways activated by cerebral ischemia are not completely understood, but are known to include activation of prodeath signaling cascades which converge on the mitochondria. This study demonstrates that overexpression of Hsp27 confers long-term neuroprotection against focal ischemic/reperfusion injury via a novel ASK1-dependent pathway. Three specific actions by Hsp27 may contribute to this neuroprotective effect: first, Hsp27 can bind directly to the kinase domain of ASK1; second, Hsp27 can inhibit ASK1 activity; third, Hsp27 can suppress the MKK4/JNK signaling cascade and neuronal cell death following ischemia. These results explore a novel mechanism by which Hsp27 confers neuronal protection and provides an upstream target for neuroprotective intervention in cerebral ischemia.
The mechanism by which Hsp27 confers cellular protection has largely been attributed to targeting pathways downstream of mitochondrial death signaling, including direct inhibition of caspases (Concannon et al., 2003; Voss et al., 2007). The present study instead demonstrated that in cortical neurons, Hsp27 functions on upstream signaling pathways instead of directly suppressing the downstream cell death-execution cascades. Two major lines of evidence support this finding. First, Bcl-x but not Hsp27 protected neurons from direct microinjection of cytochrome c or BH3 peptide. Second, Hsp27 not only inhibited activation of caspase-3 and caspase-9, but also inhibited translocation of proapoptotic Bcl-2 family members to mitochondria following either ischemia or the DNA-damaging agent bleomycin. Our data conflict with the report that Hsp27 protected sensory and motor neurons downstream of cytochrome c release and upstream of caspase-3 activation (Benn et al., 2002). However, subpopulations of sensory and motor neurons endogenously expressed much higher levels of Hsp27 compared with other neuronal systems (Plumier et al., 1997). In other neuronal populations, such as retinal neurons or PC12 cells, Hsp27 overexpression inhibited caspase activation and cytochrome c release via an unknown mechanism (Gorman et al., 2005; Whitlock et al., 2005). Furthermore, an Hsp27 mutant lacking the residues critical for binding to cytochrome c was still protective against poly(Q)-mediated cell death (Wyttenbach et al., 2002), suggesting that Hsp27 is capable of inhibiting cell death without interacting with cytochrome c. Our data support these observations by demonstrating that Hsp27 functions upstream of mitochondrial cell death stimuli in cortical neurons.
Previous reports have described Hsp27 as a “scaffolding” protein that binds to and recruits multiple signaling molecules. In non-neuronal systems, Hsp27 indirectly affected signaling cascades by modulating the interaction between component members, such as MK2 and Akt (Mearow et al., 2002; Wu et al., 2007). Cross talk between kinase signaling cascades has been well documented, and thus must be carefully considered. Several neuroprotective compounds can downregulate ASK1 activation via activation of PI3K/Akt signaling following ischemia (Xu et al., 2008). Cross talk between these two pathways can occur via Akt-dependent phosphorylation of ASK1 on Ser83, suppressing ASK1 activity (Kim et al., 2001). However, our data demonstrating that inhibition of PI3K did not affect Hsp27 neuroprotection and that Hsp27 can directly inhibit ASK1 activity indicate that Hsp27 does not appear to function through PI3K/ASK1 cross talk. ASK1 can also lead to an enhanced JNK response via p38 MAPK activation. We did not detect any alteration of p38 MAPK activation in either wild-type or Hsp27 transgenic mice, consistent with reports that p38 activation is delayed following ischemia beyond the timeframe studied here, and is mainly associated with activation of microglia. Our results therefore indicate that, although the activation of PI3K/Akt is associated with Hsp27-mediated neuroprotection, the mechanism by which Hsp27 exerts its neuroprotective effects lies primarily in direct suppression of the ASK1/JNK signaling cascade.
Activation of ASK1 occurs in ischemic injury models as well as in pharmacologically induced oxidative stress (Pan et al., 2007). ASK1 knock-out mice were remarkably less susceptible to hypoxic retinal injury, oxidative or endoplasmic reticular stress, and amyloid β- or TNF-α-induced cell death (Matsukawa et al., 2004; Kadowaki et al., 2005; Harada et al., 2006; Sekine et al., 2006). In this study, we found that Hsp27 physically interacted with the kinase domain of ASK1 and directly inhibited its activity. This finding has several implications. First, Hsp27 may also exert protective effects in other ASK1-dependent toxicity models, such as endoplasmic reticular stress, lipopolysaccharide, or other inducers of oxidative stress (Nagai et al., 2007). This is supported by experiments using the DNA-damaging reagent bleomycin, which induced cell death in ASK1/JNK-dependent manner. Second, we have demonstrated that the scaffolding model of Hsp27 function extends to binding with and inhibiting active ASK1. The activation of ASK1 is highly sensitive to the cellular redox state, thus providing a direct link between oxidative stress and kinase signaling pathways. In its unphosphorylated state, ASK1 is inactive and exists as a signalosome bound to the redox-sensitive molecule thioredoxin (Matsuzawa and Ichijo, 2008). Upon oxidation by cellular oxidants, thioredoxin dissociates from ASK1 and allows for the phosphorylation of Thr838 in the kinase domain and the formation of the active ASK1 signalosome (Saitoh et al., 1998). The observed phosphorylation of ASK1 using a Thr838-specific antibody in the presence of Hsp27 overexpression indicates that Hsp27 overexpression functions downstream of the activation of ASK1, rather than directly reduces cellular oxidative stress responsible for ASK1 activation. Activated ASK1 associates with downstream kinases via the MKK C terminus DVD domain and the ASK1 kinase domain (Takekawa et al., 2005). Thus, the observed physical association of activated ASK1 kinase domain and the N-terminal region of Hsp27 may lead to the disruption of docking and activation of downstream MKKs. Understanding the interaction between Hsp27 and ASK1 may provide the basis for the discovery of small molecule inhibitors for ASK1. Translational work is aided by the fact that the ASK1 kinase domain is highly conserved evolutionarily (Matsuzawa and Ichijo, 2008), increasing the likelihood of that a clinically useful inhibitor could be developed. Furthermore, investigation on the role that the N-terminal domain of Hsp27 plays in scaffolding functions may provide clues into the specificity and function of small heat shock proteins.
Hsp27 has multiple functions in promoting cellular survival in addition to suppression of specific death signaling pathways. For example, Hsp27 increased the solubility of protein aggregates in polyglutamine repeat models (Wyttenbach et al., 2002; Perrin et al., 2007). In the same model, Hsp27, but not phospho-mimetic mutants, was capable of protecting against poly(Q)-mediated cell death (Wyttenbach et al., 2002). The protein chaperone function of Hsp27 has been most closely associated with the unphosphorylated state of Hsp27 (Rogalla et al., 1999), whereas the anti-apoptotic actions appear to require phosphorylation of Hsp27 (Charette et al., 2000; Benn et al., 2002; de Graauw et al., 2005). Increased phosphorylation of Hsp27 was observed following ischemic preconditioning stimuli in organotypic hippocampal cultures and in vivo (Valentim et al., 2001, 2003), and phosphorylated Hsp27 prevented TNFα/Daxx-dependent cell death in 293 cells (Charette et al., 2000). Further studies examining the regulation of Hsp27 phosphorylation and its effects on ASK1 may reveal mechanistic nuances in neuronal systems.
The neuroprotective effects of another inducible heat shock protein, Hsp72, have been well documented (Garrido et al., 2006; Arya et al., 2007). In addition to its chaperone function, Hsp72 appears to directly target several cell death-execution molecules downstream of mitochondrial pathway, such as caspase-3, caspase-9, and AIF (Beere et al., 2000; Ravagnan et al., 2001; Gurbuxani et al., 2003). These downstream cell death-suppressing effects by Hsp72 likely contribute to the reported neuroprotection conferred by Hsp72 against cerebral ischemia (van der Weerd et al., 2005; Badin et al., 2006; An et al., 2008). However, the achievement of a long-term neuroprotective effect may necessitate targeting upstream signaling cascades in ways that Hsp72 may not be capable. The combined use of both Hsp27 and Hsp72 could therefore have synergistic protective effects. Dual activation of heat shock proteins is already known to occur, as both Hsp27 and Hsp70 are stimulated by and contribute to the cellular protection of bicyclol, an antihepatitis drug (Bao and Liu, 2008). The potential complementary actions of these two heat shock classes may provide a stronger therapeutic regimen against cerebral ischemia.
In summary, we have provided evidence that ASK1-dependent JNK signaling plays a critical role in mediating neuronal cell death following ischemia. Furthermore, we have demonstrated that Hsp27 is a direct inhibitor of ASK1 activity via physical association with the ASK1 kinase domain. Hsp27 overexpression conferred a long-lasting neuroprotective effect against cerebral ischemia. Together, these results suggest that Hsp27-mediated inhibition of ASK1 prodeath signaling pathway may be a promising novel neuroprotective strategy for stroke and other forms of neuronal injury.
Footnotes
This work was supported by National Institutes of Health–National Institute of Neurological Disorders and Stroke Grants NS43802, NS45048, NS36736, and NS 56118 and VA Merit Review Grant (to J.C.). Additional support was provided by the American Heart Association to R.A.S. (0725503U) and P.V. (0715254U), and by the Chinese Natural Science Foundation (Grants 30470592 and 30670642 to Y.G.). We thank Drs. Lin Zhang and Jian Yu (Pittsburgh Cancer Institute) for providing adenovirus vectors expressing Bax and PUMA. We thank Carol Culver for editorial assistance and Pat Strickler for secretarial support.
References
- An JJ, Lee YP, Kim SY, Lee SH, Lee MJ, Jeong MS, Kim DW, Jang SH, Yoo KY, Won MH, Kang TC, Kwon OS, Cho SW, Lee KS, Park J, Eum WS, Choi SY. Transduced human PEP-1-heat shock protein 27 efficiently protects against brain ischemic insult. FEBS J. 2008;275:1296–1308. doi: 10.1111/j.1742-4658.2008.06291.x. [DOI] [PubMed] [Google Scholar]
- Arya R, Mallik M, Lakhotia SC. Heat shock genes—integrating cell survival and death. J Biosci. 2007;32:595–610. doi: 10.1007/s12038-007-0059-3. [DOI] [PubMed] [Google Scholar]
- Badin RA, Lythgoe MF, van der Weerd L, Thomas DL, Gadian DG, Latchman DS. Neuroprotective effects of virally delivered HSPs in experimental stroke. J Cereb Blood Flow Metab. 2006;26:371–381. doi: 10.1038/sj.jcbfm.9600190. [DOI] [PubMed] [Google Scholar]
- Bao XQ, Liu GT. Bicyclol: a novel antihepatitis drug with hepatic heat shock protein 27/70-inducing activity and cytoprotective effects in mice. Cell Stress Chaperones. 2008;13:347–355. doi: 10.1007/s12192-008-0034-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2:469–475. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- Benn SC, Perrelet D, Kato AC, Scholz J, Decosterd I, Mannion RJ, Bakowska JC, Woolf CJ. Hsp27 upregulation and phosphorylation is required for injured sensory and motor neuron survival. Neuron. 2002;36:45–56. doi: 10.1016/s0896-6273(02)00941-8. [DOI] [PubMed] [Google Scholar]
- Bilsland J, Harper S. Caspases and neuroprotection. Curr Opin Investig Drugs. 2002;3:1745–1752. [PubMed] [Google Scholar]
- Bruey JM, Ducasse C, Bonniaud P, Ravagnan L, Susin SA, Diaz-Latoud C, Gurbuxani S, Arrigo AP, Kroemer G, Solary E, Garrido C. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat Cell Biol. 2000;2:645–652. doi: 10.1038/35023595. [DOI] [PubMed] [Google Scholar]
- Cao G, Minami M, Pei W, Yan C, Chen D, O'Horo C, Graham SH, Chen J. Intracellular Bax translocation after transient cerebral ischemia: implications for a role of the mitochondrial apoptotic signaling pathway in ischemic neuronal death. J Cereb Blood Flow Metab. 2001a;21:321–333. doi: 10.1097/00004647-200104000-00001. [DOI] [PubMed] [Google Scholar]
- Cao G, Pei W, Lan J, Stetler RA, Luo Y, Nagayama T, Graham SH, Yin XM, Simon RP, Chen J. Caspase-activated DNase/DNA fragmentation factor 40 mediates apoptotic DNA fragmentation in transient cerebral ischemia and in neuronal cultures. J Neurosci. 2001b;21:4678–4690. doi: 10.1523/JNEUROSCI.21-13-04678.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao G, Pei W, Ge H, Liang Q, Luo Y, Sharp FR, Lu A, Ran R, Graham SH, Chen J. In vivo delivery of a Bcl-xL fusion protein containing the TAT protein transduction domain protects against ischemic brain injury and neuronal apoptosis. J Neurosci. 2002;22:5423–5431. doi: 10.1523/JNEUROSCI.22-13-05423.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao G, Clark RS, Pei W, Yin W, Zhang F, Sun FY, Graham SH, Chen J. Translocation of apoptosis-inducing factor in vulnerable neurons after transient cerebral ischemia and in neuronal cultures after oxygen-glucose deprivation. J Cereb Blood Flow Metab. 2003;23:1137–1150. doi: 10.1097/01.WCB.0000087090.01171.E7. [DOI] [PubMed] [Google Scholar]
- Cao G, Xiao M, Sun F, Xiao X, Pei W, Li J, Graham SH, Simon RP, Chen J. Cloning of a novel Apaf-1-interacting protein: a potent suppressor of apoptosis and ischemic neuronal cell death. J Neurosci. 2004;24:6189–6201. doi: 10.1523/JNEUROSCI.1426-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao G, Xing J, Xiao X, Liou AK, Gao Y, Yin XM, Clark RS, Graham SH, Chen J. Critical role of calpain I in mitochondrial release of apoptosis-inducing factor in ischemic neuronal injury. J Neurosci. 2007;27:9278–9293. doi: 10.1523/JNEUROSCI.2826-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charette SJ, Lavoie JN, Lambert H, Landry J. Inhibition of Daxx-mediated apoptosis by heat shock protein 27. Mol Cell Biol. 2000;20:7602–7612. doi: 10.1128/mcb.20.20.7602-7612.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christophe M, Nicolas S. Mitochondria: a target for neuroprotective interventions in cerebral ischemia-reperfusion. Curr Pharm Des. 2006;12:739–757. doi: 10.2174/138161206775474242. [DOI] [PubMed] [Google Scholar]
- Concannon CG, Orrenius S, Samali A. Hsp27 inhibits cytochrome c-mediated caspase activation by sequestering both pro-caspase-3 and cytochrome c. Gene Expr. 2001;9:195–201. doi: 10.3727/000000001783992605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concannon CG, Gorman AM, Samali A. On the role of Hsp27 in regulating apoptosis. Apoptosis. 2003;8:61–70. doi: 10.1023/a:1021601103096. [DOI] [PubMed] [Google Scholar]
- Cregan SP, Fortin A, MacLaurin JG, Callaghan SM, Cecconi F, Yu SW, Dawson TM, Dawson VL, Park DS, Kroemer G, Slack RS. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J Cell Biol. 2002;158:507–517. doi: 10.1083/jcb.200202130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currie RW, Ellison JA, White RF, Feuerstein GZ, Wang X, Barone FC. Benign focal ischemic preconditioning induces neuronal Hsp70 and prolonged astrogliosis with expression of Hsp27. Brain Res. 2000;863:169–181. doi: 10.1016/s0006-8993(00)02133-8. [DOI] [PubMed] [Google Scholar]
- de Graauw M, Tijdens I, Cramer R, Corless S, Timms JF, van de Water B. Heat shock protein 27 is the major differentially phosphorylated protein involved in renal epithelial cellular stress response and controls focal adhesion organization and apoptosis. J Biol Chem. 2005;280:29885–29898. doi: 10.1074/jbc.M412708200. [DOI] [PubMed] [Google Scholar]
- Deshmukh M, Johnson EM., Jr Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome c. Neuron. 1998;21:695–705. doi: 10.1016/s0896-6273(00)80587-5. [DOI] [PubMed] [Google Scholar]
- Dhodda VK, Sailor KA, Bowen KK, Vemuganti R. Putative endogenous mediators of preconditioning-induced ischemic tolerance in rat brain identified by genomic and proteomic analysis. J Neurochem. 2004;89:73–89. doi: 10.1111/j.1471-4159.2004.02316.x. [DOI] [PubMed] [Google Scholar]
- Fujimura M, Morita-Fujimura Y, Murakami K, Kawase M, Chan PH. Cytosolic redistribution of cytochrome c after transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1998;18:1239–1247. doi: 10.1097/00004647-199811000-00010. [DOI] [PubMed] [Google Scholar]
- Gao Y, Signore AP, Yin W, Cao G, Yin XM, Sun F, Luo Y, Graham SH, Chen J. Neuroprotection against focal ischemic brain injury by inhibition of c-Jun N-terminal kinase and attenuation of the mitochondrial apoptosis-signaling pathway. J Cereb Blood Flow Metab. 2005;25:694–712. doi: 10.1038/sj.jcbfm.9600062. [DOI] [PubMed] [Google Scholar]
- Garrido C, Brunet M, Didelot C, Zermati Y, Schmitt E, Kroemer G. Heat shock proteins 27 and 70: anti-apoptotic proteins with tumorigenic properties. Cell Cycle. 2006;5:2592–2601. doi: 10.4161/cc.5.22.3448. [DOI] [PubMed] [Google Scholar]
- Gibson CL, Bath PM, Murphy SP. G-CSF reduces infarct volume and improves functional outcome after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2005;25:431–439. doi: 10.1038/sj.jcbfm.9600033. [DOI] [PubMed] [Google Scholar]
- Gorman AM, Szegezdi E, Quigney DJ, Samali A. Hsp27 inhibits 6-hydroxydopamine-induced cytochrome c release and apoptosis in PC12 cells. Biochem Biophys Res Commun. 2005;327:801–810. doi: 10.1016/j.bbrc.2004.12.066. [DOI] [PubMed] [Google Scholar]
- Graham SH, Chen J. Programmed cell death in cerebral ischemia. J Cereb Blood Flow Metab. 2001;21:99–109. doi: 10.1097/00004647-200102000-00001. [DOI] [PubMed] [Google Scholar]
- Gurbuxani S, Schmitt E, Cande C, Parcellier A, Hammann A, Daugas E, Kouranti I, Spahr C, Pance A, Kroemer G, Garrido C. Heat shock protein 70 binding inhibits the nuclear import of apoptosis-inducing factor. Oncogene. 2003;22:6669–6678. doi: 10.1038/sj.onc.1206794. [DOI] [PubMed] [Google Scholar]
- Harada C, Nakamura K, Namekata K, Okumura A, Mitamura Y, Iizuka Y, Kashiwagi K, Yoshida K, Ohno S, Matsuzawa A, Tanaka K, Ichijo H, Harada T. Role of apoptosis signal-regulating kinase 1 in stress-induced neural cell apoptosis in vivo. Am J Pathol. 2006;168:261–269. doi: 10.2353/ajpath.2006.050765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori K, Lee H, Hurn PD, Crain BJ, Traystman RJ, DeVries AC. Cognitive deficits after focal cerebral ischemia in mice. Stroke. 2000;31:1939–1944. doi: 10.1161/01.str.31.8.1939. [DOI] [PubMed] [Google Scholar]
- Havasi A, Li Z, Wang Z, Martin JL, Botla V, Ruchalski K, Schwartz JH, Borkan SC. Hsp27 inhibits Bax activation and apoptosis via a phosphatidylinositol 3-kinase-dependent mechanism. J Biol Chem. 2008;283:12305–12313. doi: 10.1074/jbc.M801291200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadowaki H, Nishitoh H, Urano F, Sadamitsu C, Matsuzawa A, Takeda K, Masutani H, Yodoi J, Urano Y, Nagano T, Ichijo H. Amyloid beta induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 2005;12:19–24. doi: 10.1038/sj.cdd.4401528. [DOI] [PubMed] [Google Scholar]
- Kato H, Kogure K, Liu XH, Araki T, Kato K, Itoyama Y. Immunohistochemical localization of the low molecular weight stress protein HSP27 following focal cerebral ischemia in the rat. Brain Res. 1995;679:1–7. doi: 10.1016/0006-8993(95)00198-y. [DOI] [PubMed] [Google Scholar]
- Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol. 2001;21:893–901. doi: 10.1128/MCB.21.3.893-901.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi H, Matsuzaki H, Tanaka M, Takemura Y, Kuroda S, Ono Y, Kikkawa U. Activation of protein kinase B (Akt/RAC-protein kinase) by cellular stress and its association with heat shock protein Hsp27. FEBS Lett. 1997;410:493–498. doi: 10.1016/s0014-5793(97)00541-3. [DOI] [PubMed] [Google Scholar]
- Kuan CY, Whitmarsh AJ, Yang DD, Liao G, Schloemer AJ, Dong C, Bao J, Banasiak KJ, Haddad GG, Flavell RA, Davis RJ, Rakic P. A critical role of neural-specific JNK3 for ischemic apoptosis. Proc Natl Acad Sci U S A. 2003;100:15184–15189. doi: 10.1073/pnas.2336254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakhani SA, Masud A, Kuida K, Porter GA, Jr, Booth CJ, Mehal WZ, Inayat I, Flavell RA. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 2006;311:847–851. doi: 10.1126/science.1115035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VY, Schroedl C, Brunelle JK, Buccellato LJ, Akinci OI, Kaneto H, Snyder C, Eisenbart J, Budinger GR, Chandel NS. Bleomycin induces alveolar epithelial cell death through JNK-dependent activation of the mitochondrial death pathway. Am J Physiol Lung Cell Mol Physiol. 2005;289:L521–L528. doi: 10.1152/ajplung.00340.2004. [DOI] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- Loetscher H, Niederhauser O, Kemp J, Gill R. Is caspase-3 inhibition a valid therapeutic strategy in cerebral ischemia? Drug Discov Today. 2001;6:671–680. doi: 10.1016/s1359-6446(01)01826-8. [DOI] [PubMed] [Google Scholar]
- Martin LJ. DNA damage and repair: relevance to mechanisms of neurodegeneration. J Neuropathol Exp Neurol. 2008;67:377–387. doi: 10.1097/NEN.0b013e31816ff780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsukawa J, Matsuzawa A, Takeda K, Ichijo H. The ASK1-MAP kinase cascades in mammalian stress response. J Biochem. 2004;136:261–265. doi: 10.1093/jb/mvh134. [DOI] [PubMed] [Google Scholar]
- Matsuzawa A, Ichijo H. Redox control of cell fate by MAP kinase: physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim Biophys Acta. 2008;1780:1325–1336. doi: 10.1016/j.bbagen.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Mearow KM, Dodge ME, Rahimtula M, Yegappan C. Stress-mediated signaling in PC12 cells—the role of the small heat shock protein, Hsp27, and Akt in protecting cells from heat stress and nerve growth factor withdrawal. J Neurochem. 2002;83:452–462. doi: 10.1046/j.1471-4159.2002.01151.x. [DOI] [PubMed] [Google Scholar]
- Moreau C, Cartron PF, Hunt A, Meflah K, Green DR, Evan G, Vallette FM, Juin P. Minimal BH3 peptides promote cell death by antagonizing anti-apoptotic proteins. J Biol Chem. 2003;278:19426–19435. doi: 10.1074/jbc.M209472200. [DOI] [PubMed] [Google Scholar]
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Nagai H, Noguchi T, Takeda K, Ichijo H. Pathophysiological roles of ASK1-MAP kinase signaling pathways. J Biochem Mol Biol. 2007;40:1–6. doi: 10.5483/bmbrep.2007.40.1.001. [DOI] [PubMed] [Google Scholar]
- Pan J, Wang G, Yang HQ, Hong Z, Xiao Q, Ren RJ, Zhou HY, Bai L, Chen SD. K252a prevents nigral dopaminergic cell death induced by 6-hydroxydopamine through inhibition of both mixed-lineage kinase 3/c-Jun NH2-terminal kinase 3 (JNK3) and apoptosis-inducing kinase 1/JNK3 signaling pathways. Mol Pharmacol. 2007;72:1607–1618. doi: 10.1124/mol.107.038463. [DOI] [PubMed] [Google Scholar]
- Paschen W. Endoplasmic reticulum dysfunction in brain pathology: critical role of protein synthesis. Curr Neurovasc Res. 2004;1:173–181. doi: 10.2174/1567202043480125. [DOI] [PubMed] [Google Scholar]
- Perrin V, Régulier E, Abbas-Terki T, Hassig R, Brouillet E, Aebischer P, Luthi-Carter R, Déglon N. Neuroprotection by Hsp104 and Hsp27 in lentiviral-based rat models of Huntington's disease. Mol Ther. 2007;15:903–911. doi: 10.1038/mt.sj.6300141. [DOI] [PubMed] [Google Scholar]
- Philpott KL, Facci L. MAP kinase pathways in neuronal cell death. CNS Neurol Disord Drug Targets. 2008;7:83–97. doi: 10.2174/187152708783885129. [DOI] [PubMed] [Google Scholar]
- Plumier JC, Hopkins DA, Robertson HA, Currie RW. Constitutive expression of the 27-kDa heat shock protein (Hsp27) in sensory and motor neurons of the rat nervous system. J Comp Neurol. 1997;384:409–428. [PubMed] [Google Scholar]
- Ramezani A, Hawley RG. Generation of HIV-1-based lentiviral vector particles. In: Ausubel FM, editor. Current protocols in molecular biology. New York: Wiley; 2002. pp. 16.11.11–16.22.15. [DOI] [PubMed] [Google Scholar]
- Rane MJ, Pan Y, Singh S, Powell DW, Wu R, Cummins T, Chen Q, McLeish KR, Klein JB. Heat shock protein 27 controls apoptosis by regulating Akt activation. J Biol Chem. 2003;278:27828–27835. doi: 10.1074/jbc.M303417200. [DOI] [PubMed] [Google Scholar]
- Ravagnan L, Gurbuxani S, Susin SA, Maisse C, Daugas E, Zamzami N, Mak T, Jäättelä M, Penninger JM, Garrido C, Kroemer G. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat Cell Biol. 2001;3:839–843. doi: 10.1038/ncb0901-839. [DOI] [PubMed] [Google Scholar]
- Rogalla T, Ehrnsperger M, Preville X, Kotlyarov A, Lutsch G, Ducasse C, Paul C, Wieske M, Arrigo AP, Buchner J, Gaestel M. Regulation of Hsp27 oligomerization, chaperone function, and protective activity against oxidative stress/tumor necrosis factor alpha by phosphorylation. J Biol Chem. 1999;274:18947–18956. doi: 10.1074/jbc.274.27.18947. [DOI] [PubMed] [Google Scholar]
- Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada M, Alkayed NJ, Goto S, Crain BJ, Traystman RJ, Shaivitz A, Nelson RJ, Hurn PD. Estrogen receptor antagonist ICI182,780 exacerbates ischemic injury in female mouse. J Cereb Blood Flow Metab. 2000;20:112–118. doi: 10.1097/00004647-200001000-00015. [DOI] [PubMed] [Google Scholar]
- Sekine Y, Takeda K, Ichijo H. The ASK1-MAP kinase signaling in ER stress and neurodegenerative diseases. Curr Mol Med. 2006;6:87–97. doi: 10.2174/156652406775574541. [DOI] [PubMed] [Google Scholar]
- Souhir B, Laurent P, Sonia Y, Delphine M, Marion C, Cédric R, Olivier M, Abderraouf K, Ali B. Deglycosylated bleomycin induces apoptosis in lymphoma cell via c-jun NH2-terminal kinase but not reactive oxygen species. Biochem Pharmacol. 2007;74:1445–1455. doi: 10.1016/j.bcp.2007.07.036. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Fujimura M, Morita-Fujimura Y, Kawase M, Chan PH. Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. J Neurosci. 1999;19(RC39) doi: 10.1523/JNEUROSCI.19-22-j0002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takekawa M, Tatebayashi K, Saito H. Conserved docking site is essential for activation of mammalian MAP kinase kinases by specific MAP kinase kinase kinases. Mol Cell. 2005;18:295–306. doi: 10.1016/j.molcel.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Valentim LM, Geyer AB, Tavares A, Cimarosti H, Worm PV, Rodnight R, Netto CA, Salbego CG. Effects of global cerebral ischemia and preconditioning on heat shock protein 27 immunocontent and phosphorylation in rat hippocampus. Neuroscience. 2001;107:43–49. doi: 10.1016/s0306-4522(01)00325-6. [DOI] [PubMed] [Google Scholar]
- Valentim LM, Rodnight R, Geyer AB, Horn AP, Tavares A, Cimarosti H, Netto CA, Salbego CG. Changes in heat shock protein 27 phosphorylation and immunocontent in response to preconditioning to oxygen and glucose deprivation in organotypic hippocampal cultures. Neuroscience. 2003;118:379–386. doi: 10.1016/s0306-4522(02)00919-3. [DOI] [PubMed] [Google Scholar]
- van der Weerd L, Lythgoe MF, Badin RA, Valentim LM, Akbar MT, de Belleroche JS, Latchman DS, Gadian DG. Neuroprotective effects of HSP70 overexpression after cerebral ischaemia—an MRI study. Exp Neurol. 2005;195:257–266. doi: 10.1016/j.expneurol.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Voss OH, Batra S, Kolattukudy SJ, Gonzalez-Mejia ME, Smith JB, Doseff AI. Binding of caspase-3 prodomain to heat shock protein 27 regulates monocyte apoptosis by inhibiting caspase-3 proteolytic activation. J Biol Chem. 2007;282:25088–25099. doi: 10.1074/jbc.M701740200. [DOI] [PubMed] [Google Scholar]
- Whitlock NA, Agarwal N, Ma JX, Crosson CE. Hsp27 upregulation by HIF-1 signaling offers protection against retinal ischemia in rats. Invest Ophthalmol Vis Sci. 2005;46:1092–1098. doi: 10.1167/iovs.04-0043. [DOI] [PubMed] [Google Scholar]
- Wu R, Kausar H, Johnson P, Montoya-Durango DE, Merchant M, Rane MJ. Hsp27 regulates Akt activation and polymorphonuclear leukocyte apoptosis by scaffolding MK2 to Akt signal complex. J Biol Chem. 2007;282:21598–21608. doi: 10.1074/jbc.M611316200. [DOI] [PubMed] [Google Scholar]
- Wyttenbach A, Sauvageot O, Carmichael J, Diaz-Latoud C, Arrigo AP, Rubinsztein DC. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum Mol Genet. 2002;11:1137–1151. doi: 10.1093/hmg/11.9.1137. [DOI] [PubMed] [Google Scholar]
- Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Li C, Yin XH, Zhang GY. Additive neuroprotection of GABA A and GABA B receptor agonists in cerebral ischemic injury via PI-3K/Akt pathway inhibiting the ASK1-JNK cascade. Neuropharmacology. 2008;54:1029–1040. doi: 10.1016/j.neuropharm.2008.01.014. [DOI] [PubMed] [Google Scholar]
- Zhang L, Schallert T, Zhang ZG, Jiang Q, Arniego P, Li Q, Lu M, Chopp M. A test for detecting long-term sensorimotor dysfunction in the mouse after focal cerebral ischemia. J Neurosci Methods. 2002;117:207–214. doi: 10.1016/s0165-0270(02)00114-0. [DOI] [PubMed] [Google Scholar]
- Zhang QG, Wu DN, Han D, Zhang GY. Critical role of PTEN in the coupling between PI3K/Akt and JNK1/2 signaling in ischemic brain injury. FEBS Lett. 2007;581:495–505. doi: 10.1016/j.febslet.2006.12.055. [DOI] [PubMed] [Google Scholar]
- Zheng C, Lin Z, Zhao ZJ, Yang Y, Niu H, Shen X. MAPK-activated protein kinase-2 (MK2)-mediated formation and phosphorylation-regulated dissociation of the signal complex consisting of p38, MK2, Akt, and Hsp27. J Biol Chem. 2006;281:37215–37226. doi: 10.1074/jbc.M603622200. [DOI] [PubMed] [Google Scholar]