

SUMMARY
Gastrin-releasing peptide (GRP) activates phosphatidylinositol 3-kinase (PI3-K)/Akt, an important cell survival signaling pathway, to stimulate growth of various cell types. Transforming growth factor (TGF) superfamily ligands activate intracellular Smad signaling to regulate cell growth, differentiation and apoptosis; dysregulation of the TGF-β/Smad pathway has been noted in cancer cells. Therefore, we sought to determine whether a potential cross-talk exists between the TGF-β/Smad and PI3-K pathways in the regulation of neuroblastoma cell growth. Increased Smad DNA binding was noted in SK-N-SH human neuroblastoma cells when treated with LY294002, an inhibitor of PI3-K, by transcription factor/DNA array analysis and electrophoretic mobility shift assay. LY294002 treatment resulted in Smad2 accumulation in the nuclei and an increased Smad binding element (SBE)-luciferase activity. These findings were corroborated by co-transfection with pCGNN-Δp85 plasmid, which expresses a PI3-K mutant p85 subunit. In contrast, GRP treatment decreased Smad binding activity in neuroblastoma cells. Our findings demonstrate that the PI3-K pathway negatively regulates TGF-β/Smad signaling in neuroblastoma cells. GRP-induced activation of PI3-K, resulting in neuroblastoma cell growth promotion, is potentiated by down-regulation of TGF-β/Smad signaling.
Keywords: TGF-β, Smad, PI3-K, GRP, Neuroblastoma
INTRODUCTION
As a neural crest derived endocrine tumor, neuroblastomas produce and secrete various gut peptides that affect tumor differentiation and growth [1]. In particular, gastrin-releasing peptide (GRP) is expressed abundantly in neuroblastomas with an increased expression of its cell surface GRP-receptor (GRP-R) in more advanced-stage tumors [2]. GRP, which binds to its specific cell membrane receptor, acts as an autocrine and/or paracrine growth factor to promote neuroblastoma cell growth [3, 4]; however, the exact intracellular signaling pathways involved in this process have not been clearly discerned.
The phosphatidylinositol 3-kinase (PI3-K) pathway is thought to play a crucial role in the survival of various cell types [5]. Upon stimulation, PI3-K, which is comprised of p85 catalytic and p110 regulatory subunits, activates the downstream effector, Akt, by phosphorylation via phosphoinositol lipid and PDK1 intermediates. Activated Akt, in turn, phosphorylates multiple proteins implicated in the control of the cell cycle to ultimately stimulate cell growth [5, 6]. We have recently reported that overexpression of GRP-R in neuroblastoma cells results in down-regulation of the tumor suppressor protein PTEN (phosphatase and tensin homolog deleted on chromosome ten), an endogenous negative regulator of PI3-K signaling, which then ultimately leads to increased cell growth [7]. Interestingly, potential intracellular signaling mechanisms, which may involve cross-talk between PI3-K and other signal transduction pathways during GRP-induced stimulation of neuroblastoma cell growth, have not been thoroughly explored.
Transforming growth factor (TGF)-β exerts potent anti-mitogenic and pro-apoptotic effects on certain cancer cells [8, 9]. TGF-β initiates its cellular responses by binding to distinct receptors with intrinsic serine/threonine kinase activity and subsequently activating specific downstream intracellular effectors called Smad proteins. Smads relay the signal from the cell membrane to the nucleus, where they bind to Smad binding element (SBE) sequences in the promoter regions of various target genes to regulate cell growth [10–12]. Smad proteins, as transcription factors, cooperate with other transcription factors to regulate the expression of target genes. Smad proteins 2, 3 and 4 can form heterodimers as well as complexes with other transcription factors to regulate transcriptional activity in cancers [13, 14]. Smad activation, subcellular distribution and stability are intricately regulated, and a broad array of transcription factors are known to partner with Smads [15–17]. Escape from TGF-β/Smad-induced cell growth inhibition and apoptosis is frequently observed in various cancers [18]. In addition, mutation [19] as well as gene ablation in mice [20] of certain Smads has been attributed to an increased rate of tumorigenesis. Clearly, TGF-β/Smad signaling plays a vital role in the malignant potential of a variety of cell types; however, its potential role in neuroblastoma is unknown.
In this study, we sought to evaluate a potential interaction of PI3-K and TGF-β/Smad signaling pathways in the GRP-induced stimulation of neuroblastoma cell growth. Given the tumor inhibitory actions of TGF-β/Smad signaling, we determined whether the PI3-K pathway directly inhibits DNA binding activity of Smad, thereby, potentiating cell proliferative actions of PI3-K. Our findings demonstrate that inhibition of PI3-K increases Smad2 and Smad4 nuclear translocation, Smad/SBE DNA binding and subsequent transcriptional activity of SBE.
MATERIALS AND METHODS
Plasmids and reagents
The luciferase reporter gene construct containing the SBE promoter element, which responds to TGF-β, was designed (SBE-Luc). Smad2-flag, Smad4-flag constructs were gifts from Dr. R. Derynck (University of California at San Francisco, CA). The dominant negative mutant (Δp85) of the PI3-K regulatory subunit, constructed in a vector pCGNN, was a gift from Dr. W.G. King (Harvard Medical School, Boston, MA). LY294002 compound was purchased from Cell Signaling (Beverly, MA). GRP peptide was from Bachem (Torrance, CA). TGF-β was purchased from BD Bioscience (Bedford, MA). Goat polyclonal anti-Smad2 antibody, rabbit polyclonal anti-Smad4 antibody and horseradish peroxidase (HRP)-conjugated secondary antibody were from Santa Cruz Biotechnology, Inc (Santa Cruz, CA). Anti-phospho-Smad2 (ser465/467) polyclonal antibody was from Upstate Biotechnology (Lake Placid, NY). Anti-phospho-Akt (ser473) polyclonal antibody was purchased from Cell Signaling (Beverly, MA). β-actin monoclonal antibody was from Sigma (St. Louis, MO). The secondary antibody Alexa Flour 568 goat anti-rabbit and 4′,6′-diamidino-2-phenylindole, dihydrochloride (DAPI) were purchased from Molecular Probe (Eugene, OR).
Cell culture, transfection, and reporter luciferase assays
The human neuroblastoma cell line, SK-N-SH, was purchased from American Type Cell Culture (Manassas, VA). Cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) under a humidified 5% CO2 atmosphere at 37°C. Cells were grown and transiently transfected using Lipofectamine reagent according to the manufacturer’s protocol (Invitrogen; Carlsbad, CA). Reporter luciferase activity assays were performed with Dual-Luciferase Reporter Assay (Promega; Madison, WI).
Protein/DNA array and electrophoretic mobility shift assay
Assessment of DNA binding activity in SK-N-SH cells was carried out using TranSignal™ Protein/DNA Array I according to the manufacturer’s instruction (Panomics; Redwood City, CA). Nuclear protein extracts were prepared with NE-PER Nuclear and Cytoplasmic Extraction Reagents (PIERCE; Rockford, IL). Electrophoretic mobility shift assays (EMSA) were performed with EMSA Gel-Shift Kit (Panomics) according to the manufacturer’s protocol. Biotin-labeled SBE oligonucleotides (5′–AGTATGTCTAGACTGA–3′) were mixed with nuclear proteins in a binding buffer and incubated for 20 min at room temperature. Protein/DNA complexes were resolved in 6% DNA Retardation Gels (Invitrogen; Rockville, MD).
Immunocytochemical analysis
SK-N-SH cells were plated on glass coverslips, incubated overnight and serum-starved for 24 h. Cells were treated with GRP (10−7 M), LY294002 (20 μM), or TGF-β (1 ng/ml) for 1 h. The treated cells were washed once with phosphate buffered saline (PBS) and fixed in 4% paraformaldehyde in PBS for 15 min at room temperature, then incubated in ice cold 100% methanol for 10 min at RT. Fixed cells were rehydrated in PBS for 30 min at RT, then samples blocked in 1% BSA/PBS buffer for 30 min, and incubated for 60 min in 1:100 dilution of anti-Smad2 or Smad4 antibodies. The cells were washed three times with PBS and incubated 30 min in PBS containing 1% BSA and 1:400 secondary antibody Alexa Flour 568 goat anti-rabbit, then stained with DAPI for 5 min. The cells were rinsed three times with PBS, and coverslips were mounted onto the slide glasses. The mounted cells were viewed with a fluorescence microscope using a 40X objective lens.
Immunoblots
Cell lysis buffer (Cell Signaling) containing 1 mM PMSF and protein inhibitors cocktail (Roche Applied Science; Indianapolis, IN) was used to extract total protein. Total protein (50 μg) was electrophoresed on a precast 4–12% Bis-Tris gel (Invitrogen) and electro-transferred to polyvinylidene diflouride membranes (BioRad; Hercules, CA). Blots were probed with anti-phospho-Smad2 (ser465/467) and anti-phospho-Akt (ser473) polyclonal antibodies followed by HRP-conjugated secondary antibody (1:5000 dilutions). Equal loading and transfer were confirmed by blotting the same membrane with β-actin antibody (1:5000 dilutions). Protein was visualized by enhanced chemiluminescence system (Amersham Bioscience; Arlington, IL) and autoradiography.
RESULTS
PI3-K regulation of Smad transcription factor
PI3-K is a crucial cell survival signaling pathway; aberrant expression of this pathway leads to increased cellular growth [5]. We wanted to determine whether the PI3-K pathway controls transcription factors that are known to regulate anti-apoptotic signals in neuroblastoma cells. We first performed transcription factor protein/DNA array using nuclear proteins from SK-N-SH cells treated with LY294002, an inhibitor of PI3-K, or vehicle (Fig. 1). We found that Smad/SBE binding was significantly increased by LY294002 treatment (Fig. 1A; boxed areas).
Figure 1. Smad/Smad binding element (SBE) DNA binding activity.

(A) The protein/DNA array was performed using nuclear extracts from SK-N-SH cells treated with vehicle (control) or LY294002 (20 μM), a PI3-K inhibitor. Signals outlined in boxes represent activated Smad/SBE by LY294002 treatment. (B) Nuclear protein was assayed for Smad/SBE DNA binding with a biotin-labeled oligonucleotide containing the Smad/SBE consensus sequence (AGTATGTCTAGACTGA). LY294002 treatment significantly increased DNA binding (lane 3). Treatment with wortmannin, another PI3-K inhibitor, also increased Smad/SBE DNA binding (lane 6). Competition experiments were performed with a 30-fold molar excess of the unlabeled Smad/SBE oligonucleotide.
To confirm the protein/DNA array result, EMSAs were performed. Consistent with the array data, Smad/SBE DNA binding intensity was significantly increased by LY294002 treatment (Fig. 1B, lane 3). Wortmannin, another PI3K inhibitor, produced similar effects on Smad/SBE binding intensity (Fig. 1B; lane 6). Competitor (lanes 4, 7) confirmed the specificity of this assay. The TGF-β/Smad pathway, which is altered in many tumor cells, [19–21] is commonly involved with apoptosis. The potential cross-talk between PI3-K and TGF-β/Smad pathways in cancer cell biology is unclear; this may be an important regulatory mechanism during malignant transformation of various cell types.
Conversely, GRP treatment resulted in attenuation of Smad/SBE binding activity (data not shown). Since we previously found that GRP-induced neuroblastoma cell growth is, in part, mediated by regulation of PI3-K, [7] this data is consistent with the fact that GRP may also activate PI3-K, which negatively regulates Smad/SBE binding activity in neuroblastoma cells. Taken together, these data further suggest a cross-talk mechanism between PI3-K and TGF-β/Smad signaling.
Smad2 phosphorylation is induced by inhibition of the PI3-K pathway
To demonstrate TGF-β/Smad signaling in neuroblastomas cells, we first treated SK-N-SH cells with TGF-β over a time course and determined nuclear Smad2 protein phosphorylation by Western blot analysis (Fig. 2A). TGF-β treatment resulted in phosphorylation of Smad2 in a time-dependent fashion, with its strongest expression at 60 min after treatment. It is well known that TGF-β activates the TGF-β receptor, which then activates Smad2 by phosphorylation [15]. Phosphorylated Smad2 re-localizes into the nucleus to activate transcription of target genes [15]. Figure 2B shows phosphorylation of Smad2 by TGF-β treatment. Inhibition of PI3-K by LY294002 induced Smad2 phosphorylation; inhibition of p-Akt was confirmed. These results further implicate Smad2 as the cross point or communicative core of the two signaling pathways, TGF-β/Smad and PI3-K/Akt, upon GRP stimulation.
Figure 2. TGF-β increases Smad2 protein phosphorylation.
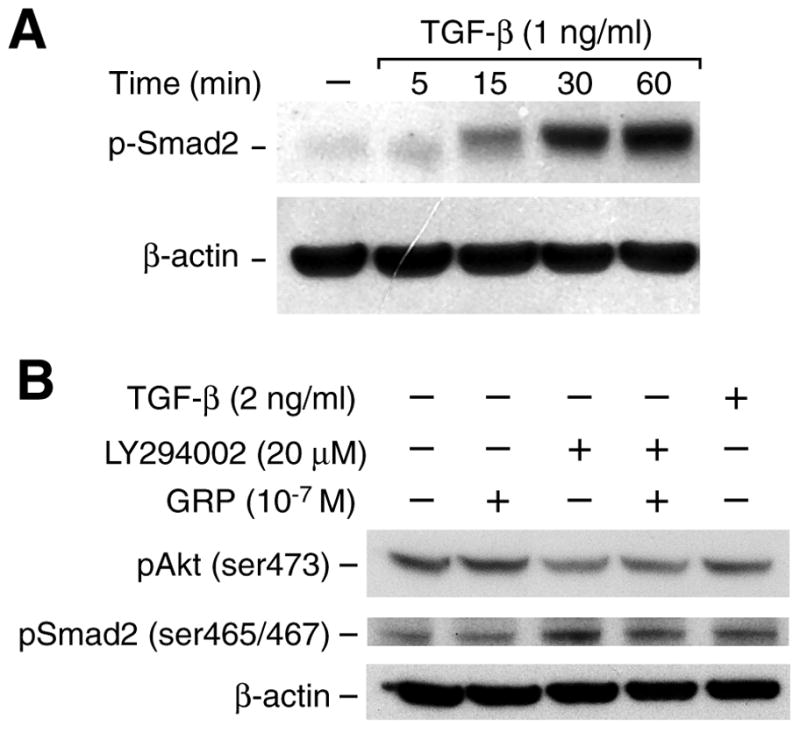
(A) Phosphorylation of Smad2 occurred in a time-dependent manner after treatment with TGF-β (1 ng/ml) demonstrating that TGF-β/Smad signaling is intact in SK-N-SH cells. (B) LY294002 treatment resulted in increased phosphorylation of Smad2 by Western blot analysis. This increase was attenuated by GRP treatment. β-actin was used as a protein loading control.
Smad2 and Smad4 nuclear translocation by PI3-K inhibition
To determine the possible point of cross-talk between the PI3-K and TGF-β signaling pathways, we next examined the effects of PI3-K/Akt on Smad2 and Smad4 intracellular nuclear localization by immunocytochemical staining analysis. Smad2 was evenly distributed in control cells; however, GRP treatment resulted in decreased Smad2 nuclear localization. Smad2 nuclear localization was noted 33% of control cell population examined, whereas, nuclear Smad2 expression was found in 27% of cells after GRP treatment (Fig. 3A). In contrast, LY294002 treatment resulted in marked nuclear accumulation of Smad2; its nuclear expression was detected in 82% of cells counted. Similarly, TGF-β treatment alone demonstrated intense nuclear localization of Smad2 (74% of cell population), thus suggesting the presence of an intact TGF-β/Smad signaling in neuroblastoma cells (Fig. 3A). Smad4 protein also accumulated in the nucleus by LY294002 treatment (Figs. 3B, 3C). These results demonstrate that inhibition of the PI3-K/Akt pathway enhances Smad2 and Smad4 protein translocation into the nucleus, whereas GRP-induced activation of PI3-K/Akt blocks this process.
Figure 3. Immunocytochemical analysis for nuclear Smad2 and Smad4 protein.
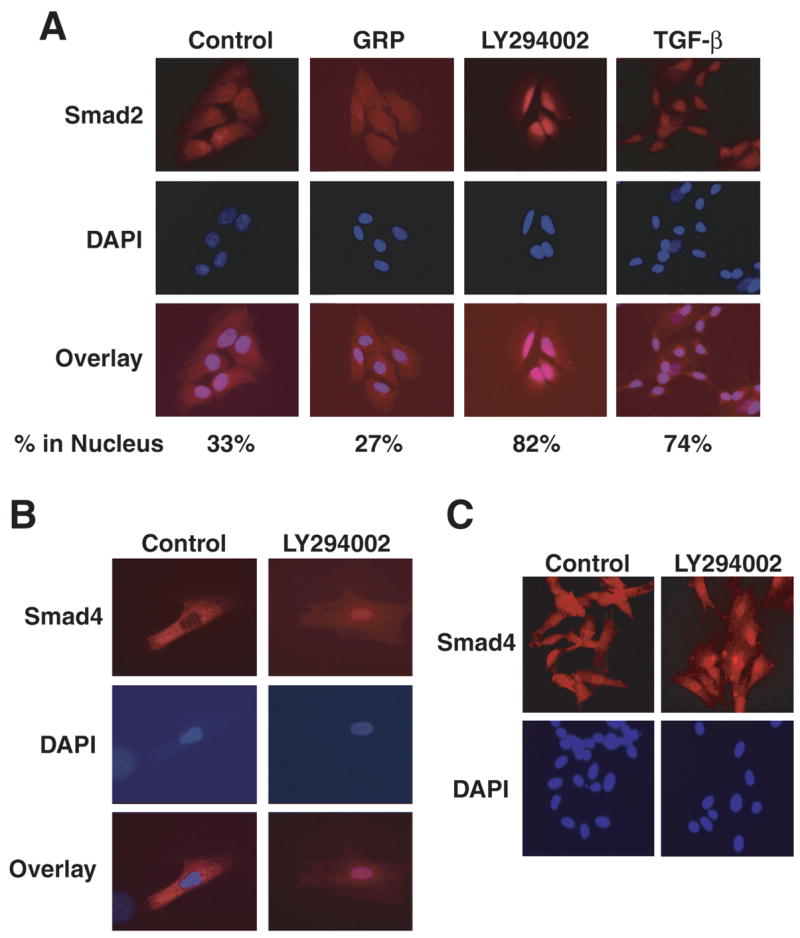
(A) GRP treatment resulted in a slightly decreased level of nuclear Smad2 protein. Inhibition of PI3-K by LY294002 significantly increased nuclear accumulation of Smad2, which was greater than treatment with TGF-β (1 ng/ml) alone. Quantitative Smad2 nuclear localization is expressed as percentage of Smad2-stained cells relative to total number of cells examined. (B and C) Similarly, LY294002 treatment resulted in intense nuclear staining of Smad4.
Induction of SBE-luciferase activity by inhibition of PI3-K
Treatment with LY294002 increased Smad2 phosphorylation and Smad2 localization into the nucleus. To further confirm these findings, we next used a SBE luciferase reporter system to determine upregulation of Smad2-mediated transcriptional activity. SBE is a specific binding site for the Smad transcription factor complex [22]; the SBE promoter element is directly regulated by Smads, therefore, the reporter luciferase activity assay directly reflects the Smad transcriptional activity [22]. The SBE luciferase construct was co-transfected into SK-N-SH cells along with the Smad2 plasmid. TGF-β treatment significantly increased Smad/SBE-regulated luciferase activity; this increase was further potentiated with LY294002 treatment (Fig. 4A). Although GRP treatment alone did not affect SBE luciferase activity, GRP attenuated LY294002-induced increases in SBE transcriptional activity, further suggesting a negative relationship between GPR-induced PI3-K pathway activation and Smad signaling in neuroblastoma cells.
Figure 4. SBE luciferase activity.

SBE-Luc was transiently transfected into SK-N-SH cells, serum was starved overnight, luciferase activity assay was carried out after another 4 h. (A) Transfection with the Smad2 plasmid and treatment with either TGF-β (2 ng/ml) or LY294002 (20 μM) resulted in increased SBE luciferase activity when compared to control cells transfected with empty vector. GRP treatment (10−7 M) attenuated LY294002-induced SBE luciferase activity. Combination of LY294002 (20 μM) and TGF-β (1 ng/ml) increased SBE luciferase activity when compared to either treatment alone. (* p<0.05 vs. TGF-β alone; † p<0.05 vs. GRP alone; triplicate trial) (B) Inhibition of the PI3-K/Akt pathway using p85 subunit dominant mutant (pCGNN-Δp85) also significantly increased SBE luciferase activity. (* p<0.05 vs. empty vector; triplicate trial).
GRP activation of cell surface G-protein coupled receptor results in the conformational change of heterotrimeric G-protein, leading to dissociation of Gβγ subunit. Gβγ subunit transduces signal to either class IB PI3-K, composed of p110γ and p101 subunits [23] or class IA PI3-K, consisting of p110 and p85 subunits, indirectly by transactivation of Src or tyrosine kinase receptor [24]. We had found that GRP treatment results in transactivation of tyrosine kinase receptor in neuroblastoma cells (data not shown); therefore, we performed co-transfection assays using the dominant negative mutant of the p85 subunit (pCGNNΔp85), which inhibits PI3-K/Akt. Consistent with data using the pharmacologic inhibitor of PI3-K/Akt, co-transfection with mutant p85 significantly increased SBE luciferase activity in neuroblastoma cells (Fig. 4B).
DISCUSSION
The potential interaction of various intracellular signaling cascades, which ultimately lead to stimulation of neuroblastoma cell growth, is relatively unknown. Based on our previous studies [7] showing that GRP-R downregulates PTEN, an endogenous negative regulator of PI3-K signaling, we sought to determine whether GRP interacts with other signaling pathways. We found that TGF-β/Smad signaling exists in neuroblastoma cells. Moreover, inhibition of PI3-K potentates the nuclear localization of Smad proteins and, ultimately, SBE-regulated luciferase gene activity. The cellular mechanisms responsible for the growth-stimulatory effects of GRP involve both activation of a key cell survival pathway, such as PI3-K, as well as negative crosstalk with TGF-β/Smad signaling in neuroblastoma cells.
The TGF-β/Smad signal transduction pathway is an important negative regulator of cell proliferation and inducer of apoptosis. Smad proteins constitute the basic components of the core intracellular signaling cascade activated by TGF-β and Smads function by carrying signals from the cell surface directly to the nucleus [25]. Dysregulation or alteration of TGF-β/Smad signaling in tumorigenesis can occur at many different levels such as mutation or transcriptional suppression of cell surface receptors, or alteration of down-stream signal transduction pathways. In this study, we found that the TGF-β/Smad signaling also exists in neuroblastoma cells, as phosphorylated Smad2 protein expression was increased with TGF-β treatment over a time course, suggesting a potential mechanism where dysregulation of TGF-β/Smad signaling contributes to neuroblastoma tumorigenesis.
A balance between PI3-K/Akt, a key cell survival pathway, and other signaling pathways regulating cell death is crucial to maintaining normal cell homeostasis. Specifically, a cross-talk between PI3-K/Akt and TGF-β has been recently explored in various cell types. PI3-K function is critical for TGF-β mediated epithelial to mesenchymal transition and cell migration [26]. Insulin-like growth factor (IGF)-1 inhibits TGF-β transcriptional responses through selective suppression of Smad3 activation via a PI3-K/Akt-dependent pathway [27]. Others have also demonstrated a functional link between the serine-threonine kinase, PKB/Akt, and Smad3 protein [28]. These recent studies have provided insights into how Smad proteins are regulated by PI3-K pathway. Our current study is, to our knowledge, the first to show that GRP-induced activation of PI3-K negatively regulates TGF-β Smad signaling in neuroblastomas. Moreover, we showed that inhibition of PI3-K/Akt resulted in increased Smad/SBE DNA binding as well as SBE luciferase activity, suggesting that a negative cross-talk exists between these cellular signaling pathways. In fact, the growth stimulatory actions of GRP in neuroblastoma cells may be significantly enhanced by GRP-induced PI3-K/Akt pathway activation along with its indirect negative control of apoptotic signaling (i.e., TGF-β Smad). Therefore, therapy targeted to pathways potentiating activation of apoptotic cellular signaling may be important for developing novel strategies for neuroblastoma cancer therapy.
Recently, cross-talk between the TGF-β/Smad signaling and other intracellular kinase pathways has also been reported [8, 29, 30]. TGF-β family members have been shown to activate JNK [8, 30] and MAPK proteins [18]. TGF-β also prevents pro-inflammatory cytokine production through inhibition of p38 MAPK and nuclear factor-κB [31]. Moreover, these cross-talk mechanisms have been shown to occur at multiple levels within the cell. Smad translocation into the nucleus may be inhibited by MAPK-dependent phosphorylation. Erk-mediated phosphorylation of Smad1, in response to TGF-β is critical for regulating Smad1 subcellular localization [30]. Whereas in the nucleus, Smads exert transcriptional regulation through functional cooperativity and physical interactions with other transcription factors, such as AP1 [8], as well as with negative co-repressors [15, 16, 25]. The findings in our current study show that the PI3-K/Akt pathway negatively regulates nuclear translocation of Smad2/4 proteins in neuroblastoma cells, as PI3-K inhibition resulted in increases in both Smad2/4 proteins in the nuclei. Clearly, further studies are required to fully elucidate the complex array of regulatory mechanisms between PI3-K and Smad nuclear translocation.
In summary, we have shown that TGF-β/Smad signaling was inhibited by GRP-induced activation of the PI3-K/Akt pathway in neuroblastomas cells. In contrast, inhibition of PI3-K activity by its selective inhibitor, LY294002, resulted in increased Smad transcriptional activity. Therefore, cross-talk exists between PI3-K/Akt and Smad signaling during GRP-induced stimulation of neuroblastoma cell growth, suggesting that an imbalance between PI3-K/Akt and TGF-β/Smad signaling may be an important mechanism of tumorigenesis in neuroblastomas. It is important to further clarify this regulatory process (i.e., how tumor cells lose the balance between proliferation and apoptosis) in order to better identify potential molecular targets for novel therapy.
Acknowledgments
The authors thank Karen Martin for manuscript preparation. This work was supported by grants RO1 DK61470, RO1 DK48498, RO1 CA104748 and PO1 DK35608 from the National Institutes of Health.
References
- 1.Maris JM, Matthay KK. Molecular biology of neuroblastoma. J Clin Oncol. 1999;17:2264–2279. doi: 10.1200/JCO.1999.17.7.2264. [DOI] [PubMed] [Google Scholar]
- 2.Kim S, Hu W, Kelly DR, Hellmich MR, Evers BM, Chung DH. Gastrin-releasing peptide is a growth factor for human neuroblastomas. Ann Surg. 2002;235:621–629. doi: 10.1097/00000658-200205000-00003. discussion 629–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aguayo, Miller Y, Boose D, Holley M, Portanova LB, Schuyler KD, Kane MA. Nonconstitutive expression of the gastrin-releasing peptide autocrine growth system in human small cell lung carcinoma NCI-H345 cells. Cell Growth Differ. 1996;7:563–572. [PubMed] [Google Scholar]
- 4.Hajri A, Balboni G, Koenig M, Garaud JC, Damge C. Gastrin-releasing peptide: in vivo and in vitro growth effects on an acinar pancreatic carcinoma. Cancer Res. 1992;52:3726–3732. [PubMed] [Google Scholar]
- 5.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 6.Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL, Franklin RA, McCubrey JA. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003;17:590–603. doi: 10.1038/sj.leu.2402824. [DOI] [PubMed] [Google Scholar]
- 7.Qiao J, Kang J, Cree J, Evers BM, Chung DH. Gastrin-releasing peptide-induced down-regulation of tumor suppressor protein PTEN (phosphatase and tensin homolog deleted on chromosome ten) in neuroblastomas. Ann Surg. 2005;241:684–691. doi: 10.1097/01.sla.0000161173.47717.71. discussion 691–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Caestecker MP, Piek E, Roberts AB. Role of transforming growth factor-beta signaling in cancer. J Natl Cancer Inst. 2000;92:1388–1402. doi: 10.1093/jnci/92.17.1388. [DOI] [PubMed] [Google Scholar]
- 9.Massague J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000;19:1745–1754. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science. 2002;296:1646–1647. doi: 10.1126/science.1071809. [DOI] [PubMed] [Google Scholar]
- 11.Hall MC, Young DA, Waters JG, Rowan AD, Chantry A, Edwards DR, Clark IM. The comparative role of activator protein 1 and Smad factors in the regulation of Timp-1 and MMP-1 gene expression by transforming growth factor-beta 1. J Biol Chem. 2003;278:10304–10313. doi: 10.1074/jbc.M212334200. [DOI] [PubMed] [Google Scholar]
- 12.Miyazono K, ten Dijke P, Heldin CH. TGF-beta signaling by Smad proteins. Adv Immunol. 2000;75:115–157. doi: 10.1016/s0065-2776(00)75003-6. [DOI] [PubMed] [Google Scholar]
- 13.Inman GJ, Hill CS. Stoichiometry of active smad-transcription factor complexes on DNA. J Biol Chem. 2002;277:51008–51016. doi: 10.1074/jbc.M208532200. [DOI] [PubMed] [Google Scholar]
- 14.Wu L, Wu Y, Gathings B, Wan M, Li X, Grizzle W, Liu Z, Lu C, Mao Z, Cao X. Smad4 as a transcription corepressor for estrogen receptor alpha. J Biol Chem. 2003;278:15192–15200. doi: 10.1074/jbc.M212332200. [DOI] [PubMed] [Google Scholar]
- 15.Attisano L, Wrana JL. Smads as transcriptional co-modulators. Curr Opin Cell Biol. 2000;12:235–243. doi: 10.1016/s0955-0674(99)00081-2. [DOI] [PubMed] [Google Scholar]
- 16.Wotton D, Massague J. Smad transcriptional corepressors in TGF beta family signaling. Curr Top Microbiol Immunol. 2001;254:145–164. [PubMed] [Google Scholar]
- 17.Zhang Y, Derynck R. Regulation of Smad signalling by protein associations and signalling crosstalk. Trends Cell Biol. 1999;9:274–279. doi: 10.1016/s0962-8924(99)01579-2. [DOI] [PubMed] [Google Scholar]
- 18.Piek E, Roberts AB. Suppressor and oncogenic roles of transforming growth factor-beta and its signaling pathways in tumorigenesis. Adv Cancer Res. 2001;83:1–54. doi: 10.1016/s0065-230x(01)83001-3. [DOI] [PubMed] [Google Scholar]
- 19.Jonson T, Albrechtsson E, Axelson J, Heidenblad M, Gorunova L, Johansson B, Hoglund M. Altered expression of TGFB receptors and mitogenic effects of TGFB in pancreatic carcinomas. Int J Oncol. 2001;19:71–81. [PubMed] [Google Scholar]
- 20.Jonson T, Gorunova L, Dawiskiba S, Andren-Sandberg A, Stenman G, ten Dijke P, Johansson B, Hoglund M. Molecular analyses of the 15q and 18q SMAD genes in pancreatic cancer. Genes Chromosomes Cancer. 1999;24:62–71. doi: 10.1002/(sici)1098-2264(199901)24:1<62::aid-gcc9>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 21.Ten Dijke P, Goumans MJ, Itoh F, Itoh S. Regulation of cell proliferation by Smad proteins. J Cell Physiol. 2002;191:1–16. doi: 10.1002/jcp.10066. [DOI] [PubMed] [Google Scholar]
- 22.He C, Chen X. Transcription regulation of the vegf gene by the BMP/Smad pathway in the angioblast of zebrafish embryos. Biochem Biophys Res Commun. 2005;329:324–330. doi: 10.1016/j.bbrc.2005.01.133. [DOI] [PubMed] [Google Scholar]
- 23.Vanhaesebroeck B, Waterfield MD. Signaling by distinct classes of phosphoinositide 3-kinases. Exp Cell Res. 1999;253:239–254. doi: 10.1006/excr.1999.4701. [DOI] [PubMed] [Google Scholar]
- 24.Sumitomo M, Milowsky MI, Shen R, Navarro D, Dai J, Asano T, Hayakawa M, Nanus DM. Neutral endopeptidase inhibits neuropeptide-mediated transactivation of the insulin-like growth factor receptor-Akt cell survival pathway. Cancer Res. 2001;61:3294–3298. [PubMed] [Google Scholar]
- 25.Derynck R, Zhang Y, Feng XH. Smads: transcriptional activators of TGF-beta responses. Cell. 1998;95:737–740. doi: 10.1016/s0092-8674(00)81696-7. [DOI] [PubMed] [Google Scholar]
- 26.Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. 2000;275:36803–36810. doi: 10.1074/jbc.M005912200. [DOI] [PubMed] [Google Scholar]
- 27.Song K, Cornelius SC, Reiss M, Danielpour D. Insulin-like growth factor-I inhibits transcriptional responses of transforming growth factor-beta by phosphatidylinositol 3-kinase/Akt-dependent suppression of the activation of Smad3 but not Smad2. J Biol Chem. 2003;278:38342–38351. doi: 10.1074/jbc.M304583200. [DOI] [PubMed] [Google Scholar]
- 28.Remy I, Montmarquette A, Michnick SW. PKB/Akt modulates TGF-beta signalling through a direct interaction with Smad3. Nat Cell Biol. 2004;6:358–365. doi: 10.1038/ncb1113. [DOI] [PubMed] [Google Scholar]
- 29.Bhowmick NA, Zent R, Ghiassi M, McDonnell M, Moses HL. Integrin beta 1 signaling is necessary for transforming growth factor-beta activation of p38MAPK and epithelial plasticity. J Biol Chem. 2001;276:4670746713. doi: 10.1074/jbc.M106176200. [DOI] [PubMed] [Google Scholar]
- 30.Mulder KM. Role of Ras and Mapks in TGFbeta signaling. Cytokine Growth Factor Rev. 2000;11:23–35. doi: 10.1016/s1359-6101(99)00026-x. [DOI] [PubMed] [Google Scholar]
- 31.Xiao YQ, Malcolm K, Worthen GS, Gardai S, Schiemann WP, Fadok VA, Bratton DL, Henson PM. Cross-talk between ERK and p38 MAPK mediates selective suppression of pro-inflammatory cytokines by transforming growth factor-beta. J Biol Chem. 2002;277:14884–14893. doi: 10.1074/jbc.M111718200. [DOI] [PubMed] [Google Scholar]