

Abstract
The solution structure of the three-heme electron transfer protein cytochrome c7 from Desulfuromonas acetoxidans is reported. The determination of the structure is obtained through NMR spectroscopy on the fully oxidized, paramagnetic form. The richness of structural motifs and the presence of three prosthetic groups in a protein of 68 residues is discussed in comparison with the four-heme cytochromes c3 already characterized through x-ray crystallography. In particular, the orientation of the three hemes present in cytochrome c7 is similar to that of three out of four hemes of cytochromes c3. The reduction potentials of the individual hemes, which have been obtained through the sequence-specific assignment of the heme resonances, are discussed with respect to the properties of the protein matrix. This information is relevant for any attempt to understand the electron transfer pathway.
Multiheme cytochromes with bis-histidine coordination are classified as class III c-type cytochromes (1). This class includes triheme, tetraheme, and octaheme cytochromes (2). They show no structural similarity with the cytochromes c (Cyt c) of other classes and are associated with the most divergent respiratory mode in sulfur or sulfate-reducing bacteria. These bacteria are able to utilize elemental sulfur or oxidized sulfur compounds as terminal electron acceptors. Their respiratory processes are exceptional in terms of the negative redox potentials involved and in the implication of the existence of several multiheme Cyt c.
Among the bacteria able to reduce sulfate to sulfide, the most characterized are the Desulfovibrio. They are ecologically versatile. In the absence of sulfate they can grow by fermentation of lactate, pyruvate, or ethanol in syntrophic association with a methanogen to utilize the hydrogen released.
The Desulfuromonas bacteria cannot reduce sulfate but obligately require sulfur as respiratory electron acceptor, using acetate as oxidizable substrate. The oxidation of acetate to CO2 is stoichiometrically linked to the reduction of elemental sulfur to sulfide. Proteins involved in this process include cytochromes and ferredoxins, the former in the periplasm while the latter are in the cytoplasm. At variance with Desulfovibrio, no hydrogenase in the periplasm of Desulfuromonas bacteria has yet been detected.
The best-studied proteins among multiheme cytochromes are Cyt c3 (13,000 kDa) found in Desulfovibrio bacteria. They contain four hemes and act as the natural electron donors and acceptors with hydrogenase and ferredoxins. The determination of a few three-dimensional structures of Cyt c3 from different sources has shown that the arrangement of the four hemes is highly conserved (3–7). Each heme exhibits a distinct redox potential in the −200- to −400-mV range.
Cyt c-555.1, also known as Cyt c7, from Desulfuromonas acetoxidans is a small (68 amino acids, ≈9,100 kDa) monomeric class III c-type cytochrome containing three hemes. Its sequence is somewhat homologous to many tetraheme Cyt c3 from Desulfovibrio organisms (8, 9). The sequence of Cyt c7 from D. acetoxidans has been aligned with those of some Cyt c3 (9). It was assumed that Cyt c7 has a deletion in the protein segment binding heme II (residues 44–67); with this assumption the sequence homology is about 38% (10). Up to now no explanation has been proposed for the reasons why one heme is missing in Cyt c7 with respect to Cyt c3. In Cyt c7, two of the heme groups have a reduction potential of −177 mV and the third of −102 mV (11).
Cyt c7 appears to be involved in the electron transfer to elemental sulfur. With regard to the metabolic processes of elemental sulfur bacteria, it was shown that Cyt c3 from Desulfovibrio may function as a sulfur oxidoreductase in vitro (12). More recently, it has been suggested that Cyt c7 would be involved in the coupled oxidation of acetate and dissimilatory reduction of Fe(III) and Mn(IV) (13) for obtaining energy for growth of these bacteria. Recent studies have demonstrated that Cyt c3 can function as a terminal Fe(III) and U(VI) reductase in Desulfovibrio (14).
Comparison among structures of different proteins of the same or similar classes has been used (15, 16) to identify common secondary structure motifs and possibly trace their evolution. The x-ray structures of four Desulfovibrio Cyt c3 are available (3–7), which provide the basis for a detailed characterization of the enzyme (17). The structure of Cyt c7 is unknown. Furthermore, almost nothing is known about the protein partners of Cyt c7 in the redox cycle. The comparison of the three-dimensional structures of Desulfovibrio Cyt c3 with that of D. acetoxidans Cyt c7 may shed light on their function.
We report here the solution structure of the fully oxidized Cyt c7. Oxidized cytochromes contain low spin iron(III) with one unpaired electron per heme. The solution structure determination of paramagnetic metalloproteins is a challenge because the presence of unpaired electrons broadens the NMR lines (18, 19), especially if three paramagnetic centers are present in a small molecule.
MATERIALS AND METHODS
NMR Sample Preparation.
Cyt c7 from D. acetoxidans was purified as reported (8). The 1H NMR samples were prepared by dissolving the lyophilized protein in 100 mM phosphate buffer at pH 6.5 to give 2–3 mM solutions.
NMR Spectroscopy.
The 1H NMR spectra were recorded on a Bruker MSL 200, a DRX 500, or an AMX 600 spectrometer, operating at 200.13 MHz, 500.13 MHz, or 600.13 MHz respectively.
Time proportional phase increment (TPPI) nuclear Overhauser effect spectroscopy (NOESY) (20, 21) spectra were recorded with presaturation of the solvent signal both during the relaxation delay and the mixing time. To optimize the detection of cross peaks involving fast-relaxing resonances, NOESY maps at 292, 300, and 308 K in 2H2O (D2O) (90% D2O/10% H2O) or in H2O (90% H2O/10% D2O) solution were recorded on the full spectral width (49.0 ppm) with recycle time of 550 ms and mixing time of 15 ms. To optimize the observation of connectivities in the diamagnetic region NOESY maps in D2O and H2O solutions were recorded on a smaller spectral width [36.2 and 17.3 ppm for NOESY and NOESY water suppression by gradient-tailored excitation (WATERGATE) (22, 23), respectively], with recycle times of 800 ms and mixing times of 25, 60, and 100 ms. Analogously, TPPI homonuclear Hartmann–Hahn (HOHAHA) (24) experiments, with presaturation during the relaxation delay, were recorded on spectral width of 49.0 ppm (recycle time of 550 ms, spin lock time of 18 ms) and 30 ppm (recycle time of 800 ms and spin lock times of 30, 60, and 90 ms). A magnitude correlated spectroscopy (COSY) (25, 26) map was recorded in H2O solution on a 30 ppm spectral width (recycle time of 800 ms).
All two-dimensional spectra consisted of 4 K data points in the F2 dimension. From 800 to 1024 experiments were recorded in the F1 dimension, using 64–192 scans per experiment. Raw data were multiplied in both dimensions by a pure cosine-squared (NOESY, HOHAHA) and a pure sine-squared (COSY) bell window function and Fourier-transformed to obtain 2048 × 2048 real data points. A polynomial base-line correction was applied in both directions.
Distance and Angle Constraints.
The volumes of the cross peaks between assigned resonances were obtained using the integration routines present in the program xeasy (27) or by manual integration. Most of the dipolar connectivities were taken from the 100-ms NOESY WATERGATE in 90% H2O/10% D2O solution at 300 K.
NOESY cross-peak intensities were converted into upper limits of interatomic distances by following the methodology of the program caliba (28).
3J(NH-Hα) were evaluated from the 100-ms NOESY WATERGATE by the infit (29) procedure available in the diana package and converted into angle constraints using the Karplus relation.
Structure Determination.
The experimental distance constraints were then used to generate protein conformers using the distance geometry program diana (28). The heme groups were constructed in analogy to x-ray data of other c-type cytochromes, and structure calculations were performed introducing a covalent link between the hemes and the sulfur of cysteine residues. An upper distance limit of 2.2 Å between the Nɛ2 of the axial histidines and the iron ion was imposed after checking that the histidine–metal coordination involves the above mentioned imidazole nitrogen in all the structures of heme proteins deposited in the Protein Data Bank.
diana calculations, including the redundant angle strategy routine (redac) (30), were performed following the procedure and with the parameters already used by us for the determination of other solution structures (31, 32). Initially 500 random structures were calculated. Stereospecific assignments of diastereotopic protons and methyl groups were obtained using the program glomsa (28). The average structure calculated from the best 20 structures obtained from diana (see below) has been energy minimized and used for comparison with the crystal structures.
Structure Analysis.
The structural analysis of the coordinates of the energy-minimized structure of whole protein, in terms of Ramachandran plots, deviation from ideal structural parameters, secondary structure elements, etc., was performed with the procheck program (33).
RESULTS AND DISCUSSION
The 1H NMR spectrum of fully oxidized Cyt c7 shows many signals shifted outside the diamagnetic envelope (Fig. 1) in agreement with the previously reported spectra (34). The downfield shifted signals are due to heme methyls and propionate α-protons and to the β-CH2 of axial histidines. The nonselective T1 values in the present protein are as low as 60 ms for the heme methyl groups. Only two ring protons of two of the coordinated histidines (i.e., His-17 and His-30) have been assigned among the broad features upfield (500- to 1000-Hz line width).
Figure 1.

Shown is the 600 MHz 1H NMR spectrum of Cyt c7 from D. acetoxidans recorded in D2O solution, 100 mM phosphate buffer (pH 6.5).
Two-dimensional NMR spectra, optimized (35) for the nuclear relaxation times of the present system, were recorded which lead to an almost complete assignment of the 1H resonances (resonances of 67 residues out of 68 were assigned, Ala-1 being the only unassigned residue) and to the evaluation of 1142 proton–proton upper limit distances. The sequence specific assignment of the heme resonances was also obtained.
From 1142 nuclear Overhauser effects (NOEs), 793 of which have been found meaningful (Fig. 2 A and B), and from 21 experimentally evaluated angle constraints, obtained analyzing the 3J(NH-Hα) coupling constants, a distance geometry family of 20 structures has been obtained through the program diana (28).
Figure 2.

Number of intra-NOEs (A) and inter-NOEs (B) per residue identified in the NMR spectra. The total height of each column represents the amount of observed experimental NOEs. The open and filled bars correspond to NOE constraints that are found to be irrelevant and meaningful, respectively. In C, diagrams of the root-mean-square deviation (rmsd) values per residue with respect to the average structure calculated on the 20-structure family for backbone (▪) and all heavy atoms are reported (○).
The root-mean-square deviation (rmsd) values with respect to the average structure for the entire protein (residues 3–67) are of 0.70 ± 0.22 and 0.98 ± 0.19 Å for the backbone and all heavy atoms, respectively, and a target function (30)—i.e., an evaluation of the disagreement with the experimental NOEs—of ≤0.41 Å2. The structure results well defined in the residue range 3–54 and 64–67 (see Fig. 2C). Indeed, if the rmsd values are calculated for these residue ranges, the values are 0.57 ± 0.30 and 0.91 ± 0.21 Å for the backbone and all heavy atoms, respectively. In the residue range 55–63 the family experiences a larger rmsd than the total value as a consequence of the lower number of NOEs per residue with respect to the rest of the protein (Fig. 2 A and B). The low number of NOEs in this part of the protein is due to the conformation of this loop which is pointing toward the exterior of the protein.
The energy-minimized average structure is shown in Fig. 3A. Two main secondary structure elements can be identified on the basis of the observed backbone NOEs (Fig. 4). Medium range NOEs between the Hα of residue i and the amide and Hβ protons of residue i+3, together with NOEs between amide–amide and Hα–amide protons of residues i and i+2 indicate the presence of a 310 helix in the 18–25 residue range. The presence of an antiparallel β-sheet involving residues 2–8 and 11–18 is clearly indicated by the presence of long range (i.e., between residues 2–8 and residues 11–18) backbone NOEs and is evident from Fig. 4B. The presence of helices in the 40–46 and 51–54 range is suggested by the presence of short and medium range sequential connectivities. A few backbone amide protons have been found to exchange slowly (i.e., their resonances are still detectable a few weeks after the sample has been dissolved in D2O solution) (Fig. 4A). The largest number of slowly exchanging amide protons is detected for the residues involved in the β-sheet motif.
Figure 3.

Stereoview of the ribbon diagram of the solution structure of Cyt c7 from D. acetoxidans (A) and of the x-ray crystal structure of Cyt c3 from Desulfovibrio desulfuricans (7) (B), displayed using molscript (36). The solution structure of Cyt c7 has been obtained from the average of the 20 solution structures of the distance geometry family, followed by energy minimization.
Figure 4.

(A) Schematic representation of the sequential connectivities involving NH, Hα, and Hβ protons. The thickness of the bar indicates the NOEs intensities. In the first line, NH resonances that have been found to exchange slowly in D2O solution are also indicated. (B) Diagonal plot of the NOEs observed between the backbone protons, which identify the secondary structure elements.
Similar secondary structure elements are also present in the x-ray structure of Cyt c3 from Desulfovibrio desulfuricans (7) (Fig. 3B), which appears to be the most similar to the present protein. A short right-handed 310 helix (His-22–Glu-26), four segments of right-handed α-helix (Glu-29–His-34, Ser-63–Ala-70, Ser-78–Lys-90, and Leu-93–Gly-99), and a short double-stranded antiparallel β-sheet (Val-9–Gly-13 and Lys-16–Phe-20) are observed in the former structure. Despite the low sequence homology found among various Cyt c3, the x-ray structures up to now reported reveal highly conserved secondary and tertiary structure elements (60–90%) (3–7, 37), where always a β-sheet and four helices are present. The main tertiary structural feature of the known Cyt c3 is the substantially unchanged relative spatial arrangement of the four heme moieties. This structural feature probably ensures the efficiency of the electron transfer process.
In Cyt c7 the cysteine residues and the ligands to the hemes belong to the same secondary structure motifs as in Cyt c3. Although Cyt c7 lacks one of the hemes (heme II), the remaining three have a relative arrangement similar to that reported for Cyt c3 (3–7). The intramolecular iron–iron and interplanar angles found in Cyt c7 are summarized in Table 1. The heme–heme distances compare well with those reported in the four available x-ray structures of Desulfovibrio Cyt c3 (3–7), where the iron–iron distance for heme I and III is 11.05 ± 0.05 Å, for heme I and IV is 17.7 ± 0.1 Å, and for heme III and IV is 12.05 ± 0.05 Å. Also the heme–heme angles are similar to those of Cyt c7: in Cyt c3 hemes I and III are almost orthogonal and I and IV approximately parallel (21 ± 1°) to each other, as found in the present protein (see Table 1). Despite the presence of the cavity produced by the missing heme II in Cyt c7, the polar propionate chains are pointing toward the outside of the protein, as it occurs in Cyt c3. Finally, the external loop constituted by residues 55–64 forms a pocket around heme III. A similar pocket has been observed also in the structures of Cyt c3. This pocket is defined by a loop which, similarly to Cyt c7, is protruding toward the exterior of the protein.
Table 1.
Intramolecular iron–iron distances (upper diagonal) and heme–heme angles (lower diagonal) in the solution structure of Cyt c7 from D. acetoxidans
| heme | Angles (°)/distances (Å)
|
||
|---|---|---|---|
| I | III | IV | |
| I | — | 11.3 | 20.5 |
| III | 84 | — | 13.3 |
| IV | 37 | 65 | — |
The heme numbering corresponds to that of four-heme Cyt c3.
The histidines are relatively well defined despite the lack of experimental constraints for four imidazole moieties, with the largest scattering of 40° experienced by His-53.
Two different midpoint redox potentials have been measured for the present protein (−102 mV for one heme and −177 mV for the other two) (11). If the protein is reduced by one electron, the hyperfine-shifted NMR signals of one heme—i.e., that with the highest reduction potential—disappear (34). On the basis of our assignment, this heme is heme IV.
Among all the tested Cyt c3, the heme with the highest reduction potential is, for three proteins from Desulfovibrio, heme IV and, in Desulfomicrobium baculatus c3, heme III (ref. 7 and references therein). These hemes are surrounded by a patch of positively charged residues (7), which accounts for the highest reduction potential.
An analysis of the distribution of charged residues around the heme iron atoms in Cyt c7 indicates that heme IV is definitely the one surrounded by the largest number of positive residues (Fig. 5). Within a sphere of 11 Å radius from the heme iron, the charge is +6 around heme IV, 0 around heme III, and −2 around heme I.
Figure 5.
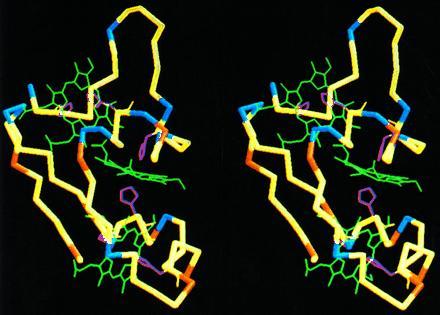
Stereoview of backbone and hemes of the energy-minimized average structure of D. acetoxidans Cyt c7. The positively and negatively charged residues are shown in blue and red, respectively. The neutral residues are shown in yellow. The three-heme groups are green, and the axial histidines are yellow. The molecule orientation is the same as in Fig. 3.
For Cyt c3, a pathway for the intramolecular electron transfer from the highest potential heme to the others has been proposed (7, 38–41): the electron goes from heme IV to heme III. Since the same structural features are all present in the solution structure of Cyt c7, a similar pathway for the electron transfer could hold for the present system.
Note Added in Proof.
After the submission of this paper, a paper by Xavier and coworkers (42) on the NMR characterization of the reduced form of Cyt c7 appeared, which reported the relative orientation of the three hemes. On that basis we interchanged the assignment of five heme proton resonances, which lead to the present orientation of hemes I and III.
Acknowledgments
We thank Prof. C. Luchimat and Dr. A. Rosato for helpful discussion. P.S. acknowledges the Italian Ministry of Foreign Affairs for a Ph.D. grant. This work has been performed with the instrumentation of the Florence Laboratory of Relaxometry and Magnetic Resonance on Paramagnetic Metalloproteins, Large Scale Facility of the European Community. We acknowledge the financial support of the Large Scale Facility of the European Union (Contract CHGE-CT94-0060) and the Italian Consiglio Nazionale delle Ricerche, Comitato Biotecnologie.
Footnotes
Abbreviations: Cyt c, cytochrome c; NOE, nuclear Overhauser effect; NOESY, NOE spectroscopy; TPPI, time proportional phase increment; WATERGATE, water suppression by gradient-tailored excitation.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, Chemistry Department, Brookhaven National Laboratory, Upton, NY 11973 (reference 1CF0).
References
- 1.Ambler R P. In: From Cyclotrons to Cytochromes. Robinson A B, Kaplan N O, editors. London: Academic; 1980. pp. 263–279. [Google Scholar]
- 2.Pettigrew G W, Moore G R. Cytochromes C: Biological Aspects. Berlin: Springer; 1987. [Google Scholar]
- 3.Haser R, Pierrot M, Frey M, Payan F, Astier J P, Bruschi M, LeGall J. Nature (London) 1979;282:806–810. doi: 10.1038/282806a0. [DOI] [PubMed] [Google Scholar]
- 4.Pierrot M, Haser R, Frey M, Payan F, Astier J P. J Biol Chem. 1982;257:14341–14348. [PubMed] [Google Scholar]
- 5.Higuchi Y, Kusunoki M, Matsuura Y, Yasuoka N, Kakudo M. J Mol Biol. 1984;172:109–139. doi: 10.1016/0022-2836(84)90417-0. [DOI] [PubMed] [Google Scholar]
- 6.Sieker L C, Jensen L H, LeGall J. FEBS Lett. 1986;209:261–264. doi: 10.1016/0014-5793(86)81124-3. [DOI] [PubMed] [Google Scholar]
- 7.Morais J, Palma P N, Frazao C, Caldeira J, LeGall J, Moura I, Moura J J G, Carrondo M A. Biochemistry. 1995;34:12830–12841. doi: 10.1021/bi00039a044. [DOI] [PubMed] [Google Scholar]
- 8.Probst I, Bruschi M, Pfennig N, LeGall J. Biochim Biophys Acta. 1977;460:58–64. doi: 10.1016/0005-2728(77)90151-7. [DOI] [PubMed] [Google Scholar]
- 9.Ambler R P. FEBS Lett. 1971;18:351–353. doi: 10.1016/0014-5793(71)80484-2. [DOI] [PubMed] [Google Scholar]
- 10.Mathews F S. Prog Biophys Mol Biol. 1985;45:1–56. doi: 10.1016/0079-6107(85)90004-5. [DOI] [PubMed] [Google Scholar]
- 11.Fiechtner M D, Kassner R J. Biochim Biophys Acta. 1979;579:269–278. doi: 10.1016/0005-2795(79)90054-0. [DOI] [PubMed] [Google Scholar]
- 12.Fauque G, Hervé D, LeGall J. Arch Microbiol. 1979;121:261–264. doi: 10.1007/BF00425065. [DOI] [PubMed] [Google Scholar]
- 13.Roden E E, Lovley D R. Appl Environ Microbiol. 1993;59:734–742. doi: 10.1128/aem.59.3.734-742.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lovley D R, Widman P K, Woodwark J C, Phillips E J P. Appl Environ Microbiol. 1993;59:3572–3576. doi: 10.1128/aem.59.11.3572-3576.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dickerson R E, Timkovich R, Almassy R J. J Mol Biol. 1976;100:473–491. doi: 10.1016/s0022-2836(76)80041-1. [DOI] [PubMed] [Google Scholar]
- 16.Ernst R R. Pure Appl Chem. 1994;66:1955–1960. [Google Scholar]
- 17.Coutinho I, Xavier A V. Methods Enzymol. 1994;243:119–140. doi: 10.1016/0076-6879(94)43011-x. [DOI] [PubMed] [Google Scholar]
- 18.Banci L, Bertini I, Luchinat C. Nuclear and Electron Relaxation. Weinheim, Germany: VCH; 1991. [Google Scholar]
- 19.Bertini I, Luchinat C. NMR of Paramagnetic Molecules in Biological Systems. Menlo Park, CA: Benjamin–Cummings; 1986. [Google Scholar]
- 20.Macura S, Wüthrich K, Ernst R R. J Magn Reson. 1982;47:351–357. [Google Scholar]
- 21.Marion D, Wüthrich K. Biochem Biophys Res Commun. 1983;113:967–974. doi: 10.1016/0006-291x(83)91093-8. [DOI] [PubMed] [Google Scholar]
- 22.Piotto M, Saudek V, Sklenar V. J Biomol NMR. 1992;2:661–666. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 23.Sklenar V, Piotto M, Leppik R, Saudek V. J Magn Reson Ser A. 1993;102:241–245. [Google Scholar]
- 24.Bax A, Davis D G. J Magn Reson. 1985;65:355–360. [Google Scholar]
- 25.Bax A, Freeman R, Morris G. J Magn Reson. 1981;42:164–168. [Google Scholar]
- 26.Bax A, Freeman R. J Magn Reson. 1981;44:542–561. [Google Scholar]
- 27.Eccles C, Güntert P, Billeter M, Wüthrich K. J Biomol NMR. 1991;1:111–130. doi: 10.1007/BF01877224. [DOI] [PubMed] [Google Scholar]
- 28.Güntert P, Braun W, Wüthrich K. J Mol Biol. 1991;217:517–530. doi: 10.1016/0022-2836(91)90754-t. [DOI] [PubMed] [Google Scholar]
- 29.Szyperski T, Güntert P, Otting G, Wüthrich K. J Magn Reson. 1992;99:552–560. [Google Scholar]
- 30.Güntert P, Wüthrich K. J Biomol NMR. 1991;1:447–456. doi: 10.1007/BF02192866. [DOI] [PubMed] [Google Scholar]
- 31.Banci L, Bertini I, Eltis L D, Felli I C, Kastrau D H W, Luchinat C, Piccioli M, Pierattelli R, Smith M. Eur J Biochem. 1994;225:715–725. doi: 10.1111/j.1432-1033.1994.00715.x. [DOI] [PubMed] [Google Scholar]
- 32.Banci L, Bertini I, Bren K L, Gray H B, Sompornpisut P, Turano P. Biochemistry. 1995;34:11385–11398. doi: 10.1021/bi00036a011. [DOI] [PubMed] [Google Scholar]
- 33.Laskowski R A, MacArthur M W, Moss D S, Thornton J M. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 34.Moura J J G, Moore G R, Williams R J P, Probst I, LeGall J, Xavier A V. Eur J Biochem. 1984;144:433–440. doi: 10.1111/j.1432-1033.1984.tb08484.x. [DOI] [PubMed] [Google Scholar]
- 35.Banci L, Bertini I, Luchinat C. Methods Enzymol. 1994;239:485–514. doi: 10.1016/s0076-6879(94)39019-3. [DOI] [PubMed] [Google Scholar]
- 36.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 37.Palma P N, Moura I, LeGall J, van Beeumen J, Wampler J E, Moura J J G. Biochemistry. 1994;33:6394–6407. doi: 10.1021/bi00187a003. [DOI] [PubMed] [Google Scholar]
- 38.Dolla A, Guerlesquin F, Bruschi M. J Mol Recognit. 1991;4:27–33. doi: 10.1002/jmr.300040105. [DOI] [PubMed] [Google Scholar]
- 39.Matias P M, Frazao C, Morais J, Coll M, Carrondo M A. J Mol Biol. 1993;234:680–699. doi: 10.1006/jmbi.1993.1620. [DOI] [PubMed] [Google Scholar]
- 40.Coletta M, Catarino T, LeGall J, Xavier A V. Eur J Biochem. 1991;202:1101–1106. doi: 10.1111/j.1432-1033.1991.tb16476.x. [DOI] [PubMed] [Google Scholar]
- 41.Picarra-Pereira M A, Turner D L, LeGall J, Xavier A V. Biochem J. 1993;294:909–915. doi: 10.1042/bj2940909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coutinho I B, Turner D L, Liu M Y, LeGall J, Xavier A V. JBIC. 1996;1:305–311. [Google Scholar]