

Abstract
Multiple sclerosis (MS) is characterized by axonal demyelination and neurodegeneration, the latter having been inadequately explored in the MS animal model experimental autoimmune encephalomyelitis (EAE). The purpose of this study was to examine the time-dependent correlation between increased calpain and caspase activities and neurodegeneration in spinal cord tissues from Lewis rats with acute EAE. An increase in TUNEL-positive neurons and internucleosomal DNA fragmentation in EAE spinal cords suggested that neuronal death was a result of apoptosis on days 8–10 following induction of EAE. Increases in calpain expression in EAE correlated with activation of pro-apoptotic proteases, leading to apoptotic cell death beginning on day 8 of EAE, which occurred before the appearance of visible clinical symptoms. Increases in calcineurin expression and decreases in phospho-Bad (p-Bad) suggested Bad activation in apoptosis during acute EAE. Increases in the Bax:Bcl-2 ratio and activation of caspase-9 showed the involvement of mitochondria in apoptosis. Further, caspase-8 activation suggested induction of the death receptor–mediated pathway for apoptosis. Endoplasmic reticulum stress leading to caspase-3 activation was also observed, indicating that multiple apoptotic pathways were activated following EAE induction. In contrast, cell death was mostly a result of necrosis on the later day (day 11), when EAE entered a severe stage. From these findings, we conclude that increases in calpain and caspase activities play crucial roles in neuronal apoptosis during the development of acute EAE.
Keywords: acute EAE, calpain, neuronal apoptosis, spinal cord
Multiple sclerosis (MS) is a T-cell-mediated autoimmune demyelinating disease of the central nervous system (CNS) with neurodegenerative components including axonal damage and neuronal and glial cell death. MS is the major disabling disease of young individuals in the United States (Martin and McFarland, 1995; Bauer et al., 1999) and can lead to fatigue, dizziness, impaired vision, pain, and paralysis of one or more limbs (Keegan and Noseworthy, 2002). Despite numerous studies, the etiology of MS remains unknown. Experimental autoimmune encephalomyelitis (EAE), an animal model of MS, has been extensively used to study the pathogenesis of MS and its treatment options (Bechtold et al., 2004). In MS, an abnormal immune response to myelin antigens causes mononuclear cell infiltration, demyelination, oligodendrocyte loss, and axonal degeneration. Currently, the consensus on the pathogenesis of MS is that autoreactive T cells activated in the periphery (Mokhtarian et al., 1994) cross the blood–brain barrier (BBB) where they encounter the CNS antigen presented to them by antigen-presenting cells (APCs; Sospedra and Martin, 2005). This process causes secondary influx of additional lymphocytes and blood-borne macrophages to the inflammatory site. The inflammatory cells and resident glial cells produce cytokines and proteases, including calpain, that individually or together contribute to the destruction of myelin and eventually cause damage to axons (Benveniste, 1997). In fact, increased calpain expression and activity in the spinal cord and optic nerve of Lewis rats with an acute form of EAE (Guyton et al., 2005a, 2005b) and in MS plaques (Shields et al., 1999a, 1999b) have been previously demonstrated. Up-regulation of calpain and caspase activity has also been implicated in apoptotic cell death in other neurodegenerative disorders, including spinal cord injury (Ray et al., 1999) and ischemia (Yamashima, 2000) and in the aging brain (Sloane et al., 2003; Hinman et al., 2004). From these and other findings, increases in calpain expression and activity have been suggested to play a crucial role in axonal damage and neuronal death in the spinal cord with the onset of clinical symptoms in EAE rats (Guyton et al., 2005a).
Although neuronal apoptosis during EAE has been previously demonstrated (Guyton et al., 2005a), the mechanisms involved in this cell death process have not been clearly elucidated. To this end, several intracellular signal transduction cascades involving proteases are thought to contribute early in this process. Elevated calpain expression and activity in the white matter of MS patients (Shields et al., 1999b) and in inflammatory cells in EAE (Shields and Banik, 1998) have implicated an important role for calpain in the pathogenesis of demyelinating disease. Although calpain inhibition has been shown to prevent cell death and reduce degradation of myelin protein in vitro (Deshpande et al., 1995; Ray et al., 1999; Das et al., 2005), it is equally important to examine the role of other proteases in the apoptosis of neurons and glial cells and axonal damage. Caspases, a family of cysteine proteases mainly involved in apoptotic pathways, are essential for the control of cell populations not only during development but also in pathology. Caspase-3, one member of the caspase family, appears to be the main effector caspase involved in neuronal apoptosis, and activation of caspase-3 has been shown to be a critical event in neuronal apoptosis; however, other caspases and signaling proteins such as calcineurin, Bax, and poly (ADP-ribose) polymerase-1 (PARP-1) may also be important.
Thus, the goal of this investigation was to characterize time-dependent alterations in apoptotic proteins as they correlate with neuron death following EAE induction in Lewis rats. Our results indicated that increases in calpain and caspase activities contributed to neuronal apoptosis with the onset EAE.
Materials and Methods
EAE Induction
Adult male Lewis rats (180–200 g) were purchased from Charles River Breeding Laboratories (Wilmington, MA), provided with water and food ad libitum, and maintained and used in the proposed experiments in accordance with the Laboratory Welfare Act and the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (Bethesda, MD). To induce EAE, rats were immunized subcutaneously with a 0.2-mL emulsion (1:1) of complete Freund's adjuvant (CFA) containing Mycobacterium tuberculosis H37Ra (10 mg/mL; Difco, Detroit, MI) and phosphate-buffered saline (PBS) containing guinea pig spinal cord (GPSC) homogenate (200 mg/mL) and myelin basic protein (MBP; 200 μg/mL). Control animals received PBS/CFA without GPSC/MBP. Two hours later, all rats received an intraperitoneal injection of Pertussis toxin (1.25 μg/rat). After induction of EAE, the rats were monitored daily for weight loss and appearance of clinical symptoms (usually occurring 8–11 days postinoculation). The clinical scores for severity of EAE symptoms were based on the following grades: 0, no change; 1, limp tail; 2, hind limb weakness with difficulty in righting; 3, one limb plegic; 4, two limbs plegic; 5, quadriplegic or moribund.
Isolation and Sectioning of Spinal Cord
Animals were sacrificed under anesthesia (ketamine, 95 mg/kg; xylazine, 5 mg/kg) on days 7, 8, 9, and 11. Lumbar spinal cord regions were surgically removed. Tissue samples were dissected on ice and immediately immersed in tissue-freezing medium (Histo Prep; Fisher Scientific, Fair Lawn, NJ) for immunofluorescent studies or fresh-frozen for Western blot analyses. Tissue samples were stored at −70°C until the experiments were performed.
Combined TUNEL and Double Immunofluorescent Labeling
Before processing, the samples were warmed to −16°C, and thin sections (5 μm) were cut using a Reichart-Jung cryostat (Cryocut 1800, Leica, Wetzlar, Germany). Sections were fixed at room temperature first in 95% ethanol for 10 min and then in 4% methanol-free formaldehyde (Polysciences, Warrington, PA) for 15 min and finally were stored in PBS (pH 7.4). The slides were saturated for 5 min with terminal deoxynucleotidyl transferase (TdT) buffer (50 μL/slide; Promega, Madison, WI) and then replaced with the TUNEL reaction mixture (50 μL/slide) containing digoxigenin (DIG) labeling mixture with DIG-11-dUTP (2.5 μL; Roche Diagnostics, Mannheim, Germany) and TdT (25 U; Promega) in TdT buffer. The slides were covered with Hybrislip hybridization coverslips (Research Products Int., Mount Prospect, IL) and incubated at 37°C in an OmniSlide thermal cycler (Hybaid Int.) for 1 hr. The TUNEL reaction was terminated by submersing the slides in a NaCl and sodium citrate solution (SSC; Promega). The slides were then washed three times with PBS to remove any unincorporated DIG-11-dUTP. The tissue was blocked for 1 hr with blocking buffer containing 2% horse serum and 2% sheep serum in PBS. Then the sections were incubated for 1 hr in blocking buffer with mouse anti-NeuN antibody (1:100). The slides were washed twice with PBS for 5 min each and then incubated for 30 min in the dark with sheep anti-DIG conjugated to FITC (1:100; Roche) and horse antimouse IgG conjugated to Texas red (1:100). The slides were washed twice with PBS and once with double-distilled water and then mounted with 1 drop of Vectashield Mounting Medium (Vector Laboratories) and coverslip. The slides were viewed under a fluorescence microscope at 400× magnification.
Analysis of DNA Fragmentation
Genomic DNA fragmentation was analyzed by agarose gel electrophoresis of DNA isolated from spinal cord tissue, as reported previously. (Ray et al., 2000). Briefly, rat spinal cord segments were homogenized in a buffer [10 mM Tris-HCl (pH 8.0), 150 mM NaCl, 50 mM EDTA] using a battery-operated homogenizer (Kontes Instruments, Vineland, NJ). The homogenates were digested in a solution (10 mM Tris-HCl, pH 8.0, 50 mM NaCl, 10 mM EDTA, 0.5% SDS, 250 ng/μL proteinase K) at 37°C for 24 hr. The digests were extracted twice with a 1:1 (v/v) mixture of phenol and chloroform and once with chloroform alone. Total genomic DNA was precipitated, dried in air, and dissolved in TE buffer [10 mM Tris-HCl (pH 8.0), 1 mM EDTA containing RNase A (50 ng/mL)] for 1 hr at 37°C. Equal amounts of the samples were loaded onto 1.6% agarose gels and electrophoresed in a TAE [40 mM Tris-acetate (pH 8.3), 1 mM EDTA] buffer. Gels were stained with ethidium bromide (1 μg/mL) and photographed on a UV (303 nm) transilluminator using Alpha Innotech (San Leandro, CA).
Protein Extraction and Western Blot Analysis
Western blotting was performed as we described previously (Das et al., 2006). The blots were washed three times (10 min each), and protein bands were detected by alkaline horseradish peroxidase (HRP)-catalyzed oxidation of luminol in the presence of H2O2 using an enhanced chemiluminescence (ECL) system (Amersham Life Science, Buckinghamshire, UK). The blots were exposed immediately to X-OMAT XAR-2 film (Eastman Kodak Co., Rochester, NY) for autoradiography. The ECL autoradiograms were scanned on a PowerLook scanner (UMAX, Fremont, CA), then imaged and digitized using Adobe Photoshop (Adobe Systems, San Jose, CA). Bands were quantitated using the NIH Image software.
Antibodies
Monoclonal antibody against α-spectrin (Affiniti, Exeter, UK) was used to measure calpain as well as caspase-3 activity. Rabbit polyclonal 80-kDa calpain and 76-kDa calpain antibodies, which were raised in our laboratory (Guyton et al., 2005b), were used to determine calpain expression and calpain activation, respectively. Monoclonal antibody against α-spectrin (Affiniti, Exeter, United Kingdom) was used to measure the activity of calpain as well as of caspase-3. Bax and Bcl-2 monoclonal antibodies (Santa Cruz Biotech, Santa Cruz, CA) were used to assess apoptotic threshold by determining the Bax:Bcl-2 ratio. All other antibodies were purchased either from Calbiochem (San Diego, CA) or Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal antibody against β-actin (clone AC-15, Sigma Chemical Co., St. Louis, MO) was used to standardize protein loading in SDS-PAGE. The secondary antibody used was HRP–conjugated goat antimouse IgG (ICN Biomedicals, Aurora, OH) except for calpain and α-spectrin, for which we used HRP-conjugated goat antirabbit IgG (ICN Biomedicals, Aurora, OH).
Statistical Analysis
Data from Western blots were analyzed for detection of significant differences between two groups by one-way analysis of variance followed by Fisher's post hoc tests using Statview software (Abacus Concepts, Berkeley, CA). Results were expressed as the mean ± the standard error of the mean (SEM) of three or more independent experiments. Differences from the control were considered significant at P ≤ 0.05.
Results
Time-Dependent Increases in Neuron-Specific Cell Death and Apoptosis in Spinal Cord of EAE Rats
One goal of the study was to determine when neuron cell death occurred in acute EAE following challenge with myelin antigens. We determined the occurrence of neuronal apoptosis with internucleosomal DNA fragmentation during the development of acute EAE in Lewis rats (Fig. 1). The studies showed a substantial increase in neuron death 11 days after challenge (grades 3–4 EAE) compared with day 9 EAE, when few cells were costained with TUNEL and NeuN (Fig. 1A). These data indicated that neuronal death occurred during the late phase of disease progression. Because neuronal death was increased in EAE spinal cord (Fig. 1A), we expected that many of these were dying via apoptotic pathways. Therefore, to confirm the occurrence of apoptotic death, we examined formation of internucleosomal DNA fragmentation, a hallmark of apoptosis, in control and EAE spinal cord samples that were collected on different days (Fig. 1B). Like the control, no DNA laddering was found in day 7 EAE spinal control. In contrast, there was increased DNA laddering in EAE spinal cord from days 8 to 11 (Fig. 1B), compared with the control. We found that apoptosis started on day 8 in EAE spinal cord (Fig. 1B), before the appearance of clinical symptoms. Our results showed a dramatic increase in internucleosomal DNA fragmentation on day 10 (grade 4 EAE; Fig 1B). In addition, a substantial number of cells were found to be necrotic on day 11 (grades 3–4), as evidenced by massive random DNA fragmentation (Fig. 1B).
Fig. 1.
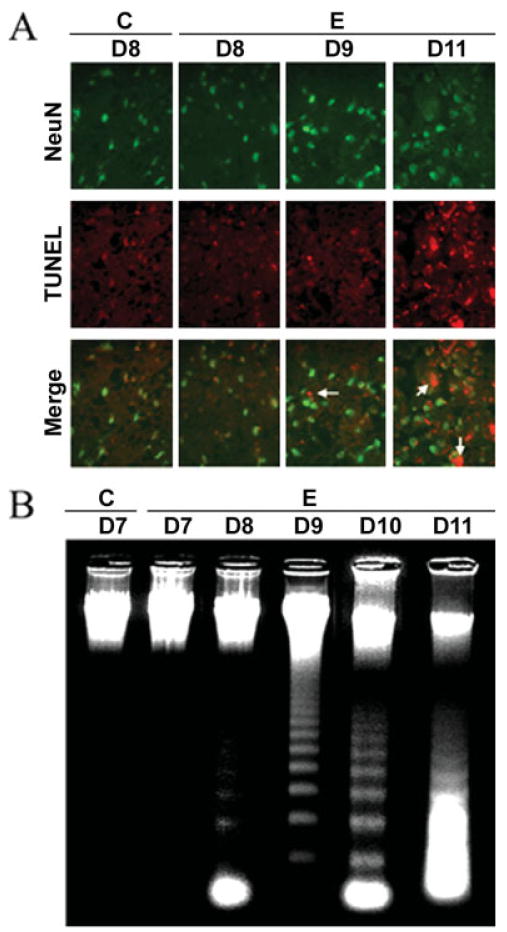
Neuronal death and DNA fragmentation in acute EAE with time. A: Immunofluorescence staining of neuron-specific cell death on days 8–11 post–EAE induction using the antibodies to NeuN (red), a neuron marker, and TUNEL staining (green), which identified cell death with anti-DIG antibody. Arrows indicate TUNEL-positive neurons (400× magnification). B: Agarose gel electrophoresis of genomic DNA for detection of DNA fragmentation (C, control; E, EAE; D7–D11, days 7–11).
Increased Calpain Expression and Activity during Acute EAE
Previous studies have demonstrated calpain expression to be up-regulated in both glial and infiltrating immune cells in the spinal cord of Lewis rats with EAE after the onset of characteristic clinical symptoms of disease (Schaecher et al., 2002). In the present study, calpain expression and activity were examined via Western blot analysis of control and EAE spinal cord tissues (Fig. 2). Calpain expression was examined using antibodies against 80-kDa inactive calpain (Fig. 2A) and 76-kDa active calpain (Fig. 2B), and calpain activity was measured by determining formation of 145-kDa Spectrin Break Down Product (SBDP; Nath et al., 1996, Wang et al., 1998a) in lumbar spinal cord tissues from control and EAE animals (grades 2–4) on different days following challenge (Fig. 2C). Calpain expression and activity were significantly (P ≤ 0.05) increased with the onset and severity of disease. On day 8 post–EAE induction, very little calpain expression and activity were seen in control and EAE tissue. In contrast, significant increases in 80-kDa inactive calpain and 76-kDa active calpain expression were observed on days 9–11 (P ≤ 0.05) in EAE spinal cord at or after onset of disease (Fig. 2A,B). There was also a gradual loss of calpastatin, the endogenous inhibitor of calpain, at the onset of disease on day 8, but this loss was greater with the severity of disease on days 9–11 compared with that in the control (Fig. 2D).
Fig. 2.
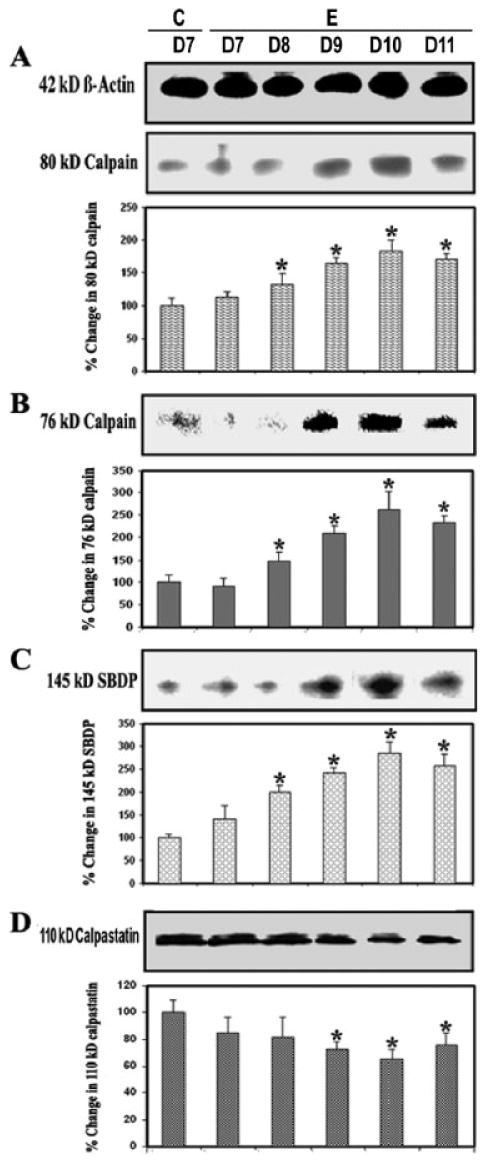
Calpain and calpastatin levels during EAE. Time-dependent changes in the expression of 80-kDa (A) and 76-kDa (B) calpain, 145-kDa SBDP (C), and 110-kDa calpastatin (D) during acute EAE. Upper panels (A–D) show representative Western blot, and lower panels show the corresponding bar graphs (n ≥ 3 animals per group). β-Actin was used as a control for equal protein loading. Data are presented as percent increase compared with the control set at 100% (C, control; E, EAE; D7–D11, days 7–11).
Up-regulated Calcineurin and Dephosphorylated Bad during Acute EAE
Calcineurin is a Ca2+/calmodulin-dependent protein phosphatase that is highly expressed in the CNS. Calcineurin is cleaved by calpain to form the activated 60-kDa calcineurin A subunit, which contains a catalytic domain, the binding site for calmodulin, and a C-terminal autoinhibitory domain (Wang et al., 1999). On activation by calpain or other signaling proteins, calcineurin dephosphorylates and activates the proapoptotic protein Bad. Based on this knowledge, we surmised that calcineurin activation caused Bad dephosphorylation to contribute to apoptosis in EAE. Compared with the control, indeed, a significant increase (P ≤ 0.05) in the 60-kDa calcineurin subunit and a decrease in phospho-Bad (p-Bad) on day 10 (grade 4 EAE) were observed (Fig. 3), suggesting that up-regulation of calcineurin dephosphorylated Bad for apoptosis during acute EAE (Fig. 3A,B). Thus, these results confirmed a role for calcineurin in activating Bad, thereby linking calcineurin to the apoptosis machinery in EAE.
Fig. 3.
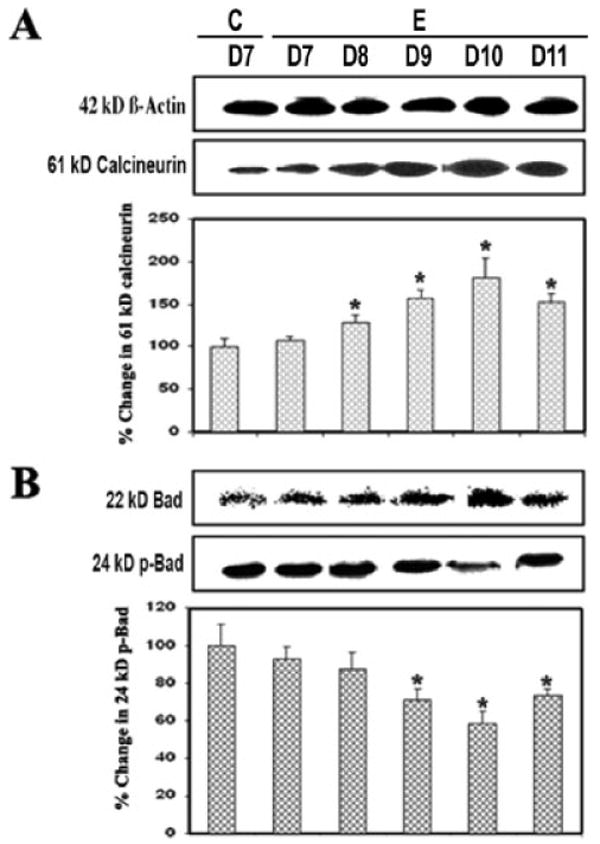
Up-regulated calcineurin and Bad dephosphorylation during EAE. Time-dependent changes in 61-kDa calcineurin (A), 22-kDa Bad, and 24-kDa p-Bad (B) expression during acute EAE. Upper panels (A–D) show representative Western blots and lower panels show the corresponding bar graphs (n ≥ 3 animals per group). β-Actin was used as a control for equal protein loading. Data are presented as percent increase compared with the control set at 100% (C, control; E, EAE; D7–D11, days 7–11).
Caspase-8 Activation and Proteolytic Cleavage of Bid in the Course of EAE
Caspase-8 plays a central role in the cytokine-mediated caspase cascade. Current in vivo observations indicated involvement of caspase-8 in cytokine-induced cell death in EAE (Fig. 4). There was a significantly (P = 0.009) greater level (1.5-fold to 2.5-fold) of caspase-8 cleaved (active) product (18-kDa fragment) generated in EAE with the onset of disease after 8 days to severe grade after 10–11 days, compared with the control (Fig. 4A). The present results showed a connection between extrinsic and intrinsic apoptotic pathways because activation of caspase-8 caused proteolytic cleavage of Bid to tBid (Fig. 4B). The level of 15-kDa tBid was also very significantly (P = 0.008) increased with the severity of disease on days 9–11, compared with the conrol (Fig 4B). Thus, the Bcl-2 family member Bid can induce apoptosis when it is activated following proteolytic cleavage by caspase-8 during induction of EAE.
Fig. 4.
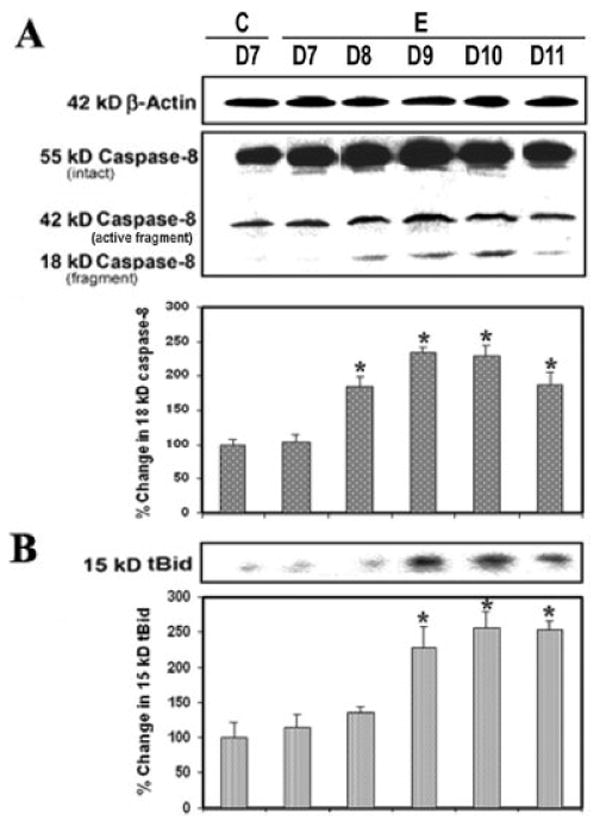
Caspase-8 activity during EAE. Time-dependent changes in 42-kDa and 18-kDa caspase-8 (A) and 15-kDa tBid (B) expression during acute EAE. Upper panels (A–D) show representative Western blots, and lower panels show the corresponding bar graphs (n ≥ 3 animals per group). β-Actin was used as a control for equal protein loading. Data are presented as percent increase compared with the control set at 100% (C, control; E, EAE;. D7–D11, day 7 to day 11).
Increase in Bax:Bcl-2 Ratio and Activation of Caspase-9 during Acute EAE
Proteins of the Bcl-2 family, including the proapoptotic protein Bax and the antiapoptotic protein Bcl-2, collectively regulate the predisposition of a cell to apoptosis through sequestration of critical activation factors or alterations of mitochondrial permeability (Das et al., 2007). We examined involvement of the mitochondrial pathway of apoptosis in acute EAE (Fig. 5). Our studies showed that Bax was significantly increased (P = 0.02) in EAE and that the elevation correlated with the development of disease (Fig. 5A). The observation of higher Bax and lower Bcl-2 expression among EAE animals suggested that mitochondrial pathway of apoptosis might play a predominant role in triggering cell death (Fig. 5A). There was a significant difference (P = 0.021) in the Bax:Bcl-2 ratio between lumbar spinal cord tissues from control and EAE (grades 2–4) on different days following challenge. Also, in mitochodria-dependent apoptosis, cytochrome c was released from mitochondria into cytosol, which could interact with Apaf-1, thereby triggering autoproteolysis of 46-kDa pro-caspase-9 to form the 37-kDa caspase-9 active fragment (Fig. 5B). A significantly (P = 0.029) greater level of caspase-9 activation was seen in the acute phase of EAE, reaching its peak level on days 9–10 post-challenge, compared with that in the control (Fig. 5B), suggesting the importance of mitochondrial regulation of the mechanism of apoptosis. Thus, the increase in the Bax:Bcl-2 ratio correlated with the increase in the caspase-9 active fragment in EAE (Fig. 5A,B).
Fig. 5.
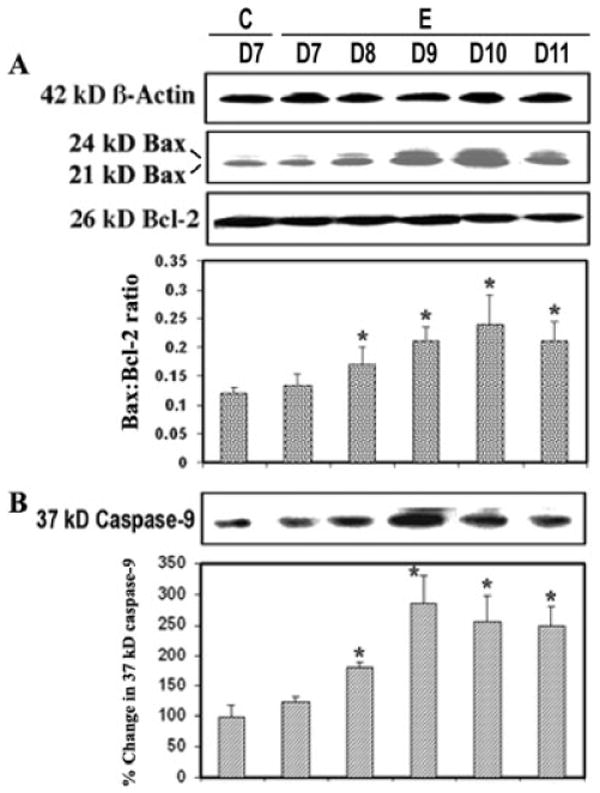
Apoptotic protein alteration during EAE. Time-dependent changes in 24-kDa and 21-kDa Bax, 26-kDa Bcl-2 (A) and 37-kDa caspase-9 (B) expression during acute EAE. Upper panels (A–D) show representative Western blots and lower panels show corresponding the bar graphs (n ≥ 3 animals per group). (β-Actin was used as a control for equal protein loading. Data are presented as percent increase compared with the control set at 100% (C, control; E, EAE; D7–D11, days 7–11).
Activation of Caspase-12 and Caspase-3 and PARP-1 Cleavage in the Course of EAE
Because both calpain and caspase-12 are activated by elevation of intracellular free [Ca2+] and both may play important roles in mediating neuronal degeneration, we examined the possible interaction between these two cysteine proteases. Our experiments demonstrated the degradation of the inactive 55-kDa pro-caspase-12 form to the 40-kDa caspase-12 active form during EAE development (Fig. 6). This activation could be carried out by the increased calpain activity in EAE (Fig. 2). The significantly increased (P = 0.011) production of the 40-kDa caspase-12 active fragment was evident at the onset of the severe stage of the disease (Fig. 6A). Also, Western blot analysis previously showed a significant decrease (P = 0.031) in calpastatin, the endogenous inhibitor of calpain (Fig. 2). Caspase-3 activation was assessed by examining the generation of the 20-kDa caspase-3 active fragment and the formation of the 120-kDa SBDP on the Western blots (Fig. 6B,C). The increased level of the 20-kDa caspase-3 active band was correlated with the severity of the disease, compared with the control (Fig. 6C). Activation of caspase-3 subsequently leads to apoptotic cell death through cleavage of a broad spectrum of central cellular target proteins in the cell nucleus, including PARP-1. Western blot analysis also showed an increase (1.5–3.5 times) in the formation of the 85-kDa PARP-1 fragment in acute EAE (Fig. 6D). These results correlated with endoplasmic reticulum stress and activation of calpain and caspase-3, suggesting the requirement of different levels of activity of these proteases for mediation of neuronal apoptosis in acute EAE.
Fig. 6.
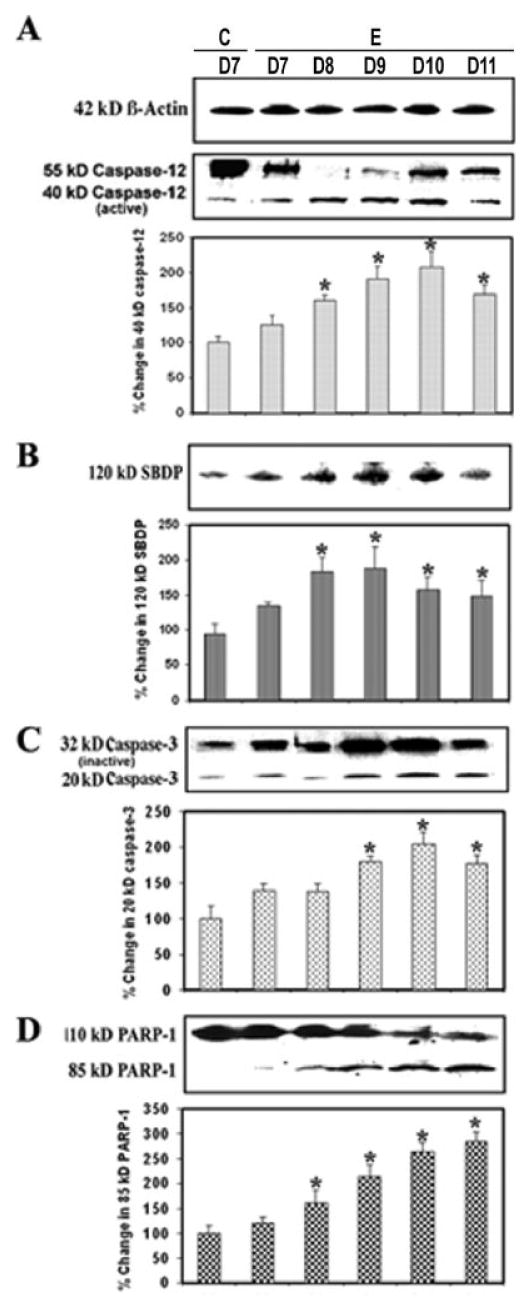
Involvement of endoplasmic reticulum and caspase-3 activity in apoptosis during EAE. Time-dependent changes in the 55- and 40-kDa caspase-12 (A), 120-kDa SBDP (B), 20-kDa caspase-3 (C), and 85-kDa PARP-1 (D) expression during acute EAE. Upper panels (A–D) show representative Western blots and lower panels show the corresponding bar graphs (n ≥ 3 animals per group). β-Actin was used as a control for equal protein loading. Data are presented as percent increase compared with the control set at 100% (C, control; E, EAE; D7–D11, days 7–11).
Discussion
The goal of this study was to determine whether time-dependent elevation of protease activity (calpain and caspases) was involved in apoptosis signaling and neurodegeneration during the development of acute EAE. The present investigation indicated that expression and activation of caspases in acute EAE spinal cord tissues were significantly up-regulated during days 9–11 post-EAE induction, which correlated with a significant increase in calpain expression, neuronal apoptosis, and axonal damage at these times (Shields et al., 1999a; Schaecher et al., 2002; Guyton et al., 2005a, 2005b; Hassen et al., 2006).
Calpain expression and activity were increased in splenic cells before the onset of the disease (Shields et al., 1999a), and increased activity was evident in infiltrating immune cells and glial cells of the spinal cord white matter and optic nerve (Shields and Banik, 1998) after the appearance of clinical symptoms. The current study demonstrated (Fig. 1) and confirmed our previous findings (Schaecher et al., 2002; Guyton et al., 2005b) that calpain expression was increased in EAE spinal cord in a time-dependent manner. This finding also suggested that calpain was involved in the neurodegenerative process in EAE and perhaps in MS. Although no significant number of TUNEL-positive neurons was observed on day 8 following challenge (Guyton et al., 2005b), increased DNA laddering, the hallmark of apoptosis, indicates occurrence of non-neuronal apoptosis in day 8 EAE spinal cord, suggesting that the apoptotic process was initiated early in the inflammatory phase in EAE development, even before the appearance of clinical symptoms. Apoptotic cell death was more evident on day 9 following challenge and was at a maximum on day 10 (Fig. 1). This finding also suggests that in the inflammatory phase, cells expressing the death mediators on day 8 are destined to die with progression of the disease. Thus, axons and neurons may be degenerated in the early phase of disease development. Such axonal degeneration has been found in EAE (Guyton, et al., 2005b, Kim et al., 2006; Budde et al., 2007) as well as in patients with early, chronic, and progressive phases of MS (Bitsch et al., 2000; Kornek et al., 2000; Kuhlmann et al., 2002). Although up-regulation of calpain occurred on day 9 post-EAE induction (Fig. 2), significant neuronal death did not occur until day 10, suggesting that calpain expression preceded neuronal loss in the acute model of EAE. Although DNA fragmentation was increased in EAE spinal cord on day 11 (grades 3–4), the severe phase of the disease, most of the cells went into the necrotic phase, as seen by random DNA fragmentation or DNA smearing. Nonetheless, these findings suggest that the neuronal death identified in EAE spinal cord was at least partially a result of an apoptotic mechanism. The current study also supports previous findings that greater neuronal death and axonal degeneration occur during the demyelinating phase of EAE and MS (Kornek et al., 2000; Guyton et al., 2005a).
This observation is supported by earlier investigations demonstrating the role of calpain in other neurodegenerative disorders, including ischemia, spinal cord injury, brain trauma, Alzheimer's disease, Parkinson's disease, and epilepsy (Ray and Banik, 2003). These results also suggested that calpain may be a potential therapeutic target for the amelioration of neurodegenerative diseases such as MS. Studies in our and other laboratories have demonstrated that treatment with calpain inhibitors decreased loss of neurofilament protein and calpain-specific spectrin breakdown and protected neurons and glial cells in spinal cord injury as well as in EAE, indicating the importance of calpain inhibition as a promising therapeutic option (Ray and Banik, 2003; Zhang et al., 2003; Guyton et al., 2005b; Hassen, et al., 2006).
In the development and progression of diseases, elevation of intracellular free [Ca2+] and calpain activation can induce apoptotic cell death through multiple mechanisms (Das et al., 2007). The finding that redistribution of Bad and apoptosis is suppressible by an inhibitory mutant of calcineurin suggests that calcineurin is a significant mediator of apoptotic death signals. Thus, the possibility that Bad activation by calcineurin activity might be involved in cell death in EAE was explored. The results suggested that calcineurin induced apoptosis by dephosphorylating Bad in EAE (Fig. 3). The interaction of calcineurin with Bad provides a Ca2+-mediated mechanism for regulating the phosphorylation state of Bad and thus making Bad a proapoptotic protein, which further confirms the participation of calcineurin in the apoptosis machinery.
Ischemic and hypoxic injuries to the nervous system have been shown to involve the release of cell death–inducing cytokines and the activation of death receptors. It is likely that these events involve caspase-8-mediated cleavage of the BH3-only protein Bid. This pathway provides evidence that activation of caspase-8 cleaves Bid to tBid. Once Bid is cleaved by caspase-8, tBid translocates from the cytosol to the mitochondria to cause mitochondrial damage, release of cytochrome c, increase in caspase activity nuclear condensation, and apoptotic cell shrinkage (Li et al., 1998; Das et al., 2006). In addition, tBid is able to activate Bax to form Bax multimers in the mitochondria, thereby inducing the release of cytochrome c. In the present study, caspase-8 activation correlated with Bid cleavage in acute EAE (Fig. 4), again demonstrating induction of cell death signals early in disease development.
Calpain has been reported to play several roles in activating factors that may be involved in apoptosis and has a variety of possible interactions with the Bcl-2 family of proteins. Calpain may activate proapoptotic Bax directly through cleavage of Bax (Choi et al., 2001). Thus, activation of Bax is an essential component of the cell death mechanism. An increased level of cleaved caspase-9 early in the apoptotic process in EAE was also demonstrated (Fig. 5). Active caspase-9 further processes other caspase members, including caspase-3, to initiate a caspase cascade, which ultimately leads to apoptosis. Inhibition of calpain has been shown to inhibit mitochondrial transition pore formation in renal cells (Gores et al., 1998). Similar mechanisms of neuronal death may also exist in EAE. Calpain interacts and modifies caspase pathways. Specifically, calpain has been shown to activate caspase-3 (Blomgren et al., 2001; Varghese et al., 2001) and caspase-12 (Nakagawa and Yuan, 2000). Caspase also has been shown to cleave calpastatin in vitro, indicating that caspase activity may also enhance calpain activity by removing calpastatin, the regulator of calpain (Wang et al., 1998b). However, the interactions between these two protease families are complex, and inhibition of calpain has also been shown to modulate caspase-3 activity (Lankiewicz et al., 2000; Neumar et al., 2003). Activation of caspase-3, the final executioner in apoptosis, which degrades the cellular target protein PARP-1, is elevated early in EAE (Fig. 6). Nevertheless, these results in particular indicate a pivotal cooperative role of calpain and caspases, generating crucial cell death signals early in disease development in EAE. This hypothesis is further supported by both in vitro and in vivo studies, in which calpain inhibition has been found to inhibit caspase-3 and to block glial and neuronal apoptosis (Das et al., 2005). For example, treatment with the calpain inhibitors E-64-d and calpeptin decreased apoptotic indicators, including internucleosomal DNA fragmentation and proapoptotic proteins in rat model of spinal cord injury (Ray et al., 1999).
The present study used the processing of pro-caspase-12 as an indirect biochemical marker for activation of the endoplasmic reticulum (ER)–mediated apoptotic pathway (Fig. 6). ER stress-induced apoptosis is dependent on this ER-resident protease (Nakagawa and Yuan, 2000), activation of which likely involves proteolytic processing and oligomeric assembly in a manner similar to all other caspases (Nakagawa and Yuan, 2000). The current study confirms that pro-caspase-12, which is enriched in ER, is processed by the family of cytosolic Ca2+-dependent cysteine proteases such as calpain during EAE (Fig. 6). These results support the concept that a rise in cytosolic free [Ca2+] and activation of calpain trigger caspase-12 activation and ER apoptotic signaling, although they do not exclude the possibility that other proteases may also be involved in activation of caspase-12 (Das et al., 2006). We propose a novel pathway of calpain-mediated caspase-12 activation induced by disturbance in intracellular Ca2+ homeostasis. These results suggest the involvement of ER stress in the apoptotic mechanism of neuronal death in EAE (Fig. 7).
Fig. 7.
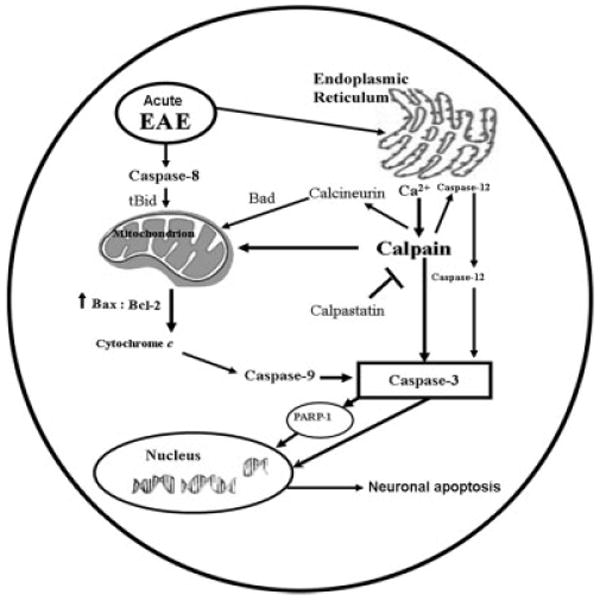
Schematic presentation of molecular mechanisms leading to activation of calpain and caspase cascades for mediation of neuronal apoptosis during acute EAE.
In conclusion, the finding of up-regulation of calpain and caspases on day 8 post-EAE induction suggests that calpain and caspase expression and activity play important and cooperative roles in apoptotic neuronal death signaling early in the development of acute EAE in Lewis rats. Later, in the severe phase of the disease, on day 11, cells die mostly as a result of necrosis. The results of this investigation also imply that inhibition of these proteases may reduce the inflammatory cascade because calpain is involved in T-cell activation (Schaecher et al., 2002; Imam et al., 2007). Also, the inhibition of these proteases may preserve and protect axons, neurons, and glia. Therefore, these proteases may be potential therapeutic targets for treatment of EAE and MS.
Acknowledgments
Contract grant sponsor: NINDS; Contract grant numbers: NS-45967, NS-57811; Contract grant sponsor: NCI; Contract grant number: CA-91460; Contract grant sponsor: NIH; Contract grant number: C06 RRO15455.
References
- Bauer J, Stadelmann C, Bancher C, Jellinger K, Lassmann H. Apoptosis of T lymphocytes in acute disseminated encephalomyelitis. Acta Neuropathol (Berl) 1999;97:543–546. doi: 10.1007/s004010051028. [DOI] [PubMed] [Google Scholar]
- Bechtold DA, Kapoor R, Smith KJ. Axonal protection using flecainide in experimental autoimmune encephalomyelitis. Ann Neurol. 2004;55:607–616. doi: 10.1002/ana.20045. [DOI] [PubMed] [Google Scholar]
- Benveniste EN. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J Mol Med. 1997;75:165–173. doi: 10.1007/s001090050101. [DOI] [PubMed] [Google Scholar]
- Bitsch A, Schuchardt J, Bunkowski S, Kuhlmann T, Brück W. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain. 2000;123:1174–1183. doi: 10.1093/brain/123.6.1174. [DOI] [PubMed] [Google Scholar]
- Blomgren K, Zhu C, Wang X, Karlsson JO, Leverin AL, Bahr BA, Mallard C, Hagberg H. Synergistic activation of caspase-3 by m-calpain after neonatal hypoxia-ischemia: a mechanism of “pathological apoptosis”? J Biol Chem. 2001;276:10191–10198. doi: 10.1074/jbc.M007807200. [DOI] [PubMed] [Google Scholar]
- Budde MD, Kim JH, Liang HF, Russell JH, Cross AH, Song SK. Axonal injury detected by in vivo diffusion tensor imaging correlates with neurological disability in a mouse model of multiple sclerosis. NMR Biomed. 2007 Epub ahead of print. [Google Scholar]
- Choi WS, Lee EH, Chung CW, Jung YK, Jin BK, Kim SU, Oh TH, Saido TC, Oh YJ. Cleavage of Bax is mediated by caspase-dependent or -independent calpain activation in dopaminergic neuronal cells: protective role of Bcl-2. J Neurochem. 2001;77:1531–1541. doi: 10.1046/j.1471-4159.2001.00368.x. [DOI] [PubMed] [Google Scholar]
- Das A, Banik NL, Ray SK. Differentiation decreased telomerase activity in rat glioblastoma C6 cells and increased sensitivity to IFN-γ and taxol for apoptosis. Neurochem Res. 2007;32:2167–2183. doi: 10.1007/s11064-007-9413-y. [DOI] [PubMed] [Google Scholar]
- Das A, Garner DP, Del Re AM, Woodward JJ, Kumar DM, Agarwal N, Banik NL, Ray SK. Calpeptin provides functional neuroprotection to rat retinal ganglion cells following Ca2+ influx. Brain Res. 2006;1084:146–157. doi: 10.1016/j.brainres.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Das A, Sribnick EA, Wingrave JM, Del Re AM, Woodward JJ, Appel SH, Banik NL, Ray SK. Calpain activation in apoptosis of ventral spinal cord 4.1 (VSC4.1) motoneurons exposed to glutamate: calpain inhibition provides functional neuroprotection. J Neurosci Res. 2005;81:551–562. doi: 10.1002/jnr.20581. [DOI] [PubMed] [Google Scholar]
- Deshpande RV, Goust JM, Hogan EL, Banik NL. Calpain secreted by activated human lymphoid cells degrades myelin. J Neurosci Res. 1995;42:259–265. doi: 10.1002/jnr.490420214. [DOI] [PubMed] [Google Scholar]
- Gores GJ, Miyoshi H, Botla R, Aguilar HI, Bronk SF. Induction of the mitochondrial permeability transition as a mechanism of liver injury during cholestasis: a potential role for mitochondrial proteases. Biochim Biophys Acta. 1998;1366:167–175. doi: 10.1016/s0005-2728(98)00111-x. [DOI] [PubMed] [Google Scholar]
- Guyton MK, Sribnick EA, Ray SK, Banik NL. A role for calpain in optic neuritis. Ann N Y Acad Sci. 2005a;1053:48–54. doi: 10.1196/annals.1344.005. [DOI] [PubMed] [Google Scholar]
- Guyton MK, Wingrave JM, Yallapragada AV, Wilford GG, Sribnick EA, Matzelle DD, Tyor WR, Ray SK, Banik NL. Upregulation of calpain correlates with increased neurodegeneration in acute experimental auto-immune encephalomyelitis. J Neurosci Res. 2005b;81:53–61. doi: 10.1002/jnr.20470. [DOI] [PubMed] [Google Scholar]
- Hassen GW, Feliberti J, Kesner L, Stracher A, Mokhtarian F. A novel calpain inhibitor for the treatment of acute experimental autoimmune encephalomyelitis. J Neuroimmunol. 2006;180:135–146. doi: 10.1016/j.jneuroim.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Hinman JD, Duce JA, Siman RA, Hollander W, Abraham CR. Activation of calpain-1 in myelin and microglia in the white matter of the aged Rhesus monkey. J Neurochem. 2004;89:430–441. doi: 10.1046/j.1471-4159.2004.02348.x. [DOI] [PubMed] [Google Scholar]
- Imam SA, Guyton MK, Haque A, Vandenbark A, Tyor WR, Ray SK, Banik NL. Increased calpain correlates with Th1 cytokine profile in PBMCs from MS patients. J Neuroimmunol. 2007;190:139–145. doi: 10.1016/j.jneuroim.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan BM, Noseworthy JH. Multiple sclerosis. Annu Rev Med. 2002;53:285–302. doi: 10.1146/annurev.med.53.082901.103909. [DOI] [PubMed] [Google Scholar]
- Kim JH, Budde MD, Liang HF, Klein RS, Russell JH, Cross AH, Song SK. Detecting axon damage in spinal cord from a mouse model of multiple sclerosis. Neurobiol Dis. 2006;21:626–632. doi: 10.1016/j.nbd.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Kornek B, Storch MK, Weissert R, Wallstroem E, Stefferl A, Olsson T, Linington C, Schmidbauer M, Lassmann H. Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am J Pathol. 2000;157:267–276. doi: 10.1016/S0002-9440(10)64537-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlmann T, Lingfeld G, Bitsch A, Schuchardt J, Brück W. Acute axonal damage in multiple sclerosis is most extensive in early disease stages and decreases over time. Brain. 2002;125:2202–2212. doi: 10.1093/brain/awf235. [DOI] [PubMed] [Google Scholar]
- Lankiewicz S, Marc Luetjens C, Truc Bui N, Krohn AJ, Poppe M, Cole GM, Saido TC, Prehn JH. Activation of calpain I converts excitotoxic neuron death into a caspase-independent cell death. J Biol Chem. 2000;275:17064–17071. doi: 10.1074/jbc.275.22.17064. [DOI] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase-8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Martin R, McFarland HF. Immunological aspects of experimental allergic encephalomyelitis and multiple sclerosis. Crit Rev Clin Lab Sci. 1995;32:121–182. doi: 10.3109/10408369509084683. [DOI] [PubMed] [Google Scholar]
- Mokhtarian F, Shi Y, Shirazian D, Morgante L, Miller A, Grob D. Defective production of anti-inflammatory cytokine, TGF-beta by T cell lines of patients with active multiple sclerosis. J Immunol. 1994;152:6003–6010. [PubMed] [Google Scholar]
- Nakagawa T, Yuan J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol. 2000;150:887–894. doi: 10.1083/jcb.150.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath R, Raser KJ, Stafford D, Hajimohammadreza I, Posner A, Allen H, Talanian RV, Yuen P, Gilbertsen RB, Wang KK. Non-erythroid α-spectrin breakdown by calpain and interleukin-1β-converting-enzyme-like protease(s) in apoptotic cells: contributory roles of both protease families in neuronal apoptosis. Biochem J. 1996;319:683–690. doi: 10.1042/bj3190683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumar RW, Xu YA, Gada H, Guttmann RP, Siman R. Cross-talk between calpain and caspase proteolytic systems during neuronal apoptosis. J Biol Chem. 2003;278:14162–14167. doi: 10.1074/jbc.M212255200. [DOI] [PubMed] [Google Scholar]
- Ray SK, Matzelle DC, Wilford GG, Hogan EL, Banik NL. E-64-d prevents both calpain up-regulation and apoptosis in the lesion and penumbra following spinal cord injury in rats. Brain Res. 2000;867:80–89. doi: 10.1016/s0006-8993(00)02260-5. [DOI] [PubMed] [Google Scholar]
- Ray SK, Shields DC, Saido TC, Matzelle DC, Wilford GG, Hogan EL, Banik NL. Calpain activity and translational expression increased in spinal cord injury. Brain Res. 1999;816:375–380. doi: 10.1016/s0006-8993(98)01128-7. [DOI] [PubMed] [Google Scholar]
- Ray SK, Banik NL. Calpain and its involvement in the pathophysiology of CNS injuries and diseases: therapeutic potential of calpain inhibitors for prevention of neurodegeneration. Curr Drug Targets CNS Neurol Disord. 2003;2:173–189. doi: 10.2174/1568007033482887. [DOI] [PubMed] [Google Scholar]
- Schaecher K, Rocchini A, Dinkins J, Matzelle DD, Banik NL. Calpain expression and infiltration of activated T cells in experimental allergic encephalomyelitis over time: increased calpain activity begins with onset of disease. J Neuroimmunol. 2002;129:1–9. doi: 10.1016/s0165-5728(02)00142-x. [DOI] [PubMed] [Google Scholar]
- Shields DC, Banik NL. Upregulation of calpain activity and expression in experimental allergic encephalomyelitis: a putative role for calpain in demyelination. Brain Res. 1998;794:68–74. doi: 10.1016/s0006-8993(98)00193-0. [DOI] [PubMed] [Google Scholar]
- Shields DC, Banik NL. Pathophysiological role of calpain in experimental demyelination. J Neurosci Res. 1999;55:533–541. doi: 10.1002/(SICI)1097-4547(19990301)55:5<533::AID-JNR1>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Shields DC, Schaecher KE, Goust JM, Banik NL. Calpain activity and expression are increased in splenic inflammatory cells associated with experimental allergic encephalomyelitis. J Neuroimmunol. 1999a;99:1–12. doi: 10.1016/s0165-5728(99)00043-0. [DOI] [PubMed] [Google Scholar]
- Shields DC, Schaecher KE, Saido TC, Banik NL. A putative mechanism of demyelination in multiple sclerosis by a proteolytic enzyme, calpain. Proc Natl Acad Sci U S A. 1999b;96:11486–11491. doi: 10.1073/pnas.96.20.11486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloane JA, Hinman JD, Lubonia M, Hollander W, Abraham CR. Age-dependent myelin degeneration and proteolysis of oligodendrocyte proteins is associated with the activation of calpain-1 in the Rhesus monkey. J Neurochem. 2003;84:157–168. doi: 10.1046/j.1471-4159.2003.01541.x. [DOI] [PubMed] [Google Scholar]
- Sospedra M, Martin R. Antigen-specific therapies in multiple sclerosis. Int Rev Immunol. 2005;24:393–413. doi: 10.1080/08830180500371256. [DOI] [PubMed] [Google Scholar]
- Varghese J, Radhika G, Sarin A. The role of calpain in caspase activation during etoposide induced apoptosis in T cells. Eur J Immunol. 2001;31:2035–2041. doi: 10.1002/1521-4141(200107)31:7<2035::aid-immu2035>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC. Ca2+-induced apoptosis through calcineurin dephosphorylation of Bad. Science. 1999;284:339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- Wang KK, Posmantur R, Nath R, McGinnis K, Whitton M, Talanian RV, Glantz SB, Morrow JS. Simultaneous degradation of αII- and βII-spectrin by caspase-3 (CPP32) in apoptotic cells. J Biol Chem. 1998a;273:22490–22497. doi: 10.1074/jbc.273.35.22490. [DOI] [PubMed] [Google Scholar]
- Wang KK, Posmantur R, Nadimpalli R, Nath R, Mohan P, Nixon RA, Talanian RV, Keegan M, Herzog L, Allen H. Caspase-mediated fragmentation of calpain inhibitor protein calpastatin during apoptosis. Arch Biochem Biophys. 1998b;356:187–196. doi: 10.1006/abbi.1998.0748. [DOI] [PubMed] [Google Scholar]
- Yamashima T. Implication of cysteine proteases calpain, cathepsin and caspase in ischemic neuronal death of primates. Prog Neurobiol. 2000;62:273–295. doi: 10.1016/s0301-0082(00)00006-x. [DOI] [PubMed] [Google Scholar]
- Zhang SX, Bondada V, Geddes JW. Evaluation of conditions for calpain inhibition in the rat spinal cord: effective postinjury inhibition with intraspinal MDL28170 microinjection. J Neurotrauma. 2003;20:59–67. doi: 10.1089/08977150360517182. [DOI] [PubMed] [Google Scholar]