

Abstract
This report is a summary of the symposium on Alcohol, Intestinal Bacterial Growth, Intestinal Permeability to Endotoxin, and Medical Consequences, organized by National Institute on Alcohol Abuse and Alcoholism, Office of Dietary Supplements, and National Institute of Diabetes and Digestive and Kidney Diseases of National Institutes of Health in Rockville, Maryland, October 11, 2006. Alcohol exposure can promote the growth of Gram negative bacteria in the intestine which may result in accumulation of endotoxin. In addition, alcohol metabolism by Gram negative bacteria and intestinal epithelial cells can result in accumulation of acetaldehyde, which in turn can increase intestinal permeability to endotoxin by increasing tyrosine phosphorylation of tight junction and adherens junction proteins. Alcohol-induced generation of nitric oxide may also contribute to increased permeability to endotoxin by reacting with tubulin, which may cause damage to microtubule cytoskeleton and subsequent disruption of intestinal barrier function. Increased intestinal permeability can lead to increased transfer of endotoxin from the intestine to the liver and general circulation where endotoxin may trigger inflammatory changes in the liver and other organs. Alcohol may also increase intestinal permeability to peptidoglycan which can initiate inflammatory response in liver and other organs. In addition, acute alcohol exposure may potentiate the effect of burn injury on intestinal bacterial growth and permeability. Decreasing the number of Gram negative bacteria in the intestine can result in decreased production of endotoxin as well as acetaldehyde which is expected to decrease intestinal permeability to endotoxin. In addition, intestinal permeability may be preserved by administering epidermal growth factor, L-glutamine, oats supplementation, or zinc thereby preventing the transfer of endotoxin to the general circulation. Thus reducing the number of intestinal Gram negative bacteria and preserving intestinal permeability to endotoxin may attenuate alcoholic liver and other organ injuries.
Keywords: alcohol, bacterial growth, intestinal permeability, endotoxin, acetaldehyde, nitric oxide
INTRODUCTION
Chronic alcohol consumption is associated with the development of various medical disorders such as alcoholic liver diseases (ALD), pancreatitis, cardiomyopathy, acute respiratory distress syndrome (ARDS), and brain injury in some individuals. Endotoxin appears to play a central role in the initiation of alcohol-induced tissue/organ damage and its role is most convincing for liver injury. Endotoxin is a lipopolysaccharide (LPS) derived from the cell wall of Gram negative bacteria residing in the intestine. The connection between alcohol, endotoxin and liver injury became apparent when researchers observed elevated plasma endotoxin levels in patients affected with ALD (Bode et al., 1987; Fukui et al., 1991). This connection was further confirmed when administration of LPS caused the progression of fatty liver into necro-inflammatory changes in a rat model of alcoholic liver injury (Bhagwandeen et al., 1987; Pennington et al., 1997). In addition, elimination of intestinal bacteria or suppression of intestinal Gram negative bacteria attenuated alcoholic liver injury in rats by reducing plasma endotoxin levels (Adachi et al., 1995; Nanji et al., 1994). Normally, only a trace amount of endotoxin is absorbed from the intestine through the intestinal epithelial lining to the portal vein that carries it to the liver where it is cleared by Kupffer cells. However, excess amount of endotoxin reaching to the liver can activate Kupffer cells to initiate liver inflammation (Thurman, 1998; Wheeler et al., 2001). Furthermore, excess amount of endotoxin in the general circulation may trigger injury to other organs. Elevated levels of plasma endotoxin in response to alcohol ingestion may result from: 1) excessive production of endotoxin in the intestine through overgrowth of Gram negative bacteria; 2) increased permeability of the intestine to endotoxin; and 3) delayed clearance of endotoxin by Kupffer cells. Suppressing the growth of intestinal Gram negative bacteria and/or restoring/preserving intestinal integrity and thereby blocking the transfer of endotoxin from the intestine to general circulation may be logical steps in preventing or attenuating alcohol-associated tissue/organ injuries. Therefore, understanding the underlying mechanisms by which alcohol promotes intestinal bacterial growth and increases intestinal permeability to endotoxin may help design strategies for the prevention or treatment of alcohol/endotoxin-associated medical disorders.
The National Institute on Alcohol Abuse and Alcoholism (NIAAA) in collaboration with Office of Dietary Supplements (ODS) and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) of National Institutes of Health (NIH) organized a symposium on Alcohol, Intestinal Bacterial Growth, Intestinal Permeability to Endotoxin, and Medical Consequences in Rockville, Maryland, October 11, 2006. The following topics were discussed by nine speakers: 1. Role of Endotoxin in Alcoholic and Non-Alcoholic Liver Injury (David A. Brenner); 2. Alcohol, Immune System, and Intestinal Bacterial Growth (Mashkoor A. Choudhry); 3. Probiotics, Intestinal Bacterial Overgrowth, and Peptidoglycan (R. Balfour Sartor); 4. The Leaky Epithelial Barrier in Intestinal Diseases (Jerrold R Turner); 5. Alcohol, Intestinal Permeability, and Endotoxemia (J. Christian Bode and Christiane Bode); 6. Effects of Acetaldehyde and L-glutamine on Intestinal Permeability to Endotoxin (Radhakrishna Rao); 7. Role of Zinc in Preserving Intestinal Integrity in Alcohol-Intoxicated Mice (James Kang); and 8. Effects of Nitric Oxide and Oats Supplementation on Alcohol-Induced Leaky Gut (Ali Keshavarzian). The following is a summary of the symposium.
SUMMARY
Alcohol and Intestinal Bacterial Growth
The intestine of normal human subjects is a habitat for various types of bacteria. Overgrowth of Gram negative bacteria can result in increased production of endotoxin which can escape into portal circulation leading to increased plasma levels of endotoxin. Only a few studies are available in literature investigating relationship between alcohol consumption and bacterial growth in the small intestine. Increased number of microorganisms, including Gram negative coliform bacteria, was reported in the proximal part of small intestine of chronic alcoholics; however, no correlation was found between the number or types of microorganisms in the jejunum and the type or degree of liver disease in the alcoholics (Bode et al., 1984). Bacterial over growth was also reported in the duodenum of alcoholics (Hauge et al., 1997). In another study, the incidence of intestinal bacterial overgrowth was three times higher in alcoholics with ALD than that in control subjects with no history of alcohol abuse (Bode et al., 1993). These studies suggest that chronic alcohol abuse can promote bacterial growth in the intestine; however, further studies are required to answer the following questions. Does alcohol affect bacterial growth in a dose-dependent manner? How does alcohol affect bacterial growth at a mechanistic level? Is bacterial growth associated with increased amount of endotoxin production? What types of bacteria are affected by alcohol?
Suppression of Bacterial Growth by Probiotics
Suppressing the growth of Gram negative bacteria in the intestine may reduce the amount of endotoxin which in turn may attenuate endotoxin-associated organ damage. In this regard, use of probiotic bacteria, otherwise known as beneficial bacteria, provides a viable option. The most numerous probiotic bacteria normally inhabiting the small intestine are species of Lactobacilli whereas in the colon the majority is mainly Bifidobacteria. These beneficial bacteria have been shown to attenuate the growth of Gram negative bacteria. Feeding of Lactobacillus acidophilus strain NP51 was found to reduce the number of Escherichia coli O157:H7 in the fecal samples of beef cattle (Younts-Dahl et al., 2005; Peterson et al., 2007). Lactobacillus strains, LAP5 and LF33, obtained from swine and poultry, respectively, were shown to inhibit the growth of Escherichia coli and Salmonella typhimurium in an in vitro culture system (Tsai et al., 2005). Enteral administration of Lactobacillus R2LC attenuated bacteremia and endotoxemia associated with intra-abdominal infection in rats (Thorlacius et al., 2003). Bifidobacterium animalis MB5 and Lactobacillus rhamnosus GG were shown to protect intestinal Caco-2 cells from the inflammation-associated response induced by Escherichia coli K88 (Roselli et al., 2006). Finally, in an intragastric infusion model of alcoholic liver injury, feeding of rats with Lactobacillus GG reduced plasma levels of endotoxin and severity of liver injury (Nanji et al., 1994). Whether probiotic treatment can attenuate ALD in humans needs further investigation.
The Leaky Epithelial Barrier in Intestinal Diseases
Intestinal diseases are associated with leaky epithelial barrier and increased intestinal permeability to toxic agents, such as endotoxin. This may result in increased transfer of endotoxin from the intestine into liver and general circulation, which in turn may initiate injury to liver and other organs. Therefore, an intact intestinal barrier primarily formed by the epithelium is critical to normal physiological function and prevention of diseases. The space between adjacent epithelial cells (the paracellular space) is sealed primarily by tight junctions, which control the barrier function of an intact intestinal epithelium. Studies showing an association of thin actin-like filaments within the apical cytoplasm of small intestinal epithelial cells with tight junctions suggest that tight junctions are intimately related to the perijunctional cytoskeleton (Hull and Staehelin, 1979; Madara, 1987). Severing of actin filaments by fungal-derived cytochalasins disrupted tight junction barrier function and structure (Bentzel et al., 1980). Further studies showed that cytochalasin caused morphological condensation of perijunctional actin (Madara et al., 1987), suggesting that cytochalasin-induced condensation and associated contraction of perijunctional actin may have been responsible for tight junction disruption. Exposure of intestinal epithelial monolayer to actin-depolymerizing drugs removed one transmembrane protein - occludin — from the tight junction by endocytosis at the precise time that barrier function was disrupted (Shen and Turner, 2005). Thus, occludin endocytosis may be an important marker of tight junction regulation mediated by the cytoskeleton.
Evidence suggests that intestinal epithelial barrier function is regulated by physiological stimuli. Using isolated segments of hamster small intestine, it was shown that that Na+ - coupled nutrient (glucose, alanine, or leucine) transport triggers contraction of perijunctional actomyosin, thereby increasing intestinal tight junction permeability and enhancing absorption of nutrients (Madara and Pappenheimer, 1987). The role of Na+glucose transport in the regulation of epithelial tight junction permeability was further confirmed using Caco-2 intestinal epithelial cells transfected with the intestinal Na+-glucose cotransporter, wherein the activation of the co-transporter increased and inactivation decreased the permeability of the epithelial cells (Turner et al., 1997). Further research showed that the phosphporylation of myosin light-chain (MLC) catalyzed by myosin light-chain kinase (MLCK) was necessary for Na+-glucose co-transport-induced tight junction permeability regulation since pharmacological inhibition of MLCK prevented both MLC phosphorylation and regulation of tight junction barrier function in cultured intestinal epithelial cells (Turner et al., 1997).
Intestinal epithelial barrier function can be compromised in intestinal disease. This was initially recognized in patients with Crohn’s disease (Hollander, 1988; Ukabam, 1983), but has subsequently been reported in patients or experimental models of a spectrum of inflammatory, immune-mediated, and infectious intestinal diseases (Clayburgh et al., 2004). Tumor necrosis factor-α (TNF-α) appears to play an important role in regulating intestinal epithelial barrier function since TNFα-neutralizing antibodies restored barrier function in Crohn’s disease patients (Suenaert et al., 2002). Exposure of cultured epithelial monolayers to TNF-α in vitro caused epithelial barrier loss (Mullin et al., 1992; Marano et al., 1998). In another study, exposure of cultured epithelial monolayers to TNF-α increased MLC phosphorylation which was associated with impaired barrier function (Zolotarevsky et al., 2002). In this study, specific inhibition of MLCK prevented MLC phosphorylation and restored barrier function in TNF-α-treated monolayers (Zolotarevsky et al., 2002). Thus TNF-α appears to increase MLC phosphorylation by increasing the expression of MLCK (Wang et al., 2005; Ma et al., 2005). In inflammatory bowel disease patients the extent of increased MLCK expression and MLC phosphorylation correlates directly with the magnitude of active inflammation, suggesting a relationship between MLCK expression and disease severity (Blair et al., 2006).
Alcohol and Intestinal Permeability
Several researchers have investigated the effect of alcohol on intestinal permeability to various molecules including endotoxin. In rats, chronic alcohol feeding by gavage increased permeability of the intestinal mucosa to macromolecules such as hemoglobin (Bungert, 1973) and horseradish peroxidase (Worthington et al., 1978). Intestinal permeability assessed by the lactulose/mannitol (L/M) ratio was significantly increased in rats administered ethanol by intragastic infusion (Mathurin et al., 2000). An ethanol-induced increase in intestinal permeability was also reported in human subjects when smaller molecules were used as a permeability probe. For example, an increased absorption of 51Cr EDTA was found in subjects chronically abusing alcohol (Bjarnason et al., 1984) and an increase in absorption of polyethylene glycol (PEG) 400 was observed after the oral administration of alcohol to healthy volunteers (Robinson et al., 1981). To determine whether alcohol can increase intestinal permeability to macromolecules, permeability to PEG with different molecular masses (Mr 400, Mr 1500, Mr 4000, and Mr 10000) was measured in recently drinking alcoholics with different stages of ALD (Parlesak et al., 2000). The permeability to PEG Mr 400 was found to be unchanged when compared to healthy controls, whereas the permeability to PEG Mr 1500 and Mr 4000 were distinctly enhanced and the prevalence of increased permeability to PEG 10000 was more than 10-fold higher in alcoholics (Parlesak et al., 2000). Direct evidence for increased gut permeability to endotoxin by alcohol was obtained in a rat study where intragastric administration of LPS (5 mg/kg) to alcohol-fed rats significantly increased portal vein endotoxin level after 2 hours (Mathurin et al., 2000). No such increases were observed in pair-fed controls. In another rat study, plasma endotoxin levels in long-term ethanol-fed rats were higher than those in control rats after intragastric administration of high-dose endotoxin (20 mg/kg) (Tamai et al., 2002). Furthermore, intestinal permeability to fluorescein isothiocyanate-labeled dextran with an average molecular weight of 4000 D (FD4) was increased by long-term ethanol administration (Tamai et al., 2002). Similar results were obtained in a mice study where animals were administered 1 mg/kg bacterial LPS by intragastirc gavage one hour after administration of alcohol by gavage (Lambert et al., 2003). Ethanol alone, but not LPS alone, significantly increased plasma endotoxin level, and ethanol+LPS caused further significant increase in plasma endotoxin levels (Lambert et al., 2003). These studies clearly indicate that alcohol can increase intestinal permeability to various macromolecules including endotoxin.
Alcohol and Intestinal Mucosal Damage
Acute administration of alcohol to rodents or dogs at concentrations corresponding to those found in commonly available alcoholic beverages (≥ 4%, v/v) leads to mucosal damage in the upper small intestine with a loss of epithelium at the tips of the villi, haemorrhagic erosions and even haemorrhage in the lamina propria (Bode et al., 2001). Similar lesions were observed in volunteers 2-3 hours after ingestion of alcohol (15-20%, v/v) and in recently drinking alcohol abusing subjects (Bode et al., 2001). In rats, administration of 20% ethanol by gavage for 4 hours resulted in hemorrhagic erosions of the proximal small intestine with epithelial cell loss (Tamai et al., 2000). Similarly, administration of 20% ethanol to rats for 14 days by gavage resulted in significant mucosal disruption of ileum as evidenced by subepithelial edema of villous tips, cellular infiltration of the villi and exfoliation (Napolitano et al., 1995). In mice, acute ethanol exposure (single oral dose of 6 g/kg) caused sever injury to the mucosal lining of ileum of the small intestine (Lambert et al., 2003). This included breaches in the epithelial layer of the villi, submucosal blebbing, and ulceration of the villi (Lambert et al., 2003). The mucosal damage caused by alcohol might result in an impaired intestinal barrier function, enabling toxins of gut-inhabiting bacteria such as endotoxins to enter the systemic circulation and to contribute to liver injury after alcohol consumption.
Role of Acetaldehyde in Increasing Intestinal Permeability
Acetaldehyde is the first and most toxic metabolite of ethanol metabolism. In the gastrointestinal tract, acetaldehyde can be generated from ethanol through mucosal and/or bacterial alcohol dehydrogenase (ADH) (Seitz and Oneta, 1998). During chronic ethanol administration, high concentration of acetaldehyde can be detected in the mucosa of large intestine. In rats, colonic luminal concentrations of acetaldehyde were significantly increased after ethanol intake (132.6 +/- 31.6 micromole/L versus 20.8 +/- 1.4 micromole/L) and antibiotic treatment attenuated this increase to some extent (86.2 +/- 10.9 micromol/L) (Ferrier et al., 2006), indicating a role of bacteria in the generation of acetaldehyde. Other investigators have reported further higher concentrations of acetaldehyde ranging from 0.12 mM to 3 mM in rat colonic lumen after ethanol administration (Rao et al, 2004). Accumulation of acetaldehyde in the colonic lumen could be due to low efficiency of bacterial aldehyde dehydrogenase (ALDH) to metabolize acetaldehyde in the colon (Nosova et al., 1998).
Increasing evidence suggest that acetaldehyde can increase intestinal permeability to endotoxin (Rao, 1998; Rao et al., 2004; Ferrier et al., 2006). Acetaldehyde in the concentration of 99 to 760 micromolar increased paracellular permeability in a human colonic epithelial (Caco-2) cell monolayer (Rao, 1998). The paracellular permeability was assessed by measuring transepithelial electrical resistance, sodium chloride dilution potential, and unidirectional flux of labeled mannitol. However, ethanol up to 5% concentration failed to increase paracellular permeability, suggesting that ethanol metabolism to acetaldehyde is required for the action. This was confirmed by another study where ethanol up to 3% showed no significant effect on paracellular permeability in Caco-2 cell monolayers (Ma et al., 1999). This study showed that ethanol increased paracellular permeability at 7.5% concentration. Subsequent studies by Banan et al., (1999, 2000) showed that ethanol increased paracellular permeability at doses 2.5% and above by reducing the cell viability. The importance of ethanol metabolism into acetaldehyde was further confirmed when acetaldehyde, and not ethanol, was shown to increase mucosal permeability in proximal rat colonic strips mounted to Ussing chambers in vitro (Ferrier et al., 2006). Further mechanistic studies showed that acetaldehyde-induced increase in paracellular permeability is associated with redistribution of tight junction proteins (occludin and ZO1) and adherens junction (E-cadherin and β-catenin) proteins from the intercellular junctions into the intracellular compartments (Atkinson and Rao, 2001; Seth et al., 2004; Sheth et al., 2004).
Researchers further investigated the role of protein tyrosine phosphorylation in acetaldehyde-induced disruption of epithelial tight junctions in the Caco-2 cell monolayer (Atkinson and Rao, 2001). Acetaldehyde increased tyrosine phosphorylation of ZO-1, E-cadherin, and beta-catenin, which was associated with a significant reduction in protein tyrosine phosphatase (PTP) activity without affecting tyrosine kinase activity. Treatment with acetaldehyde resulted in a 97% loss of PTP1B activity and a partial reduction of PTP1C and PTP1D activities. These results suggest that acetaldehyde inhibits PTP to increase protein tyrosine phosphorylation of tight junction and adherens junction proteins, which may result in disruption of the tight junctions and adherens junctions via redistribution of their proteins.
Since interactions between E-cadherin, β-catenin and PTP1B are crucial for the organization of adherens junctions and epithelial cell-cell adhesion and subsequent maintenance of intestinal epithelial barrier, researchers further investigated the effect of acetaldehyde on the interactions between E-cadherin, β-catenin and PTP1B in Caco-2 cell monolayers (Sheth et al., 2007). In this study, investigators demonstrated that acetaldehyde induced disruption of interactions between E-cadherin, β-catenin and PTP1B by a phosphorylation-dependent mechanism, suggesting that this may be a mechanism whereby acetaldehyde impairs intestinal permeability.
Role of Nitric Oxide in Increasing Intestinal Permeability
The role of nitric oxide (NO) in intestinal barrier dysfunction was recognized when researchers observed increased expression of inducible nitric oxide synthase (iNOS) in concert with intestinal barrier dysfunction in inflamed human colonic mucosa (Singer et al., 1996; Kimura et al., 1998). In human Caco-2 monolayers, ethanol (2.5 to 15%) significantly increased expression of iNOS that was associated with increased nitration of tubulin and disruption of barrier function as measured by apical-to-basolateral flux of fluroscent marker (Banan et al., 1999). The role of nitric oxide in mediating the effect of ethanol on intestinal barrier function was further confirmed in another Coca-2 monolayer study (Banan et al., 2000). In this study, ethanol exposure (1%, 2.5%, and 15%) increased expression of iNOS and increased production of NO and superoxide. These findings were associated with nitration and oxidation of tubulin, decreased levels of stable polymerized tubulin, increased levels of disassembled tubulin, and damage to microtubule cytoskeleton. In addition, ethanol significantly disrupted epithelial barrier function as determined by measuring apical-to-basolateral flux of a fluorescent marker. The effects of ethanol were mimicked by peroxynitrite (a product of the reaction of NO with superoxide anions). Pretreatment with iNOS inhibitor (L-N6-1-iminoethyl-lysine), peroxynitrite scavengers (urate or L-cysteine) or superoxide anion scavenger (superoxide dismutase) attenuated the damage caused by ethanol. These results suggest that NO and superoxide anion generated in response to ethanol exposure may react with each other to form peroxynitrite which in turn may cause damage to microtubule cytoskeleton and subsequently disrupt intestinal barrier function. It was further demonstrated that NF-kappaB is required for oxidant-induced iNOS upregulation and for the consequent nitration and oxidation of cytoskeleton (Banan et al., 2004). These researchers further showed that epidermal growth factor (EGF) may protect intestinal barrier function by stabilizing cytoskeleton via down regulating the activity of iNOS (Banan et al., 2003).
Combined Effect of Alcohol and Burn Injury on Intestinal Bacterial Growth and Barrier Function
Researchers have investigated the combined effect of alcohol and burn injury on intestinal bacterial growth and barrier function. The reason for this two-hit insult is that alcohol intoxication is recognized as a major problem in post-burn injury pathogenesis. According to an estimate nearly one million people are affected with burn injuries every year within the United States and nearly half of these injuries occur under the influence of ethanol intoxication (Messingham et al., 2002; Choudhry and Chaudry, 2006). The effects of acute ethanol intoxication combined with burn injury were determined on intestinal bacterial growth, intestinal permeability, intestinal immune defense, and bacterial translocation (Choudhry et al., 2002; Kavanaugh et al., 2005; Li et al., 2006). Ethanol was administered to rats by gavage (5 ml of 20% ethanol) four hours prior to burn injury. An analysis of various parameters on days one and two after injury revealed that the combined insult of ethanol and burn injury significantly increased intestinal permeability and the number of bacteria in mesenteric lymph nodes. Furthermore, the combined insult caused a significant decrease in intestinal T cell functions and their numbers in intestinal mucosa (Choudhry et al., 2002; Kavanaugh et al., 2005). Additionally, the finding of Kavanaugh et al. (2005) showed that the combined insult significantly increases bacterial growth in the small intestine on day two after injury. The ethanol alone group exhibited an increase in intestinal permeability on day one and not on day two (Choudhry et al., 2002; Kavanaugh et al., 2005; Li et al., 2006). No change in other parameters was noted in the ethanol alone group. In contrast, as compared to sham, rats receiving burn injury alone did not exhibit a significant difference in intestinal permeability and intestinal T cell function on day one after injury. However, on day two, the burn alone group exhibited a tendency of an increase in bacterial growth (though not significant), intestinal permeability and number of bacteria in the mesenteric lymph nodes, and a decrease in intestinal T cell functions. Nonetheless, the changes in these above parameters were more severe in the group of rats that has undergone a combined insult of alcohol and burn injury (Choudhry et al., 2002; Kavanaugh et al., 2005). These results suggest that acute alcohol intoxication potentiated the effect of burn injury on all of the parameters listed above including intestinal bacterial growth and permeability.
Preservation of Intestinal Epithelial Barrier Function
Preservation of intestinal epithelial barrier function may prevent the diffusion of colonic luminal endotoxin and other toxic products into portal vein, which in turn may attenuate endotoxin-induced organ/tissue injury. Several researchers have investigated the role of the following factors in the preservation of intestinal epithelial barrier function disrupted by alcohol.
A. Role of epidermal growth factor
Epidermal growth factor (EGF) is known to promote growth and differentiation of gastrointestinal mucosa and provide protection against injurious agents. The role of EGF in the protection of epithelial barrier function from acetaldehyde was evaluated in Caco-2 intestinal epithelial cell monolayer (Sheth et al., 2004). EGF administration significantly reduced acetaldehyde-induced increases in paracellular permeability to inulin and LPS in a time- and dose-dependent manner. EGF prevented acetaldehyde-induced reorganization of occludin, zonula occludens-1 (ZO-1), E-cadherin, and β-catenin from the cellular junctions to the intracellular compartments. In addition, EGF prevented acetaldehyde-induced reorganization of actin cytoskeleton and the interaction of occludin, ZO-1, E-cadherin, and β-catenin with the actin cytoskeleton. These results indicate that EGF can preserve intestinal paracellular permeability by attenuating acetaldehyde-induced disruption of tight junctions and adherens junctions and by preventing acetaldehyde-induced redistribution of actin cytoskeleton and its interaction with occludin, ZO-1, E-cadherin, and β-catenin. Further investigation revealed that EGF protects the tight junction barrier function from acetaldehyde by PLCγ1, calcium, PKCβI, and PKCε-dependent mechanisms (Suzuki et al., 2007). EGF-mediated prevention of acetaldehyde-induced increase permeability was attenuated by knock down of PLCγ1 by shRNA. EGF induces membrane translocation of PKCε and PKCβI by PLCγ1-dependent mechanism, and this translocation of PKCε and PKCβI was required for prevention of acetaldehyde-induced increase in permeability. A similar protective role of EGF on acetaldehyde-induced disruption of tight junctions and adherens junctions was also demonstrated in human colonic mucosa (Basuroy et al., 2005).
B. Role of L-Glutamine
Glutamine, a non-essential amino acid, has been shown to improve intestinal barrier function in experimental biliary obstruction (White et al., 2005) and in patients with systemic infections (De-Souza and Greene, 2005). Role of L-glutamine in the protection of intestinal epithelium from acetaldehyde-induced disruption of barrier function was evaluated in Caco-2 cell monolayer (Seth et al., 2004). L-Glutamine reduced the acetaldehyde-induced decrease in transepithelilal electrical resistance and increase in permeability to inulin and lipopolysaccharide in a time- and dose-dependent manner. L-glutamine reduced the acetaldehyde-induced redistribution of occludin, ZO-1, E-cadherin, and β-catenin from the intercellular junctions to the intracellular compartments. L-glutamine reduced acetaldehyde-induced dissociation of occludin, ZO-1, E-cadherin, and β-catenin from the actin cytoskeleton. These researchers further demonstrated that L-glutamine protects the intestinal barrier function by an EGF receptor-dependent mechanism. A similar protective role of L-glutamine on acetaldehyde-induced disruption of tight junctions and adherens junctions was also demonstrated in human colonic mucosa (Basuroy et al., 2005).
C. Role of oats supplementation
Chronic administration of ethanol (8 g/kg/day) to rats for 10 weeks by gavage induced gut leakiness as assessed by urinary excretion of lactulose and mannitol, which was associated with increased levels of blood endotoxin as well as liver injury. Oat supplementation ameliorated all of these changes (Keshavarzian et al., 2001). Studies are required to understand the mechanisms by which oat supplement preserves intestinal permeability.
D. Role of zinc
Zinc is an essential trace element involved in many physiological functions. Zinc deficiency has been reported in patients with inflammatory bowl disease (Solomons et al., 1977; Hendricks and Walker, 1988), which is associated with increased intestinal permeability. On the other hand, zinc supplementation has been found to preserve intestinal permeability in patients with Crohn’s disease (Sturniolo et al., 2001) and in rats with experimental colitis (Sturniolo et al., 2002). Ethanol appears to impair the absorption of zinc from the intestine. In rats, chronic ethanol ingestion significantly impaired zinc absorption in the ileum, which is the most active area of zinc uptake in the intestine (Antonson and Vanderhoof, 1983). Similarly in mice, acute alcohol treatment decreased zinc concentration in the ileum (Lambert et al., 2004).
Using a mouse model of acute alcohol toxicity mimicking human binge drinking (Carson and Pruett, 1996), researchers investigated whether zinc supplementation could preserve intestinal permeability that is increased by alcohol consumption. Preserving intestinal permeability may block the transfer of endotoxin to the circulation and prevent alcoholic liver injury. Mice were treated with three intragastric doses of zinc sulfate at 5 mg zinc element/kg each dosing, with an interval of 12 hours, prior to acute ethanol challenge with a single oral dose of 6 g/kg ethanol. Alcohol caused severe damage to the small intestine as determined by morphological and permeability changes. Serum endotoxin levels were significantly higher in alcohol-treated mice compared with control animals. Mice treated with ethanol and subsequently loaded with exogenous LPS showed a further increase in serum endotoxin levels compared with mice exposed to ethanol alone. These findings demonstrate that ethanol increased the transfer of endogenous as well as exogenous endotoxin from the intestine to the general circulation. Increased serum endotoxin levels were associated with liver injury as detected by an elevation in serum alanine aminotransferase (ALT) activity, fatty liver and hepatic degenerative necrotic foci along with a significant increase in hepatic TNF-α levels (Lambert et al., 2003; Lambert et al., 2004). Zinc supplementation attenuated ethanol-induced increases in serum endotoxin levels, serum ALT activity, and hepatic TNF-α levels. These changes were associated with prevention of ethanol-induced liver injury (Lambert et al., 2003; Lambert et al., 2004). These results suggest that zinc brought these changes, at least in part, by preventing ethanol-induced transfer of endotoxin from intestine to circulation which could be ascribed to preservation of intestinal morphology and permeability. The mechanism by which zinc preserves intestinal morphology and permeability is a subject of investigation.
Alcohol and Plasma Endotoxin Levels
Alcohol-induced increases in intestinal bacterial growth and intestinal permeability to endotoxin is expected to result in elevated plasma endotoxin levels. An association between alcohol consumption and plasma endotoxin levels has been investigated in humans as well as in animals. Plasma endotoxin levels were significantly elevated in patients affected with different stages of ALD - fatty liver, hepatitis, and cirrhosis when compared with healthy control subjects (Fukui et al., 1991; Parlesak et al., 2000; Schafer et al., 2002). Amount of alcohol appeared to make significant impact on endotoxin levels and liver injury since in these studies healthy control subjects consumed less than 20 g/d of alcohol compared to more than 60 g/d of alcohol by ALD patients. Furthermore, plasma endotoxin levels were also significantly elevated in actively drinking alcoholics without evidence of liver disease (Bode et al., 1987; Nolan, 1989), suggesting that alcohol itself can increase endotoxin levels presumably via increasing intestinal permeability to endotoxin. Endotoxemia was reversible in the majority of patients with alcoholic fatty liver and in about 50% of patients with mild alcoholic hepatitis within 1 week following the cessation of alcohol intake (Fukui et al., 1991), further suggesting a direct role of alcohol in increasing plasma endotoxin levels. Liver disease itself may increase plasma endotoxin levels since they were significantly elevated in non-alcoholic cirrhotic patients (Bode et al., 1987; Fukui et al., 1991). However, plasma endotoxin levels were significantly higher in patients with alcoholic cirrhosis than in patients with non-alcoholic cirrhosis (Bode et al., 1987; Fukui et al., 1991), thus implicating alcohol in increasing endotoxin levels.
The relationship between alcohol consumption and plasma endotoxin levels has been shown in animals with alcoholic liver injury. Serum endotoxin levels were elevated in rats with alcoholic liver injury developed after ethanol administration for 10 weeks by gavage (Keshavarzian et al., 2001). Similarly, plasma endotoxin levels were significantly elevated in rats with alcoholic liver injury developed in response to intragastric infusion of ethanol for 3-4 weeks (Nanji et al., 1994; Adachi et al., 1995). In mice, plasma endotoxin levels were significantly elevated 1.5 hour after intragastric administration of 6g/kg ethanol by gavage, and this was associated with significant small intestinal injury (Lambert et al., 2003). These mice developed significant liver injury 6 hours after ethanol administration, suggesting that alcohol is the causal factor for endotoxemia. Both human and animal studies suggest that alcohol mediates the transfer of endotoxin from intestine to the liver that results in the elevation of plasma enotoxin levels.
Role of Endotoxin in Alcoholic Liver Injury
Increasing evidence suggests that gut-derived endotoxin plays a central role in the initiation and perhaps progression of alcoholic liver injury. The evidence for the role of endotoxin in alcoholic liver injury was primarily obtained from animal studies. In rats exposed to chronic ethanol in a liquid diet, administration of endotoxin led to the progression of liver injury from fatty liver to necro-inflammatory changes (Bhagwandeen et al., 1987; Pennington et al., 1997; Apte et al., 2005). In an intragstric infusion rat model of alcoholic liver injury, sterilization of intestine by antibiotics significantly attenuated liver injury by reducing plasma levels of endotoxin (Adachi et al., 1995), and in the same model, similar results were obtained by simultaneous feeding of probiotic lactobacillus GG bacteria (competitive inhibitor of Gram negative bacteria) to the rats (Nanji et al., 1994).
Endotoxin initiates liver injury via activation of hepatic Kupffer cells because inactivation of Kupffer cells with gadolinium chloride significantly attenuated liver injury caused by chronic ethanol exposure (Adachi et al., 1994). Endotoxin after binding with LPS-binding protein activates Kupffer cells through two types of receptors, CD-14 and Toll-like receptors-4 (TLR-4). CD-14 is a surface receptor without a cytoplasmic domain and thus it lacks the ability to transduce LPS-induced cytoplasmic signal across a cell membrane. On the other hand, TLR-4 is a transmembrane protein with a cytoplasmic domain which is capable of transducing LPS-induced cytoplasmic signal across a cell membrane. The finding that alcoholic liver injury is blocked in both CD-14 (Yin et al., 2000) and TLR-4 deficient mice (Uesugi et al., 2001) suggests that both of these receptors are required to initiate liver injury caused by alcohol. Binding of LPS to these receptors on Kupffer cells initiates a cascade of events leading to free radical generation, nuclear factor kappaB (NF-kB) activation, and production of inflammatory mediators such as cytokines (e.g., TNF-α), chemokines, and adhesion molecules (Thurman, 1998; Wheeler et al., 2001; Park et al., 2006; Thakur et al., 2006; Nagata et al., 2007). These mediators eventually initiate necro-inflammatory and fibrotic changes in the liver.
Researchers have discovered that in addition to activating Kupffer cells, LPS may also activate hepatic stellate cells (HSCs) which are the main fibrogenic cell type in the injured liver. Culture-activated human HSCs expressed LPS receptors CD-14 and TLR-4, and stimulation of these cells with LPS induced NF-kB activation and up-regulated gene expression of chemokines - interleukin-8 (IL-8) and monocyte chemoattractant protein-1 (MCP-1) - and adhesion molecules (intercellular adhesion molecules and vascular cellular adhesion molecules -1) (Paik et al., 2003). Activated mouse HSCs expressed TLR-4 and CD-14 and responded to LPS with an up regulation of extracellular signal-regulated kinase phosphorylation and IL-6, transforming growth factor-β1, and MCP-1 secretion (Brun et al., 2005). In quiescent rat hepatic stellate cells, LPS stimulated the synthesis of proinflammatory cytokines TNF-α, IL-6, and IL-1 (Thirunavukkarasu et al., 2005). These results suggest that LPS may directly activate HSCs, thereby contributing to liver fibrosis. Endotoxin may also contribute to the development of other alcohol-associated tissue/organ injuries such as pancreatitis (Wang et al., 2005), ARDS (Ryffel et al., 2005), and brain injury (Duncan et al., 2005).
Peptidoglycan: intestinal permeability and liver toxicity
In addition to endotoxin, intestinal bacteria may release many other TLR ligands that activate innate immune cells such as Kupffer cells. Peptidoglycan and flagellin, which bind to TLR-2 and TLR-5, respectively, have been implicated in the pathogenesis of inflammation of the liver and intestine. Peptidoglycan is present in all types of bacteria residing in the intestinal tract. The Gram positive bacteria have a very thick peptidoglycan layer whereas the Gram negative bacteria have a relatively thin layer. Limited information is available on the role of alcohol in intestinal permeability to peptidoglycan. In a rat study, plasma peptidoglycan concentration of portal blood was significantly increased 24 hour after the acute administration of 10 ml/kg of 20% ethanol (Tabata et al., 2002), suggesting a role of alcohol in increasing intestinal permeability to peptidoglycan and possible pathological consequences. In another study, injection of peptidoglycan increased TNF-α mRNA expression more in the livers of ethanol-fed mice than in control mice. In addition, peptidoglycan induced liver inflammatory infiltrate in ethanol-fed mice but not in control mice (Gustot et al., 2006). These studies suggest that alcohol may not only increase intestinal permeability to peptidoglycan, but also works synergistically with it to induce liver injury. Experimental jejunal bacterial overgrowth in rats leads to increased portal blood concentrations of peptidoglycan that induces hepatic inflammation and steatosis (Lichtman et al., 1992). Peptidoglycan activates TNF-α production by Kupffer cells. Further studies are required to confirm the role of alcohol in increasing intestinal permeability to peptidoglycan and its medical consequences.
Conclusions
The mechanisms whereby alcohol exposure leads to increased intestinal permeability to endotoxin and subsequent injury to the liver and other organs have been presented in Figure 1. Alcohol exposure can promote the growth of Gram negative bacteria in the intestine which may result in accumulation of endotoxin. In addition, alcohol metabolism by Gram negative bacteria and intestinal epithelial cells can result in accumulation of acetaldehyde, which in turn can increase intestinal permeability to endotoxin. Alcohol-induced generation of nitric oxide may also contribute to increased permeability to endotoxin. Increased intestinal permeability to endotoxin leads to increased transfer of endotoxin from the intestine to the portal vein which carries endotoxin to the liver where it binds to Kupffer cells and initiates a cascade of events leading to TNF-α production and liver injury. Endotoxin that escapes to general circulation may induce injury to other organs. A part of TNF- α produced in the liver may reach to intestine via bile duct or general circulation and further increase intestinal permeability to endotoxin.
Figure 1. Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and organ injury: a summary.

Alcohol exposure can promote the growth of Gram negative bacteria in the intestine which may result in accumulation of endotoxin. In addition, alcohol metabolism by Gram negative bacteria and intestinal epithelial cells can result in accumulation of acetaldehyde, which in turn can increase intestinal permeability to endotoxin. Alcohol-induced generation of nitric oxide may also contribute to increased permeability to endotoxin. Increased intestinal permeability to endotoxin leads to increased transfer of endotoxin from the intestine to the portal vein which carries endotoxin to the liver where it binds to Kupffer cells and initiates a cascade of events leading to tumor necrosis factor-α (TNF-α) production and liver injury. Endotoxin that escapes to general circulation may induce injury to other organs. A part of TNF- α produced in the liver may reach to intestine via bile duct or general circulation and further increase intestinal permeability to endotoxin.
Acetaldehyde may increase intestinal permeability to endotoxin by a following mechanism comprised of multiple steps (Figure 2). Acetaldehyde has been shown to decrease the activity of protein tyrosine phosphatase in the intestinal epithelial paracellular space. This results in the increased tyrosine phosphorylation of tight junction proteins (occludin and ZO-1) and adherens junction proteins (E-cadherin and β-catenin). Increased phosphorylation of these proteins leads to their redistribution from intercellular junctions to intracellular compartments and that probably results in increased intestinal permeability to endotoxin.
Figure 2. Role of acetaldehyde in increasing intestinal permeability to endotoxin: a proposed mechanism.

Acetaldehyde may increase intestinal permeability to endotoxin by decreasing the activity of protein tyrosine phosphatase in the intestinal epithelial paracellular space. This results in the increased tyrosine phosphorylation of tight junction proteins (occludin and ZO-1) and adherens junction proteins (E-cadherin and β-catenin). Increased phosphorylation of these proteins leads to their redistribution from intercellular junctions to intracellular compartments and that probably results in increased intestinal permeability to endotoxin.
Alcohol may also increase intestinal permeability by increasing the production of nitric oxide, via up-regulating iNOS activity, and superoxide (Figure 3). These radicals can react with each other to form peroxynitrite which in turn can react with tubulin leading to damage to microtubule cytoskeleton, disruption of barrier function, and increased intestinal permeability.
Figure 3. Role of nitric oxide in increasing intestinal permeability to endotoxin: a proposed mechanism.

Alcohol may also increase intestinal permeability by increasing the production of nitric oxide, via up-regulating inducible nitric oxide synthase (iNOS) activity, and superoxide. These radicals can react with each other to form peroxynitrite which in turn can react with tubulin leading to damage to microtubule cytoskeleton, disruption of barrier function, and increased intestinal permeability.
In intestinal inflammatory diseases, TNF-α may play an important role in impairing intestinal barrier function by upregulating the activity of MLCK and associated increased phosphorylation of MLC (Figure 4).
Figure 4. Role of TNF-α in intestinal barrier function.
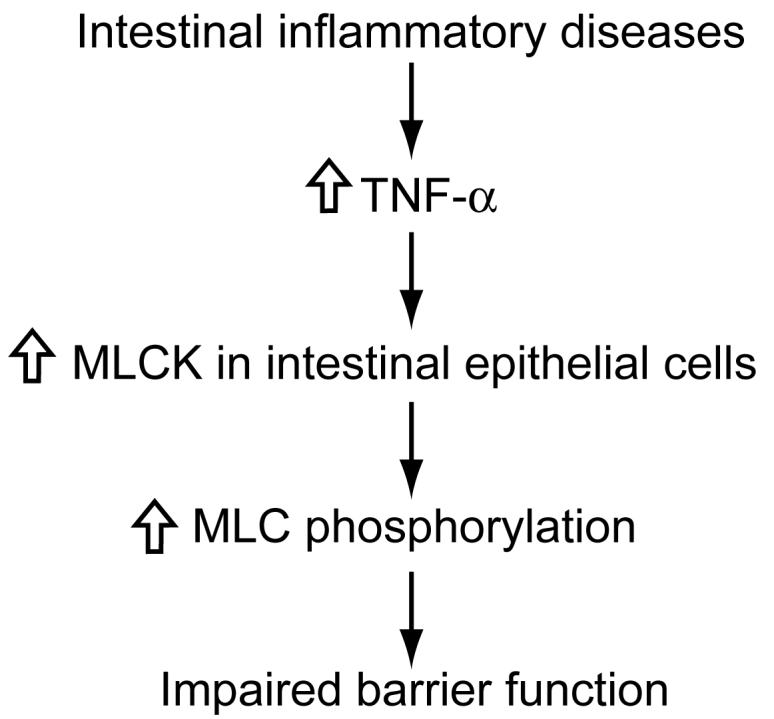
In intestinal inflammatory diseases, TNF-α may play an important role in impairing intestinal barrier function by upregulating the activity of myosin light-chain kinase (MLCK) and associated increased phosphorylation of myosin light-chain (MLC).
Decreasing plasma endotoxin levels may attenuate alcoholic liver injury as well as alcohol-associated injury of other organs. This can be accomplished by reducing the number of Gram negative bacteria in the intestine or preserving intestinal permeability to endotoxin (Figure 5). The number of Gram negative bacteria can be reduced by feeding of probiotics such as Lactobacillus GG or Bifidobacteria. This can result in decreased production of endotoxin as well as acetaldehyde which is expected to decrease intestinal permeability to endotoxin. In addition, intestinal permeability may be preserved by administering EGF, L-glutamine, oats supplementation, or zinc which is expected to prevent the transfer of endotoxin to the general circulation.
Figure 5. Strategies for attenuating plasma endotoxin levels in alcoholic liver diseases.

Plasma endotoxin levels may be attenuated by reducing the number of Gram negative bacteria in the intestine or preserving intestinal permeability to endotoxin. The number of Gram negative bacteria can be reduced by feeding probiotics such as Lactobacillus GG or Bifidobacteria. This can result in decreased production of endotoxin as well as acetaldehyde which is expected to decrease intestinal permeability to endotoxin. In addition, intestinal permeability may be preserved by administering epidermal growth factor (EGF), L-glutamine, oat supplementation, or zinc which is expected to prevent the transfer of endotoxin to the general circulation.
Alcohol may also increase intestinal permeability to peptidoglycan, which can initiate inflammatory response in liver and other organs.
Acute alcohol exposure may potentiate the effect of burn injury on intestinal bacterial growth and permeability.
Future Directions
Investigating mechanisms whereby alcohol promotes intestinal bacterial growth. Is it dose-dependent? Is bacterial growth associated with increased amount of endotoxin production? What types of bacteria grow in the presence of alcohol? Does alcohol promote bacterial growth directly or indirectly through suppression of immune system?
Developing effective and safe strategies to prevent production of endotoxin in the intestine. This may include direct killing of Gram negative bacteria or suppressing their growth by probiotics or prebiotics.
Developing strategies for accelerating acetaldehyde metabolism in the intestine. This may include down-regulating the activity of ADH or up-regulating the activity of ALDH through various mechanisms.
Further discern the underlying mechanisms by which acetaldehyde makes intestinal epithelium more permeable to endotoxin. For example, is it an effect of acetaldehyde itself or an effect of acetaldehyde adducts such as acetaldehyde-protein adducts or acetaldehyde-malondialdehyde-protein adducts?
Understand the relative and interactive role of acetaldehyde, nitric oxide, and TNF-α in impairing intestinal barrier function.
Developing safe and effective agents that will preserve the integrity of intestine and thus prevent the transfer of endotoxin to portal vein.
Understanding the underlying mechanisms by which zinc, oat supplementation, and L-glutamine preserve intestinal permeability to endotoxin.
Examining the ability of alcohol to increase intestinal permeability to other toxic bacterial products such as peptidoglycan and flagellin.
Acknowledgements
Studies reported in this manuscript were supported in part by the National Institutes of Health (R21 AA015979, R01 AA015731, R01 HL059225, R01 H063760, R01 AA013745, R01 AA012307, and R01 DK055532).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adachi Y, Bradford BU, Gao W, Bojes HK, Thurman RG. Inactivation of Kupffer cells prevents early alcohol-induced liver injury. Hepatology. 1994;20:453–460. [PubMed] [Google Scholar]
- Adachi Y, Moore LE, Bradford BU, Gao W, Thurman RG. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology. 1995;108:218–224. doi: 10.1016/0016-5085(95)90027-6. [DOI] [PubMed] [Google Scholar]
- Antonson DL, Vanderhoof JA. Effect of chronic ethanol ingestion on zinc absorption in rat small intestine. Dig. Dis. Sci. 1983;28:604–608. doi: 10.1007/BF01299920. [DOI] [PubMed] [Google Scholar]
- Apte UM, Banerjee A, McRee R, Wellberg E, Ramaiah SK. Role of osteopontin in hepatic neutrophil infiltration during alcoholic steatohepatitis. Toxicol. Appl. Pharmacol. 2005;207:25–38. doi: 10.1016/j.taap.2004.12.018. [DOI] [PubMed] [Google Scholar]
- Atkinson KJ, Rao RK. Role of protein tyrosine phosphorylation in acetaldehyde-induced disruption of epithelial tight junctions. Am. J. Physiol. Gastrointest. Liver Physiol. 2001;280:G1280–G1288. doi: 10.1152/ajpgi.2001.280.6.G1280. [DOI] [PubMed] [Google Scholar]
- Banan A, Choudhary S, Zhang Y, Fields JZ, Keshavarzian A. Ethanol-induced barrier dysfunction and its prevention by growth factors in human intestinal monolayers: evidence for oxidative and cytoskeletal mechanisms. J. Pharmacol. Exp. Ther. 1999;291:1075–1085. [PubMed] [Google Scholar]
- Banan A, Fields JZ, Decker H, Zhang Y, Keshavarzian A. Nitric oxide and its metabolites mediate ethanol-induced microtubule disruption and intestinal barrier dysfunction. J. Pharmacol. Exp. Ther. 2000;294:997–1008. [PubMed] [Google Scholar]
- Banan A, Zhang LJ, Shaikh M, Fields JZ, Farhadi A, Keshavarzian A. Key role of PLC-gamma in EGF protection of epithelial barrier against iNOS upregulation and F-actin nitration and disassembly. Am. J. Physiol. Cell Physiol. 2003;285:C977–993. doi: 10.1152/ajpcell.00121.2003. [DOI] [PubMed] [Google Scholar]
- Banan A, Zhang LJ, Shaikh M, Fields JZ, Farhadi A, Keshavarzian A. Novel effect of NF-kappaB activation: carbonylation and nitration injury to cytoskeleton and disruption of monolayer barrier in intestinal epithelium. Am. J. Physiol. Cell Physiol. 2004;287:C1139–C1151. doi: 10.1152/ajpcell.00146.2004. [DOI] [PubMed] [Google Scholar]
- Basuroy S, Sheth P, Mansbach CM, Rao RK. Acetaldehyde disrupts tight junctions and adherens junctions in human colonic mucosa: protection by EGF and L-glutamine. Am. J. Physiol. Gastrointest. Liver Physiol. 2005;289:G367–G375. doi: 10.1152/ajpgi.00464.2004. [DOI] [PubMed] [Google Scholar]
- Bentzel CJ, Hainau B, Ho S, Hui SW, Edelman A, Anagnostopoulos T, Benedetti EL. Cytoplasmic regulation of tight-junction permeability: effect of plant cytokinins. Am. J. Physiol. 1980;239:C75–C89. doi: 10.1152/ajpcell.1980.239.3.C75. [DOI] [PubMed] [Google Scholar]
- Bhagwandeen BS, Apte M, Manwarring L, Dickeson J. Endotoxin induced hepatic necrosis in rats on an alcohol diet. J. Pathol. 1987;152:47–53. doi: 10.1002/path.1711520107. [DOI] [PubMed] [Google Scholar]
- Bjarnason I, Peters TJ, Wise RJ. The leaky gut of alcoholism: possible route of entry for toxic compounds. Lancet. 1984;1(8370):179–182. doi: 10.1016/s0140-6736(84)92109-3. [DOI] [PubMed] [Google Scholar]
- Blair SA, Kane SV, Clayburgh DR, Turner JR. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab. Invest. 2006;86:191–201. doi: 10.1038/labinvest.3700373. [DOI] [PubMed] [Google Scholar]
- Bode JC, Bode C, Heidelbach R, Dürr HK, Martini GA. Jejunal microflora in patients with chronic alcohol abuse. Hepatogastroenterology. 1984;31:30–34. [PubMed] [Google Scholar]
- Bode C, Kolepke R, Schäfer K, Bode JC. Breath hydrogen excretion in patients with alcoholic liver disease--evidence of small intestinal bacterial overgrowth. Z. Gastroenterol. 1993;31:3–7. [PubMed] [Google Scholar]
- Bode C, Kugler V, Bode JC. Endotoxemia in patients with alcoholic and non-alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. J. Hepatol. 1987;4:8–14. doi: 10.1016/s0168-8278(87)80003-x. [DOI] [PubMed] [Google Scholar]
- Bode JC, Parlesak A, Bode C. Gut-derived bacterial toxins (endotoxin) and alcoholic liver disease. In: Agarwal DP, Seitz HK, editors. Alcohol in Health and Disease. M Dekker; New York-Basel: 2001. pp. 369–386. [Google Scholar]
- Brun P, Castagliuolo I, Pinzani M, Palú G, Martines D. Exposure to bacterial cell wall products triggers an inflammatory phenotype in hepatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2005;289:G571–578. doi: 10.1152/ajpgi.00537.2004. [DOI] [PubMed] [Google Scholar]
- Bungert HJ. Absorption of hemoglobin and hemoglobin iron in alcohol-induced liver injury. Digestion. 1973;9:293–308. doi: 10.1159/000197455. [DOI] [PubMed] [Google Scholar]
- Carson EJ, Pruett SB. Development and characterization of a binge drinking model in mice for evaluation of the immunological effects of ethanol. Alcohol. Clin. Exp. Res. 1996;20:132–138. doi: 10.1111/j.1530-0277.1996.tb01055.x. [DOI] [PubMed] [Google Scholar]
- Choudhry MA, Chaudry IH. Alcohol intoxication and post-burn complications. Front. Biosci. 2006;11:998–1005. doi: 10.2741/1857. [DOI] [PubMed] [Google Scholar]
- Choudhry MA, Fazal N, Goto M, Gamelli RL, Sayeed MM. Gut-associated lymphoid T cell suppression enhances bacterial translocation following alcohol and burn injury. Am. J. Physiol. 2002;282:G937–G947. doi: 10.1152/ajpgi.00235.2001. [DOI] [PubMed] [Google Scholar]
- Clayburgh DR, Shen L, Turner JR. A porous defense: the leaky epithelial barrier in intestinal disease. Lab. Invest. 2004;84:282–291. doi: 10.1038/labinvest.3700050. [DOI] [PubMed] [Google Scholar]
- De-Souza DA, Greene LJ. Intestinal permeability and systemic infections in critically ill patients: effect of glutamine. Crit. Care Med. 2005;33:1125–1135. doi: 10.1097/01.ccm.0000162680.52397.97. [DOI] [PubMed] [Google Scholar]
- Duncan JR, Cock ML, Suzuki K, Scheerlinck JP, Harding R, Rees SM. Chronic endotoxin exposure causes brain injury in the ovine fetus in the absence of hypoxemia. J. Soc. Gynecol. Investig. 2006;13:87–96. doi: 10.1016/j.jsgi.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Ferrier L, Berard F, Debrauner L, Chabo C, Langella P, Bueno L, Fioramonti J. Impairment of intestinal barrier by ethanol involves enteric microflora and mast cell activation in rodents. Am. J. Pathol. 2006;168:1148–1154. doi: 10.2353/ajpath.2006.050617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui H, Brauner B, Bode JC, Bode C. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: reevaluation with an improved chromogenic assay. J. Hepatol. 1991;12:162–169. doi: 10.1016/0168-8278(91)90933-3. [DOI] [PubMed] [Google Scholar]
- Gustot T, Lemmers A, Moreno C, Nagy N, Quertinmont E, Nicaise C, Franchimont D, Louis H, Deviére J, Le Moine O. Differential liver sensitization to toll-like receptor pathways in mice with alcoholic fatty liver. Hepatology. 2006;43:989–1000. doi: 10.1002/hep.21138. [DOI] [PubMed] [Google Scholar]
- Hauge T, Persson J, Danielsson D. Mucosal bacterial growth in the upper gastrointestinal tract in alcoholics (heavy drinkers) Digestion. 1997;58:591–595. doi: 10.1159/000201507. [DOI] [PubMed] [Google Scholar]
- Hendricks KM, Walker WA. Zinc deficiency in inflammatory bowel disease. Nutr. Rev. 1988;46:401–408. doi: 10.1111/j.1753-4887.1988.tb05381.x. [DOI] [PubMed] [Google Scholar]
- Hollander D. Crohn’s disease--a permeability disorder of the tight junction? Gut. 1988;29:1621–1624. doi: 10.1136/gut.29.12.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull BE, Staehelin LA. The terminal web. A reevaluation of its structure and function. J. Cell Biol. 1979;81:67–82. doi: 10.1083/jcb.81.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanaugh MJ, Clark C, Goto M, Kovacs EJ, Gamelli RL, Sayeed MM, Choudhry MA. Effect of acute alcohol ingestion prior to burn injury on intestinal bacterial growth and barrier function. Burns. 2005;31:290–296. doi: 10.1016/j.burns.2004.09.021. [DOI] [PubMed] [Google Scholar]
- Keshavarzian A, Choudhary S, Holmes EW, Yong S, Banan A, Jakate S, Fields JZ. Preventing gut leakiness by oats supplementation ameliorates alcohol-induced liver damage in rats. J. Pharmacol. Exp. Ther. 2001;299:442–448. [PubMed] [Google Scholar]
- Kimura H, Hokari R, Miura S, Shigematsu T, Hirokawa M, Akiba Y, Kurose I, Higuchi H, Fujimori H, Tsuzuki, et al. Increased expression of an inducible isoform of nitric oxide synthase and the formation of peroxynitrite in colonic mucosa of patients with active ulcerative colitis. Gut. 1998;42:180–187. doi: 10.1136/gut.42.2.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Zhou Z, Wang L, Song Z, McClain CJ, Kang YJ. Prevention of alterations in intestinal permeability is involved in zinc inhibition of acute ethanol-induced liver damage in mice. J. Pharmacol. Exp. Ther. 2003;305:880–886. doi: 10.1124/jpet.102.047852. [DOI] [PubMed] [Google Scholar]
- Lambert JC, Zhou Z, Wang L, Song Z, McClain CJ, Kang YJ. Preservation of intestinal structural integrity by zinc is independent of metallothionein in alcohol-intoxicated mice. Am. J. Pathol. 2004;164:1959–1966. doi: 10.1016/S0002-9440(10)63756-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Rana SN, Schwacha MG, Chaudry IH, Choudhry MA. A novel role for IL-18 in corticosterone-mediated intestinal damage in a two hit rodent model of alcohol intoxication and injury. J. Leukoc. Biol. 2006;80:364–375. doi: 10.1189/jlb.1205745. [DOI] [PubMed] [Google Scholar]
- Lichtman SN, Okoruwa EE, Keku J, Schwab JH, Sartor RB. Degradation of endogenous bacterial cell wall polymers by the muralytic enzyme mutanolysin prevents hepatobiliary injury in genetically susceptible rats with experimental intestinal bacterial overgrowth. J. Clin. Invest. 1992;90:1313–1322. doi: 10.1172/JCI115996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma TY, Boivin MA, Ye D, Pedram A, Said HM. Mechanism of TNF-{alpha} modulation of Caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. Am.J. Physiol. Gastrointest. Liver Physiol. 2005;288:G422–G430. doi: 10.1152/ajpgi.00412.2004. [DOI] [PubMed] [Google Scholar]
- Ma TY, Nguyen D, Bui V, Nguyen H, Hoa N. Ethanol modulation of intestinal epithelial tight junction barrier. Am. J. Physiol. Gastrointest. Liver Physiol. 1999;276:G965–G974. doi: 10.1152/ajpgi.1999.276.4.G965. [DOI] [PubMed] [Google Scholar]
- Madara JL. Intestinal absorptive cell tight junctions are linked to cytoskeleton. Am. J. Physiol. 1987;253(1 Pt 1):C171–C175. doi: 10.1152/ajpcell.1987.253.1.C171. [DOI] [PubMed] [Google Scholar]
- Madara JL, Moore R, Carlson S. Alteration of intestinal tight junction structure and permeability by cytoskeletal contraction. Am. J. Physiol. 1987;253:C854–C861. doi: 10.1152/ajpcell.1987.253.6.C854. [DOI] [PubMed] [Google Scholar]
- Madara JL, Pappenheimer JR. Structural basis for physiological regulation of paracellular pathways in intestinal epithelia. J. Membr. Biol. 1987;100:149–164. doi: 10.1007/BF02209147. [DOI] [PubMed] [Google Scholar]
- Marano CW, Lewis SA, Garulacan LA, Soler AP, Mullin JM. Tumor necrosis factor-alpha increases sodium and chloride conductance across the tight junction of CACO-2 BBE, a human intestinal epithelial cell line. J. Membr. Biol. 1998;161:263–274. doi: 10.1007/s002329900333. [DOI] [PubMed] [Google Scholar]
- Mathurin P, Deng QG, Keshavarzian A, Choudhary S, Holmes EW, Tsukamoto H. Exacerbation of alcoholic liver injury by enteral endotoxin in rats. Hepatology. 2000;32:1008–1017. doi: 10.1053/jhep.2000.19621. [DOI] [PubMed] [Google Scholar]
- Messingham KA, Faunce DE, Kovacs EJ. Alcohol, injury, and cellular immunity. Alcohol. 2002;28:137–149. doi: 10.1016/s0741-8329(02)00278-1. [DOI] [PubMed] [Google Scholar]
- Mullin JM, Laughlin KV, Marano CW, Russo LM, Soler AP. Modulation of tumor necrosis factor-induced increase in renal (LLC-PK1) transepithelial permeability. Am. J. Physiol. 1992;263:F915–F924. doi: 10.1152/ajprenal.1992.263.5.F915. [DOI] [PubMed] [Google Scholar]
- Nagata K, Suzuki H, Sakaguchi S. Common pathogenic mechanism in development progression of liver injury caused by non-alcoholic or alcoholic steatohepatitis. J. Toxicol. Sci. 2007;32:453–468. doi: 10.2131/jts.32.453. [DOI] [PubMed] [Google Scholar]
- Nanji AA, Khettry U, Sadrzadeh SM. Lactobacillus feeding reduces endotoxemia and severity of experimental alcoholic liver (disease) Proc. Soc. Exp. Biol. Med. 1994;205:243–247. doi: 10.3181/00379727-205-43703. [DOI] [PubMed] [Google Scholar]
- Napolitano LM, Koruda MJ, Zimmerman K, McCowan K, Chang J, Meyer AA. Chronic ethanol intake and burn injury: evidence for synergistic alteration in gut and immune integrity. J. Trauma. 1995;38:198–207. doi: 10.1097/00005373-199502000-00008. [DOI] [PubMed] [Google Scholar]
- Nolan JP. Intestinal endotoxins as mediators of hepatic injury - an idea whose time has come again. Hepatology. 1989;10:887–891. doi: 10.1002/hep.1840100523. [DOI] [PubMed] [Google Scholar]
- Nosova T, Jokelainen K, Kaihovaara P, Heine R, Jousimies-Somer H, Salaspuro M. Characteristics of aldehyde dehydrogenases of certain aerobic bacteria representing human colonic flora. Alcohol Alcohol. 1998;33:273–280. doi: 10.1093/oxfordjournals.alcalc.a008391. [DOI] [PubMed] [Google Scholar]
- Paik YH, Schwabe RF, Bataller R, Russo MP, Jobin C, Brenner DA. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology. 2003;37:1043–1055. doi: 10.1053/jhep.2003.50182. [DOI] [PubMed] [Google Scholar]
- Park PH, Thakur V, Pritchard MT, McMullen MR, Nagy LE. Regulation of Kupffer cell activity during chronic ethanol exposure: role of adiponectin. J. Gastroenterol. Hepatol. 2006;21(Suppl 3):S30–33. doi: 10.1111/j.1440-1746.2006.04580.x. [DOI] [PubMed] [Google Scholar]
- Parlesak A, Schäfer C, Schütz T, Bode JC, Bode C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J.Hepatol. 2000;32:742–747. doi: 10.1016/s0168-8278(00)80242-1. [DOI] [PubMed] [Google Scholar]
- Pennington HL, Hall PM, Wilce PA, Worrall S. Ethanol feeding enhances inflammatory cytokine expression in lipopolysaccharide-induced hepatitis. J. Gastroenterol. Hepatol. 1997;12:305–313. doi: 10.1111/j.1440-1746.1997.tb00426.x. [DOI] [PubMed] [Google Scholar]
- Peterson R,E, Klopfenstein TJ, Erickson GE, Folmer J, Hinkley S, Moxley RA, Smith DR. Effect of Lactobacillus acidophilus strain NP51 on Escherichia coil O157:H7 fecal shedding and finishing performance in beef feedlot cattle. J. Food Prot. 2007;70:287–291. doi: 10.4315/0362-028x-70.2.287. [DOI] [PubMed] [Google Scholar]
- Rao RK. Acetaldehyde-induced increase in paracellular permeability in Caco-2 cell monolayer. Alcohol. Clin. Exp. Res. 1998;22:1724–1730. [PubMed] [Google Scholar]
- Rao RK, Seth A, Sheth P. Recent Advances in Alcoholic Liver Disease I. Role of intestinal permeability and endotoxemia in alcoholic liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2004;286:G881–G884. doi: 10.1152/ajpgi.00006.2004. [DOI] [PubMed] [Google Scholar]
- Robinson GM, Orrego H, Israel Y, Devenyi P, Kapur BM. Low-molecular-weight polyethylene glycol as a probe of gastrointestinal permeability after alcohol ingestion. Dig. Dis. Sci. 1981;26:971–977. doi: 10.1007/BF01314757. [DOI] [PubMed] [Google Scholar]
- Roselli M, Finamore A, Britti MS, Mengheri E. Probiotic bacteria Bifidobacterium animalis MB5 and Lactobacillus rhamnosus GG protect intestinal Caco-2 cells from the inflammation-associated response induced by enterotoxigenic Escherichia coli K88. Br J. Nutr. 2006;95:1177–1184. doi: 10.1079/bjn20051681. [DOI] [PubMed] [Google Scholar]
- Ryffel B, Couillin I, Maillet I, Schnyder B, Paesen GC, Nuttall P, Weston-Davies W. Histamine scavenging attenuates endotoxin-induced acute lung injury. Ann. N Y Acad. Sci. 2005;1056:197–205. doi: 10.1196/annals.1352.034. [DOI] [PubMed] [Google Scholar]
- Schäfer C, Parlesak A, Schütt C, Bode JC, Bode C. Concentrations of lipopolysaccharide-binding protein, bactericidal/permeability-increasing protein, soluble CD14 and plasma lipids in relation to endotoxaemia in patients with alcoholic liver disease. Alcohol Alcohol. 2002;37:81–86. doi: 10.1093/alcalc/37.1.81. [DOI] [PubMed] [Google Scholar]
- Seitz HK, Oneta CM. Gastrointestinal alcohol dehydrogenases. Nutr. Rev. 1998;56:52–60. doi: 10.1111/j.1753-4887.1998.tb01692.x. [DOI] [PubMed] [Google Scholar]
- Seth A, Basuroy S, Sheth P, Rao RK. L-Glutamine ameliorates acetaldehyde-induced increase in paracellular permeability in Caco-2 cell monolayer. Am. J. Physiol. Gastrointest. Liver Physiol. 2004;287:G510–G517. doi: 10.1152/ajpgi.00058.2004. [DOI] [PubMed] [Google Scholar]
- Shen L, Turner JR. Actin depolymerization disrupts tight junctions via caveolae-mediated endocytosis. Mol. Biol. Cell. 2005;16:3919–3936. doi: 10.1091/mbc.E04-12-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth P, Seth A, Atkinson KJ, Gheyi T, Kale G, Giorgianni F, Desiderio DM, Li C, Naren A, Rao RK. Acetaldehyde dissociates the PTP1B-E-cadherin-beta-catenin complex in Caco-2 cell monolayers by a phosphorylation-dependent mechanism. Biochem. J. 2007;402:291–300. doi: 10.1042/BJ20060665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth P, Seth A, Thangavel M, Basuroy S, Rao RK. Epidermal growth factor prevents acetaldehyde-induced paracellular permeability in Caco-2 cell monolayer. Alcohol. Clin. Exp. Res. 2004;28:797–804. doi: 10.1097/01.alc.0000125358.92335.90. [DOI] [PubMed] [Google Scholar]
- Singer II, Kawka DW, Scott S, Weidner JR, Mumford RA, Riehl TE, Stenson WF. Expression of inducible nitric oxide synthase and nitrotyrosine in colonic epithelium in inflammatory bowel disease. Gastroenterology. 1996;111:871–885. doi: 10.1016/s0016-5085(96)70055-0. [DOI] [PubMed] [Google Scholar]
- Solomons NW, Rosenberg IH, Sandstead HH, Vo-Khactu KP. Zinc deficiency in Crohn’s disease. Digestion. 1977;16:87–95. doi: 10.1159/000198059. [DOI] [PubMed] [Google Scholar]
- Sturniolo GC, Di Leo V, Ferronato A, D’Odorico A, D’Incá R. Zinc supplementation tightens “leaky gut” in Crohn’s disease. Inflamm. Bowel Dis. 2001;7:94–98. doi: 10.1097/00054725-200105000-00003. [DOI] [PubMed] [Google Scholar]
- Sturniolo GC, Fries W, Mazzon E, Di Leo V, Barollo M, D’inca R. Effect of zinc supplementation on intestinal permeability in experimental colitis. J. Lab. Clin. Med. 2002;139:311–315. doi: 10.1067/mlc.2002.123624. [DOI] [PubMed] [Google Scholar]
- Suenaert P, Bulteel V, Lemmens L, Noman M, Geypens B, Van Assche G, Geboes K, Ceuppens JL, Rutgeerts P. Anti-tumor necrosis factor treatment restores the gut barrier in Crohn’s disease. Am. J. Gastroenterol. 2002;97:2000–2004. doi: 10.1111/j.1572-0241.2002.05914.x. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Seth A, Rao RK. Role of PLCγ-induced activation of PKCε and PKCβI in EGF-mediated protection of tight junctions from acetaldehyde in Caco-2 cell monolayers. J. Biol. Chem. 2007 doi: 10.1074/jbc.M709141200. In press. [DOI] [PubMed] [Google Scholar]
- Tabata T, Tani T, Endo Y, Hanasawa K. Bacterial translocation and peptidoglycan translocation by acute ethanol administration. J. Gastroenterol. 2002;37:726–731. doi: 10.1007/s005350200118. [DOI] [PubMed] [Google Scholar]
- Tamai H, Horie Y, Kato S, Yokoyama H, Ishii H. Long-term ethanol feeding enhances susceptibility of the liver to orally administered lipopolysaccharides in rats. Alcohol. Clin. Exp. Res. 2002;26(8 Suppl):75S–80S. doi: 10.1097/01.ALC.0000026981.32386.FD. [DOI] [PubMed] [Google Scholar]
- Tamai H, Kato S, Horie Y, Ohki E, Yokoyama H, Ishii H. Effect of acute ethanol administration on the intestinal absorption of endotoxin in rats. Alcohol. Clin. Exp. Res. 2000;24:390–394. [PubMed] [Google Scholar]
- Thakur V, Pritchard MT, McMullen MR, Wang Q, Nagy LE. Chronic ethanol feeding increases activation of NADPH oxidase by lipopolysaccharide in rat Kupffer cells: role of increased reactive oxygen in LPS-stimulated ERK1/2 activation and TNF-alpha production. J. Leukoc. Biol. 2006;79:1348–1356. doi: 10.1189/jlb.1005613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirunavukkarasu C, Uemura T, Wang LF, Watkins SC, Gandhi CR. Normal rat hepatic stellate cells respond to endotoxin in LBP-independent manner to produce inhibitor(s) of DNA synthesis in hepatocytes. J. Cell Physiol. 2005;204:654–665. doi: 10.1002/jcp.20366. [DOI] [PubMed] [Google Scholar]
- Thorlacius H, Nobaek S, Wang XD, Andersson R, Molin G, Bengmark S, Jeppsson B. Lactobacilli attenuate bacteremia and endotoxemia associated with severe intra-abdominal infection. Surgery. 2003;134:467–473. doi: 10.1067/s0039-6060(03)00246-0. [DOI] [PubMed] [Google Scholar]
- Thurman RG. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am. J. Physiol. 1998;275:G605–G611. doi: 10.1152/ajpgi.1998.275.4.G605. [DOI] [PubMed] [Google Scholar]
- Tsai CC, Hsih HY, Chiu HH, Lai YY, Liu JH, Yu B, Tsen HY. Antagonistic activity against Salmonella infection in vitro and in vivo for two Lactobacillus strains from swine and poultry. Int. J. Food Microbiol. 2005;102:185–194. doi: 10.1016/j.ijfoodmicro.2004.12.014. [DOI] [PubMed] [Google Scholar]
- Turner JR, Rill BK, Carlson SL, Carnes D, Kerner R, Mrsny RJ, Madara JL. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am. J. Physiol. 1997;273:C1378–C1385. doi: 10.1152/ajpcell.1997.273.4.C1378. [DOI] [PubMed] [Google Scholar]
- Uesugi T, Froh M, Arteel GE, Bradford BU, Thurman RG. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology. 2001;34:101–108. doi: 10.1053/jhep.2001.25350. [DOI] [PubMed] [Google Scholar]
- Ukabam SO, Clamp JR, Cooper BT. Abnormal small intestinal permeability to sugars in patients with Crohn’s disease of the terminal ileum and colon. Digestion. 1983;27:70–74. doi: 10.1159/000198932. [DOI] [PubMed] [Google Scholar]
- Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J. Pathol. 2005;166:409–419. doi: 10.1016/s0002-9440(10)62264-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wu L, Wu K, Zhang R, Dong Y. Roles of endotoxin-related signaling molecules in the progression of acute necrotizing pancreatitis in mice. Pancreas. 2005;31:251–257. doi: 10.1097/01.mpa.0000175179.62916.17. [DOI] [PubMed] [Google Scholar]
- Wheeler MD, Kono H, Yin M, Nakagami M, Uesugi T, Arteel GE, Gabele E, Rusyn I, Yamashina S, Froh M, et al. The role of Kupffer cell oxidant production in early ethanol-induced liver disease. Free Radic. Biol. Med. 2001;31:1544–1549. doi: 10.1016/s0891-5849(01)00748-1. [DOI] [PubMed] [Google Scholar]
- White JS, Hoper M, Parks RW, Clements WD, Diamond T. Glutamine improves intestinal barrier function in experimental biliary obstruction. Eur. Surg. Res. 2005;37:342–347. doi: 10.1159/000090334. [DOI] [PubMed] [Google Scholar]
- Worthington BS, Meserole L, Syrotuck JA. Effect of daily ethanol ingestion on intestinal permeability to macromolecules. Am. J. Dig. Dis. 1978;23:23–32. doi: 10.1007/BF01072571. [DOI] [PubMed] [Google Scholar]
- Yin M, Bradford B,U, Wheeler M,D, Uesugi T, Froh M, Goyert SM, Thurman RG. Reduced early alcohol-induced liver injury in CD14-deficient mice. J. Immunol. 2001;166:4737–4742. doi: 10.4049/jimmunol.166.7.4737. [DOI] [PubMed] [Google Scholar]
- Younts-Dahl SM, Osborn GD, Galyean ML, Rivera JD, Loneragan GH, Brashears MM. Reduction of Escherichia coli O157 in finishing beef cattle by various doses of Lactobacillus acidophilus in direct-fed microbials. J. Food Prot. 2005;68:6–10. doi: 10.4315/0362-028x-68.1.6. [DOI] [PubMed] [Google Scholar]
- Zolotarevsky Y, Hecht G, Koutsouris A, Gonzalez DE, Quan C, Tom J, Mrsny RJ, Turner JR. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology. 2002;123:163–172. doi: 10.1053/gast.2002.34235. [DOI] [PubMed] [Google Scholar]