

Abstract
In the synthesis of inorganic polyphosphate (polyP) from ATP by polyphosphate kinase (PPK; EC 2.7.4.1) of Escherichia coli, an N—P-linked phosphoenzyme was previously identified as the intermediate. The phosphate is presumed to be linked to N3 of the histidine residue because of its chemical stabilities and its resemblance to other enzymes known to contain N3-phosphohistidine. Tryptic digests of [32P]PPK contain a predominant 32P-labeled peptide that includes His-441. Of the 16 histidine residues in PPK of E. coli, 4 are conserved among several bacterial species. Mutagenesis of these 4 histidines shows that two (His-430 and His-598) are unaffected in function when mutated to glutamine, whereas two others (His-441 and His-460) mutated to glutamine or alanine fail to be phosphorylated, show no enzymatic activities, and fail to support polyP accumulation in cells bearing these mutant enzymes.
Keywords: autophosphorylation, histidine phosphorylation, site-directed mutagenesis
Inorganic polyphosphate (polyP) is a linear polymer of orthophosphate residues linked by high-energy phosphoanhydride bonds (1). PolyP has been conserved in nature, having been found in all organisms from bacteria to mammals (1). The ubiquitous occurrence of polyP suggests a number of important physiological roles, some of which have been demonstrated (2).
In Escherichia coli, polyphosphate kinase (PPK; EC 2.7.4.1) is responsible for polyP synthesis. The homotetrameric enzyme catalyzes the reversible transfer of the terminal phosphate from ATP to form polyP (3–5). An intermediate is the autophosphorylated PPK, in which a phosphate is presumably linked to histidine through an N—P bond (3). The ppk gene cloned and sequenced from E. coli (6), Klebsiella aerogenes (7), and Neisseria meningitidis (8) shows a 93% identity between the PPK of E. coli and that of Klebsiella and a 46% identity of E. coli and Neisseria PPKs (Fig. 1).
Figure 1.

C-terminal segments of PPK from three different sources. N.m, N. meningitidis PPK (8); K.a, K. aerogenes PPK (7); E.c., E. coli PPK (6). The boxed sequences represent regions of identity. Amino acids marked with an asterisk indicate the residues changed by site-directed mutagenesis.
In this report, we have identified the histidine residues that are at the active site in E. coli PPK by site-specific mutagenesis. Analysis of the mutant PPK proteins indicate that the conserved His-441 and His-460 residues are important for PPK function; sequencing of a tryptic phosphopeptide has identified His-441 as the predominant phosphorylated residue. In each case, alteration of these residues to either glutamine or alanine rendered the kinase protein incapable of autophosphorylation and enzymatic activity. Additionally, cells expressing the mutant PPK protein were unable to synthesize detectable amounts of polyP.
MATERIALS AND METHODS
Assay for PPK Activity.
To measure polyP synthesis (forward reaction), the assay mixture (10 μl) contained: 50 mM Hepes–KOH (pH 7.2), 40 mM (NH4)2SO4, 4 mM MgCl2, 2 mM creatine phosphate, 20 μg/ml creatine kinase, 1 mM [γ-32P]ATP (20 cpm/pmol), and purified PPK as indicated. Products were analyzed as described earlier (9) after incubation at 37°C for various periods of time. To measure ATP synthesis (reverse reaction), the ATP was replaced by 1 mM ADP and [32P]polyP, 350 μM (as Pi residues), prepared as described (10). The ATP-regenerating system was absent from the reverse reaction.
32P-Phosphoenzyme Formation.
The reaction mixture contained 50 mM Hepes-KOH (pH 7.2), 40 mM (NH4)2SO4, 10 mM MgCl2, 5 μM [γ-32P]ATP (105 cpm/pmol), and purified PPK (3). After incubation on ice for 10 min, the reaction was terminated by addition of EDTA to 35 mM. The phospho-PPK was precipitated with 2 vol of chilled acetone and then 2 vol of chilled ethanol to remove excess [γ-32P]ATP; the pellet was dissolved in SDS/PAGE sample buffer and electrophoresed on an SDS/4-15% polyacrylamide gradient gel. Phospho-PPK was transferred onto an Immobilon polyvinylidene difluoride (PVDF) membrane (11) and visualized by staining with Ponceau S, and the phospho-PPK band was excised from the membrane and used for analysis.
Chemical Stability of the Phospho-PPK Bond.
Samples containing ≈3 pmol of [32P]phospho-PPK were electrophoresed and transferred to an Immobilon PVDF membrane, and the [32P]phospho-PPK bands were excised after Ponceau S staining. The phospho-PPK bands were incubated in 0.2 M sodium citrate buffer (pH 2.4) at 42°C, in 50 mM potassium phosphate (pH 7.0) at 25°C, in 2 M NaOH (pH 13.5) at 42°C, or in 0.4 M hydroxylamine (pH 7.0) at 25°C for up to 2 h, rinsed in 50 mM potassium phosphate (pH 7.0), and examined for residual radioactivity. First-order rate constants were estimated from linear regression analysis of these data.
Phosphoamino Acid Analysis.
The [32P]phospho-PPK was hydrolyzed in 3 M KOH at 105°C for 3 h in a volume of 20 μl. The solution was neutralized by addition of HClO4. The KClO4 precipitate was removed by centrifugation. To the supernatant were added 5 μl of 15 mg/ml l-histidine and 5 μl of phosphohistidine as prepared by the phosphoramidate method (12). The reaction of phosphoramidate with histidine has been reported to result in formation of N1- and N3-phosphohistidine when carried out for 15 min (12). A 5-μl sample was spotted on a silica gel 60-Å K6 plate (Whatman), and TLC was performed with chloroform/methanol/ammonia (17%, wt/vol) (2:2:1, vol/vol) in the first dimension and with phenol/water (75:25, vol/vol) in the second dimension (13). The plates, after drying, were sprayed with ninhydrin (2% in ethanol) to detect positions of the standards histidine and phosphohistidine and then exposed to x-ray film.
Isolation and Sequencing of the Phosphopeptide from PPK.
To [32P]phospho-PPK (2 nmol) were added urea and dithiothreitol to final concentrations of 8 M and 2 mM, respectively. After incubation at 37°C for 15 min, carboxymethylation was carried out by addition of 5 mM iodoacetamide and incubation at 25°C for 15 min. The protein was lyophilized after dialyzing against distilled water for 3 h. The phospho-PPK was then resuspended in 8 M urea, diluted 8-fold with 25 mM Tris·HCl (pH 8.0), and digested with trypsin at 37°C for 3 h. The digest was applied to a Vydac C18 reverse-phase HPLC column (0.46 × 25 cm) equilibrated with 20 mM triethylamine (TEA)·HOAc, pH 5.8 (14). Isocratic elution with the equilibration buffer yielded two radioactive peaks: the first corresponded to [32P]Pi and the second to a 32P-peptide. Three separate runs were carried out to fractionate the peptides from the entire sample. The 32P-peptide-containing fractions were pooled, lyophilized, and after resuspension in distilled water, applied back on the column as a single sample. The 32P-peptide was dephosphorylated in 80% (wt/vol) trifluoroacetic acid and subjected to sequence analysis using an automated gas-phase sequencer.
Plasmid Construction and Site-Directed Mutagenesis.
Standard methods were used for all DNA manipulations (15). A template for mutagenesis was created by cloning the ppk gene in M13 mp8 in two steps. Briefly, pBC10 (6) was digested with NcoI and BstBI, and then blunted by DNA polymerase I (large fragment) extension. The resultant 2.5-kb fragment was ligated into pQE30 (Qiagen) that had been digested with SmaI and dephosphorylated with shrimp alkaline phosphatase. A 2.1-kb fragment of ppk, containing the sites of mutagenesis obtained by digestion with EcoRI, was ligated into M13 mp8, also digested with EcoRI, to create M13ppk. Site-directed mutagenesis was performed as described (16). Six mutants were obtained: H430Q, H441Q, H460Q, H598Q, H441A, and H460A. The site of mutation was confirmed in each instance by dideoxynucleotide sequencing.
Purification of Recombinant PPK.
The E. coli strain M15 (pREP) was transformed with recombinant PPK and the several mutant PPK-containing plasmids. Cells were grown in 50 ml of Luria–Bertani medium (LB; 1% tryptone/0.5% yeast extract/1% NaCl, pH 7.5) plus ampicillin (100 μg/ml) and kanamycin (25 μg/ml) to an OD600 of ≈0.8. Cells were induced with isopropyl β-d-thiogalactoside (IPTG; 50 μM) and grown for an additional 2 h at 25°C. Cells were harvested, washed, and resuspended in buffer A [50 mM Tris·HCl, pH 7.5/10% (vol/vol) glycerol] containing lysozyme (200 μg/ml). The heated lysozyme extract was centrifuged at 16,000 × g for 10 min, and the pellet was resuspended by sonication in buffer A containing 5 mM MgCl2 and 10 μg/ml each of DNase I and RNase A. To extract the PPK from the membranes, solid KCl was added to 1 M, followed by addition of 1/10 vol of 1 M Na2CO3. The mixture was stirred for 30 min at 4°C and then sonicated for 1 min. Insoluble material was removed by centrifugation at 40,000 rpm for 15 min. The supernatant was desalted by passing through a Sephadex G-25 column equilibrated with buffer B (buffer A containing 0.2 M KCl and 0.1% Triton X-100). The eluate was applied to a column of Ni–nitrilotriacetic acid-agarose (Qiagen; 1 ml of gel per 7.5 mg of protein) equilibrated with buffer B; the column was sequentially washed with two bed volumes each of buffer B, followed by buffer B containing 0.01 M imidazole and 0.25 M imidazole. Bound PPK was eluted with 0.5 M imidazole in buffer B. The active fraction was desalted by passage through a Sephadex G-25 column equilibrated with buffer A containing 10 mM KCl, 1 mM dithiothreitol, and 0.1% Triton X-100.
Native PPK (non-histidine-tagged) was purified from E. coli strain W3110/pBC10 as previously described (6).
Other Methods.
Protein concentrations were determined by the method of Bradford (17) with bovine serum albumin as a standard. SDS/PAGE was performed as described (18). Urea/polyacrylamide gels for polyP samples were run as described (19). Extraction and estimation of polyP will be described elsewhere (D. Ault-Richè and A.K., unpublished work).
RESULTS
General Properties of PPK.
Many samples of polyP synthesized by PPK from ATP have a remarkably uniform length of 750 ± 50 residues as determined by urea/PAGE. The reaction is highly processive inasmuch as no intermediate-size chains are seen throughout the course of the reaction (data not shown). Furthermore, addition of radiolabeled short-chain oligomers ranging in length from 2 to ≈400 residues were not extended in a reaction mixture in which unlabeled ATP produced polyP750 as visualized by toluidene blue staining following electrophoresis on a urea/15% polyacrylamide gel (Fig. 2). Thus PPK fails to accept polyP chains as primers in its processive synthesis of polyP from ATP.
Figure 2.

PPK does not elongate polyP oligomers to synthesize polyP750. Activity was measured in a 70-μl assay mixture. Unlabeled ATP replaced [γ-32P]ATP, and [32P]polyP oligomers were added at a final concentration of 75 μM (as Pi residues). At various time intervals, 10-μl samples were removed and the reaction was stopped by addition of 25 mM EDTA. Samples were electrophoresed on a urea/15% polyacrylamide gel, and the gel was exposed to x-ray film. Unlabeled polyP was visualized by toluidene blue staining. (A) Autoradiogram of the polyacrylamide gel. The four lanes on the left represent the indicated standards: Std., polyP750 subjected to partial acid hydrolysis. (B) Quantitation of polyP750 in the toluidene blue-stained gel. The amount of polyP was estimated by using a standard curve plotted from known amounts of polyP750 subjected to laser densitometry after electrophoresis and toluidene blue staining of a polyacrylamide gel.
In the reverse direction, PPK processively transfers Pi from polyP to ADP and generates ATP; no intermediate-size chains were detected. The Km for polyP750 (as polymer) is 60 nM and that for ADP is 360 μM.
Phosphorylated Residues in Phospho-PPK.
The extent of phosphorylation of PPK by ATP depends on the ATP concentration over the range of 5 μM to 1 mM (Table 1).
Table 1.
Stoichiometry of phosphorylation of PPK at different ATP concentrations
| ATP, mM | Pi, mol/mol of PPK monomer |
|---|---|
| 0.005 | 0.20 |
| 0.05 | 0.60 |
| 0.5 | 1.25 |
| 1.0 | 2.90 |
The phosphoryl-protein linkages were stable at neutral and alkaline pH but unstable in acid and hydroxylamine (Table 2), a behavior characteristic of a histidinyl phosphate (20, 21). The first-order rate constants (Table 2) for phospho-PPK hydrolysis in citrate buffer, pH 2.4 at 42°C and in 0.4 M hydroxylamine, pH 7.0, resembled those reported for hydrolysis of 3-phosphohistidine in phosphoenzyme I of the bacterial phosphoenolpyruvate:sugar phosphotransferase system (PTS) (22) and for phospho-CheA (23). This was confirmed by comigration of a phosphoamino acid from an alkaline hydrolysate of 32P-labeled 6-phosphofructo-2-kinase prepared as described (24) with the phosphohistidine from PPK (data not shown). Phosphorylation of 6-phosphofructo-2-kinase has been reported to occur exclusively on the N3 position of a histidine residue (24). To confirm the identity of the phosphorylated residue, the phospho-PPK was hydrolyzed under basic conditions and the hydrolysate was analyzed by two-dimensional TLC. The 32P-labeled amino acid comigrated with phosphohistidine standard and was the sole phosphorylated residue in the alkaline hydrolysate.
Table 2.
Chemical stability of phospho-PPK bond
| Condition | Stability of
product synthesized from ATP, bonds cleaved per min
|
|
|---|---|---|
| Synthesized at 0.005 mM | Synthesized at 1.0 mM | |
| pH 2.4 | 0.025 | 0.014 |
| pH 7.0 | 0.000 | 0.000 |
| pH 13.5 | 0.000 | 0.000 |
| NH2OH | 0.012 | 0.015 |
Isolation and Sequencing of a Phosphohistidine-Containing Peptide from PPK.
[32P]Phospho-PPK was denatured and alkylated as described in Materials and Methods and digested with trypsin. The peptides were fractionated on a C18 reverse-phase column at pH 5.8 due to the acid lability of the phosphohistidyl bond. The highly charged nature and small size of the 32P-labeled peptide resulted in its elution with 20 mM TEA·HOAc. A tryptic phosphopeptide obtained after chromatography was dephosphorylated and sequenced to yield a tetrapeptide with the sequence Ile-His-Ala-Lys.
Purification of Recombinant PPK (rPPK).
For overexpression of PPK, the gene was inserted into a vector (pQE30), resulting in a plasmid designated pQE30PPK. This plasmid allowed an IPTG-inducible production of rPPK activity. A 6- to 10-fold overproduction of rPPK compared with uninduced cells was obtained after a 2-h incubation at 25°C after induction with 50 μM IPTG added to cells at a density of 4 × 108 cells per ml. Linkage of rPPK to a fusion sequence including six histidine residues at the N terminus of PPK enables this histidine array to facilitate purification using nickel-affinity chromatography (25).
Purification of rPPX from E. coli (Table 3) involved extraction of the protein from the membrane fraction by sonication in the presence of 1 M KCl and 0.1 M Na2CO3. Binding to a nickel-agarose column yielded a preparation with a final specific activity of 107 units/mg of protein. The 3-fold lower value compared with that of the previously reported PPK from wild-type cells (3) could be a result of the perturbation of the native enzyme structure. The enzyme was >95% homogeneous as judged by densitometric analysis of the Coomassie blue-stained gel (Fig. 3).
Table 3.
Purification of rPPK
| Fraction | Activity, units × 10−6 | Specific activity, units × 10−6/mg of protein |
|---|---|---|
| Lysate pellet | 15.2 | 1.0 |
| Solubilized membrane | 16.1 | 1.2 |
| Ni-agarose | 6.9 | 9.2 |
Figure 3.
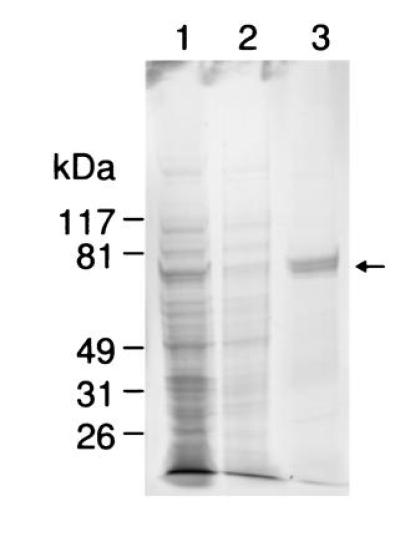
PAGE analysis of purified rPPK. After electrophoresis on an SDS/4–15% polyacrylamide gradient gel, proteins were visualized by Coomassie blue staining. Purification steps of rPPK as described in Materials and Methods: lane 1, lysate pellet (25 μg); lane 2, solubilized membrane fraction (5 μg); lane 3, nickel-affinity-purified rPPK (5 μg). Arrow indicates position of rPPK.
In Vitro Analysis of Mutated rPPK.
The mutant rPPKs were individually expressed and purified from E. coli using the protocol described in Materials and Methods for purification of rPPK. The mutants were purified to ≈90% homogeneity (data not shown). Analysis of each rPPK included its ability to be autophosphorylated, to synthesize polyP from ATP, and to transfer a phosphate from polyP to ADP to make ATP.
Autophosphorylation with 5 μM [γ-32P]ATP indicated that two of the mutants (H441Q and H460Q) failed to autophosphorylate, while mutations of H430 and H598 had little effect on the ability of rPPK to form a phosphoenzyme (Table 4).
Table 4.
Enzymatic activities of wild-type and mutant rPPKs
| Protein | PolyP synthesis, units × 10−6/mg | ATP synthesis, units × 10−6/mg | Autophosphorylation, mol of Pi/mol of PPK monomer |
|---|---|---|---|
| rPPK | 9.2 | 1.0 | 0.19 |
| H430Q | 9.7 | 1.1 | 0.15 |
| H441Q | <0.01 | 0.007 | <0.0001 |
| H460Q | <0.01 | 0.005 | <0.0001 |
| H598Q | 8.3 | 1.3 | 0.10 |
| H441A | <0.01 | 0.03 | <0.0001 |
| H460A | <0.05 | 0.03 | <0.0001 |
Mutants lacking the ability to autophosphorylate were also incapable of synthesizing polyP from ATP, as well as phosphorylating ADP by polyP to make ATP (Table 4). To show that the lack of enzymatic activity of these mutants was not due to the charge effect of substituting glutamine for histidine, alanine was substituted for histidine at positions 441 and 460. These alanine mutant proteins had properties similar to those seen for H441Q and H460Q, confirming the importance of these histidine residues for enzyme activity.
PolyP Content in Cells Overexpressing Wild-Type and Mutant rPPKs.
PolyP was extracted after IPTG induction from cells containing either the wild-type rPPK plasmid or one of the mutant rPPK plasmids. The extraction procedure separated polyP primarily from ATP by using glass milk after cell lysis in 6 M guanidine hydrochloride. The polyP content of extracts was measured by using the PPK conversion of ADP to ATP and estimating the amount of ATP formed by measuring the light released after a luciferase reaction. The detailed procedure for extraction and estimation of polyP from unlabeled cells will be published elsewhere (D. Ault-Richè and A.K., unpublished work).
Cells expressing the nonmutated rPPK contained 330 nmol of polyP (as Pi residues) per mg of cell protein. Cells synthesizing the PPK mutants H441Q and H460Q had virtually no polyP, while the H430Q and H598Q mutants had polyP levels comparable to those of the wild-type rPPK-expressing cells (Table 5).
Table 5.
PolyP content in E. coli expressing wild-type and mutated rPPK
| Protein expressed | PolyP content,* nmol/mg of cell protein |
|---|---|
| rPPK | 330 |
| H430Q | 311 |
| H441Q | 0.29 |
| H460Q | 0.17 |
| H598Q | 220 |
| H441A | 0.08 |
| H460A | 0.08 |
As Pi residues
DISCUSSION
PPK of E. coli is a membrane-bound homotetramer of 80-kDa subunits that catalyzes (i) the polymerization of the terminal (γ) phosphate of ATP into long-chain polyP, and (ii) the depolymerization of polyP by ADP to generate ATP (3–5). The reaction
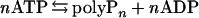 |
goes to near completion in the polymerization direction when ADP is trapped (regenerated to ATP) and in the reverse direction when ADP is supplied in excess; the rate of the reverse reaction under optimal conditions is only about one-tenth that of the forward reaction.
The reactions in either direction are highly processive. The product of polymerization is a remarkably monodisperse long chain of 750 (±50) residues; intermediate chain lengths are neither detected nor extended when supplied as substrates (Fig. 2). Whereas shortened chains are not detected in the depolymerization reaction, intermediate chain lengths can be used—though less effectively than the long chain—in converting ADP to ATP. The basis for the rather uniform length of the polyP product remains to be determined.
An early event in the polymerization is the autophosphorylation of PPK; the phosphoryl-enzyme appears to be a valid intermediate inasmuch as the phosphate can be chased into polyP by ATP or back into ATP by ADP. The phosphate is linked to N3 of one or more histidine residues. Of the 16 histidine residues in PPK, 4 are conserved as judged on the basis of comparison of the sequence of E. coli PPK with that of the PPK of K. aerogenes and of N. meningitidis. Mutagenesis of these four histidines has revealed that two are essential for autophosphorylation, for enzymatic activities, and for accumulation of polyP in cells. Just how these histidines are disposed at the active site and participate in the actions of PPK requires further investigation.
Among other issues that need additional study is the question as to whether the tetrameric state of PPK is essential for its activity and also whether it may provide a structural basis for the size of its polyP product. The membrane association of PPK (6) is also intriguing and may provide clues to the mechanism of its action as well as to the multiple functions that polyP can perform in the cell (2). The dynamic turnover of polyP in E. coli in response to stringencies and other factors relies largely on its synthesis by PPK balanced by its hydrolysis by the principal exopolyphosphatase (PPX) expressed in the same operon (6). Under what circumstances and to what extent PPK converts polyP to ATP are items that deserve attention.
Acknowledgments
The authors thank Dana Ault-Richè for estimations of polyP, Leroy Bertsch for critical evaluation of the manuscript, Alan J. Smith for assistance with sequence analysis of the 32P-labeled tryptic peptide, and Drs. Simon J. Pilkis and M. Raafat El-Maghrabi for a gift of 6-phosphofructo-2-kinase and fructose 2,6-bis[32P]phosphate. K.H.A. was supported by a fellowship from the National Institutes of Health.
Footnotes
Abbreviations: IPTG, isopropyl β-d-thiogalactoside; polyP, inorganic polyphosphate; PPK, polyphosphate kinase; rPPK, recombinant PPK; TEA, triethylamine.
References
- 1.Kulaev I S. The Biochemistry of Inorganic Polyphosphates. New York: Wiley; 1979. [DOI] [PubMed] [Google Scholar]
- 2.Kornberg A. J Bacteriol. 1995;177:491–496. doi: 10.1128/jb.177.3.491-496.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahn K, Kornberg A. J Biol Chem. 1990;265:11734–11739. [PubMed] [Google Scholar]
- 4.Kornberg A, Kornberg S R, Simms E S. Biochim Biophys Acta. 1956;20:215–227. doi: 10.1016/0006-3002(56)90280-3. [DOI] [PubMed] [Google Scholar]
- 5.Kornberg S R. Biochim Biophys Acta. 1957;26:294–300. doi: 10.1016/0006-3002(57)90008-2. [DOI] [PubMed] [Google Scholar]
- 6.Akiyama M, Crooke E, Kornberg A. J Biol Chem. 1992;267:22556–22561. [PubMed] [Google Scholar]
- 7.Kato J, Yamamoto T, Yamada K, Ohtake H. Gene. 1993;137:237–242. doi: 10.1016/0378-1119(93)90013-s. [DOI] [PubMed] [Google Scholar]
- 8.Tinsley C R, Gotschlich E C. Infect Immun. 1995;63:1624–1630. doi: 10.1128/iai.63.5.1624-1630.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crooke E, Akiyama M, Rao N N, Kornberg A. J Biol Chem. 1994;269:6290–6295. [PubMed] [Google Scholar]
- 10.Keasling J, Bertsch L, Kornberg A. Proc Natl Acad Sci USA. 1993;90:7029–7033. doi: 10.1073/pnas.90.15.7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Towbin H, Staehlin T, Gordon J. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei Y-F, Matthews H R. Methods Enzymol. 1991;200:388–414. doi: 10.1016/0076-6879(91)00156-q. [DOI] [PubMed] [Google Scholar]
- 13.Niederweiser A. Methods Enzymol. 1972;25:60–99. doi: 10.1016/S0076-6879(72)25008-X. [DOI] [PubMed] [Google Scholar]
- 14.Hess J F, Bourret K B, Simon M I. Nature (London) 1988;336:139–143. doi: 10.1038/336139a0. [DOI] [PubMed] [Google Scholar]
- 15.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 16.Messing J. Methods Enzymol. 1983;101:20–78. doi: 10.1016/0076-6879(83)01005-8. [DOI] [PubMed] [Google Scholar]
- 17.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 18.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 19.Kumble K D, Kornberg A. J Biol Chem. 1995;270:5818–5822. doi: 10.1074/jbc.270.11.5818. [DOI] [PubMed] [Google Scholar]
- 20.Anthony R S, Spector L B. J Biol Chem. 1972;247:2120–2125. [PubMed] [Google Scholar]
- 21.Hultquist D E, Moyer R W, Boyer P D. Biochemistry. 1966;5:322–331. doi: 10.1021/bi00865a041. [DOI] [PubMed] [Google Scholar]
- 22.Weigel N, Kukuruzinska M A, Nakazawa A, Waygood E B, Roseman S. J Biol Chem. 1982;257:14477–14491. [PubMed] [Google Scholar]
- 23.Wylie D, Stock A, Wong C-Y, Stock J. Biochem Biophys Res Commun. 1988;151:891–896. doi: 10.1016/s0006-291x(88)80365-6. [DOI] [PubMed] [Google Scholar]
- 24.Pilkis S J, Walderhaug M, Murray K, Beth A, Venkataramu S D, Pilkis J, El-Maghrabi M R. J Biol Chem. 1983;258:6135–6141. [PubMed] [Google Scholar]
- 25.Janknecht R, de Martynoff G, Lou J, Hipskind R A, Nordheim A, Stunnenberg H G. Proc Natl Acad Sci USA. 1991;88:8972–8976. doi: 10.1073/pnas.88.20.8972. [DOI] [PMC free article] [PubMed] [Google Scholar]