

Abstract
Suppression of androgen production and function provides palliation but not cure in men with prostate cancer (PCa). Therapeutic failure and progression to hormone refractory prostate cancer (HRPC) are often accompanied by molecular alterations involving the androgen receptor (AR). In this study, we report novel forms of AR alteration that are prevalent in HRPC. Through in silico sequence analysis and subsequent experimental validation studies, we uncovered 7 AR variant transcripts lacking the reading frames for the ligand-binding domain, due to splicing of “intronic” cryptic exons to the upstream exons encoding the AR DNA binding domain. We focused on the two most abundantly expressed variants, AR-V1, and AR-V7, for more detailed analysis. AR-V1 and AR-V7 mRNA demonstrated an average 20-fold higher expression in HRPC (n=25) when compared to hormone naïve PCa (n=82) (p<0.0001). Among the hormone naïve PCa, higher expression of AR-V7 predicted biochemical recurrence following surgical treatment (p=0.012). Polyclonal antibodies specific to AR-V7 detected the AR-V7 protein frequently in HRPC specimens but rarely in hormone naïve PCa specimens. AR-V7 was localized in the nuclei of cultured PCa cells under androgen-depleted conditions, and constitutively active in driving the expression of canonical androgen responsive genes, as revealed by both AR reporter assays and expression microarray analysis. These results suggest a novel mechanism for the development of HRPC that warrants further investigation. In addition, as expression markers for lethal PCa, these novel AR variants may be explored as potential biomarkers and therapeutic targets for advanced PCa.
Keywords: Hormone refractory prostate cancer, androgen receptor, cryptic exon, premature termination codon
Introduction
Prostate cancer (PCa) depends upon androgenic signaling for growth and survival. Androgens exert their cellular and physiological effects through binding to the androgen receptor (AR), a member of the steroid hormone receptor family of transcription factors (1). The human AR gene is located on chromosome Xq11–12 and spans approximately 180 kb of DNA with eight known exons. The prototype AR protein contains a number of functional domains (2). The N-terminal domain (NTD), encoded by exon 1, constitutes nearly 60% of the 110KD full-length protein and is the transcriptional regulatory region of the protein. The central DNA-binding domain (DBD) is encoded by exon 2 and exon 3, while exons 4–8 code for the C-terminal ligand-binding domain (LBD). Androgen binding to the AR LBD allows entry of the ligand-bound receptor into the nucleus and subsequent transcriptional regulation of androgen responsive genes (3). Hormonal therapies for advanced PCa target AR-mediated functions by suppressing the production of androgens and/or androgen binding to the AR LBD. Although these therapies often result in a period of clinical regression, they are not curative due to progression to hormone refractory PCa (HRPC) for which effective therapeutic options are limited (4).
AR mediated functions are not completely abrogated by the existing hormone therapies. HRPC continues to depend on AR-mediated functions, but bypasses the requirement for physiological levels of androgens (5, 6). Molecular alterations involving AR itself, such as AR overexpression and gain-of-function AR LBD mutations, are common in HRPC and allow for continued AR-mediated genomic functions under the presence of reduced or altered ligands (5,6). Despite the established clinical relevance of these well-characterized AR alterations in HRPC, a few previous studies have suggested an alternative mechanism for HRPC and investigated the putative role of AR variants lacking the AR LBD (ARΔLBD) (7,8). These variants, arising from somatic nonsense mutations (7), or posttranslational AR proteolysis (8), were functionally active in the absence of androgens (7,8). While cellular production of such ARΔLBD isoforms appears to be most effective in achieving complete androgen-independence, their precise origins and relevance to HRPC were less well substantiated. In a recent study, Dehm et al. employed the RACE (rapid amplification of cDNA ends) technique and reported a novel exon within AR intron 2 (9). Splicing of this novel exon introduced a premature stop codon upstream of exon 3 in the AR transcript that would encode an AR protein lacking the second zinc finger of the DNA binding domain (DBD) and LBD if translated (9). In the present study, we performed a comprehensive in silico sequence analysis of the human AR genomic locus and uncovered multiple novel ARΔLBD variants with intact coding potential for the full AR NTD and AR DBD. These novel AR transcripts were overexpressed in HRPC and one of the most abundant variants, AR-V7, was expressed at elevated levels in a subset of hormone naïve PCa that recurred after surgical treatment. Moreover, for the first time we generated variant-specific antibody against the LBD truncated AR-V7, and detected this AR variant protein frequently in HRPC specimens. The expression pattern and the validated androgen-independent function of these novel AR variants potentially added another level of detail to the molecular mechanism of HRPC that could ultimately impact on the management of patients with advanced prostate cancer.
Materials and Methods
Human prostate tissue samples
Hormone naïve prostate tissue specimens used in this study (n=82) were collected and fresh frozen at the time of radical retropubic prostatectomy (RRP), from 1993 to 2001, at the Johns Hopkins Hospital. Prostate specimens were processed as described previously prior to RNA extraction (10). HRPC specimens were either collected at the time of the transurethral resection of the prostate (TURP) operation in patients who failed hormone therapies (n=4), or metastatic HRPC tissues (n=21) collected from 20 patients who died from PCa, as part of the Johns Hopkins Autopsy Study of lethal PCa (JHASPC) (11) (Supplemental Table I). The use of surgical and autopsy specimens for molecular analysis was approved by the Johns Hopkins Medicine Institutional Review Boards.
Cloning and sequencing of AR variants
First Strand cDNA synthesis was performed using 500 ng total RNA, 0.5 μg oligo (dT), and 200 units of SuperScript II reverse transcriptase (Invitrogen, Carlsbad, CA) in a volume of 20μL. PCR products derived from the primer pairs (Supplemental Table II) were cloned into TopoTA vector (Invitrogen, Carlsbad, California) and subjected to sequencing analysis using the Applied Biosystem 3730x1 DNA analyzer. To facilitate the amplification and sequencing of GC-rich AR NTD, DMSO (10%) was added in the PCR reaction for full-length variant cloning, and subsequent sequencing analysis.
AR variant mRNA expression analysis
For semi-quantitative RT-PCR analysis, 2.5% of the cDNA product from 500ng input total RNA was used for each sample and each transcript. For real-time quantitative RT-PCR, 0.125% of the cDNA product was used in the iQ SYBR Green Supermix assays (Bio-Rad, Hercules, CA). Given the highly variable expression of many genes among clinical specimens, we analyzed previously published expression microarray data and identified SF3A3, which encodes a splicing factor, as a reference gene for normalization, due to its stable expression levels among various prostate specimens including HRPC, primary PCa, normal prostate samples, and cell lines (12). Only primer pairs with validated amplification specificity were used (Supplemental Table II). Following validation of equal amplification efficiencies for both target transcripts and SF3A3, the average threshold cycle (Ct) numbers from reactions run in triplicate were used for comparative threshold analysis. For presentation purposes and for comparison among different figures, all expression values were Log2 transformed with measurable values for the RRP cases centered at zero.
AR variant Protein Analysis
Whole cell lysates were prepared using RIPA buffer (Pierce, Rockford, IL) according to the vendor’s recommendations. Nuclear and cytosolic extracts were prepared using the Nuclear and Cytoplasmic Extraction Reagents (Pierce). Protein samples were resolved on 4–12% gradient SDS-PAGE gels, and subjected to standard immunoblot analysis with anti-AR(N20) (Santa Cruz Biotechnology, Santa Cruz, CA), anti-AR-V7, or anti-β-actin (Sigma-Aldrich, St. Louis, MO) antibodies. The mouse polyclonal anti-AR-V7 antibody was developed using the c-terminal peptide (CKHLKMTRP) specific to the AR-V7 protein by a commercial vendor (A&G Pharmaceutical, Columbia, MD). For immunoprecipitation (IP), a total of 300 μg input whole cell lysates from cell lines or human tissues were precipitated with 4 μg of monoclonal anti-AR(441) (Santa Cruz) or control mouse IgG, followed by the addition of protein G-agarose (GE-Healthcare, Piscataway, NJ), and subjected to standard immunoblot analysis.
Luciferase Reporter Assay
pEGFP-AR and pEGFP-Q640X which contain the full-length prototype AR and AR Q640X LBD truncated mutant cDNA were kind gifts of Dr. Jocelyn Céraline (Université Strasbourg, Strasbourg, France). The cDNA encoding the full length AR-V7 was inserted into the pEGFP-C3 vector to express the GFP-AR-V7 fusion protein. Each of these constructs was co-transfected together with the PSAP1 luciferase reporter plasmid and pRL-CMV plasmid, an internal Renilla luciferase transfection control. Transfected cells were cultured in phenol red-free RPMI1640 containing 10% charcoal stripped serum (CSS) for 24 hrs, and cultured for another 24 hrs in the absence or presence of R1881 (NEN, Boston, MA), before being harvested and subjected to the Dual-Luciferase Reporter Assay (Promega, Madison, WI).
Statistical Analysis
All data were analyzed using Stata™ v10.0 statistical analyses software (Stata Corporation, College Station, TX). The Mann-Whitney test was used to evaluate distribution difference across two groups. Cox proportional hazard regression was used to identify significant prognostic factors for prediction of PCa progression free-survival. The proportional hazard assumption was verified by examination of residual plots and schoenfeld residuals. Log rank was used to test equality of survivor functions across two groups. Statistical significance in this study was set as p ≤0.05.
Results and Discussion
Identification of cryptic AR exons
We performed BLAST searches of the approximately 170 kb AR intron sequences against the NCBI human Expressed Sequence Tag (EST) database. High quality hits (99% identity) were found in intron 1 (6 hits), intron 2 (3 hits), and intron 3 (3 hits), but not in the remaining 4 introns (Supplemental Table III). These transcribed “intronic” genomic fragments, considered as putative cryptic exons, were not spliced as currently annotated and therefore their exon-intron junctions were undefined. Because a functional AR would most likely retain the AR DBD encoded by exon 2 and exon 3, we focused on the 3 putative cryptic exons in intron 3 to determine whether and how they were joined (i.e., spliced) with the upstream exon 3, and their potential to disrupt the AR open reading frame (ORF). We designed primers (P1, P2, P3) (Supplemental Table II) to amplify and sequence mRNA transcripts containing exons encoding AR DBD and the putative cryptic exons. The detection and subsequent sequencing of the amplicons derived from the CWR22Rv1 cells confirmed that all 3 cryptic exons (CE1, CE2, CE3) were joined with exon 3 (Figure 1A). These sequencing results were used to construct 7 AR transcript variants, named AR-V1 to AR-V7, each containing one of the 3 original cryptic exons (Figure 1A). Analysis of transcripts containing cryptic exon 1 (CE1) also uncovered an additional cryptic exon in intron 2, named CE4 (Figure 1A), that was spliced in both AR-V3 and AR-V4 (Figure 1A). The genomic position of CE4 is identical to the novel exon recently published by Dehm et al. but the specific sequence reported (9) differed from the consensus sequences we detected in the two CE4 containing variants (AR-V3 and AR-V4) (Figure 1A). CWR22Rv1 is a human PCa cell line derived from a serially transplanted PCa xenograft that relapsed after castration-induced regression, and is known to have a unique duplicated exon 3 (13). The duplicated exon 3 was reflected in AR-V2 and AR-V4 transcripts (Figure 1A). AR-V5 and AR-V6 contained cryptic exon 2 (CE2) and differed by a contiguous 80bp sequence at the 5′ junction of CE2, due to alternative 5′ splicing sites spaced 80bp apart in CE2 (not shown). Of importance, all 7 AR variants harbor premature termination codons (PTC) downstream of AR DBD, generating AR LBD truncated AR proteins if translated (Figure 1A).
Figure 1. Cloning of novel AR variants.

A. Novel AR variants lacking LBD generated by splicing of 4 cryptic exons. The 8 canonical exons of the AR gene were represented by numbered open boxes and shown (not to scale) in relation to the genomic positions of the 4 cryptic exons (CE1 to CE4) in shaded boxes. The identical forward primer, P1/P2/P3(F), in exon 2 was paired with 3 reverse primers (P1R, P2R, P3R) (Supplemental Table II) designed based on GenBank entries for the three transcribed genomic fragments in intron 3 (supplemental Table III). Sequencing of the amplicons (from CWR22Rv1 cells) defined the 5′ junctions of CE1, CE2, and CE3, and 5′ and 3′ junctions of CE4, as marked by vertical lines with the corresponding genomic coordinates (Human Genome Assembly March 2006, HG18). Note there were 4 CE1 containing variants (AR-V1, V2, V3, V4), and that the two CE2-containing variants (AR-V5, V6) differed by an 80bp contiguous 5′ extension in CE2. Stop codons were marked with the arrowheads in the schematically illustrated transcripts. The 7 translated protein sequences corresponding to the 7 transcripts were shown, starting from the last 4 amino acids encoded by exon 3 (AR-V1, V2, V4, V5, V6, V7) or exon 2 (AR-V3), and followed by variable lengths of variant specific sequences in light gray that matched the cryptic exons
B. Detection of the AR variant transcripts by semi-quantitative RT-PCR in clinical prostate specimens using the same sets of P1, P2 and P3 primers. Note that AR-V3, with expected size of 1126 bp by P1 primer set (Supplemental Table II), was not detected in these clinical specimens. HRPC (autopsy): metastatic HRPC samples from autopsies; HRPC (TURP): HRPC samples from transurethral resection of prostate (TURP); PCa (RRP): Hormone Naïve PCa from radical retropubic prostatectomy (RRP) specimens
C. Amplification of full-length coding region for AR-V1 and AR-V7, using primer sets P4 and P5 (Supplemental Table II), from one HRPC autopsy sample, one TURP sample, and the CWR22RV1 cell line. An identical forward primer, P4(F) and P5(F) located upstream of the translation start codon in exon 1, were paired with reverse primers, P4(R) and P5(R), located downstream of the stop codon in cryptic exon 1 and cryptic exon 3.
Cloning of the Full-length ORFs of AR-V1 and AR-V7
Semi-quantitative RT-PCR analysis in a small set of clinical specimens detected the variant transcripts prevalently in HRPC samples (Figure 1B). The full-length open reading frames (ORF) of AR-V1 and AR-V7 were then amplified from two clinical HRPC specimens and CWR22Rv1 cells (Figure 1C). Sequence analysis of the full-length amplicons confirmed the intact ORF of AR NTD and DBD, and thus the transcript structure for AR-V1 and AR-V7. Due to their relative lower abundance (Figure 1B), AR-V5 and AR V-6 were not further pursued for full-length ORF cloning. AR-V2 and AR-V4 were specific to CWR22Rv1 (not shown) due to the presence of exon 3 duplication and therefore also not pursued further. AR-V3 harbors a stop codon in CE4 and would lack the second zinc finger of AR DBD encoded by exon 3. Such variants may not be functional according to a previous study (14), though the study by Dehm et al. suggested otherwise (9). In addition, the full-length ORF for AR-V3 so far has not been detected in sequenced clones (data not shown). For these reasons, only AR-V1 and AR-V7 were pursued further.
Expression analysis of AR-V1 and AR-V7
HRPC specimens expressed consistently higher levels of AR-V1, AR-V7 and the prototype AR detected using optimized primer sets specific to each target transcript (Figure 2A). Expression of the prototype AR can be readily detected at 28 PCR cycles while detection of the AR variants required 38 cycles, suggesting the lower abundance of the AR variants, at mRNA levels, relative to the prototype AR (Figure 2A). Quantitative real-time RT-PCR of AR-V1, AR-V7 and prototype AR was performed on an expanded series of human prostate tissues (n=124) and cell lines (n=9, Supplemental Figure 1). Expression levels of AR-V1, AR-V7, and prototype AR were significantly higher in HRPC (n=25) than in hormone naïve PCa (n=82) (p<0.0001, Mann-Whitney test). Adjusted for amplification efficiency, the average expression values for prototype AR (Supplemental Figure 2A), AR-V1 (Supplemental Figure 2B) and AR-V7 (Figure 2B) were elevated by 11, 22 and 20 fold, respectively, when compared to hormone naïve PCa. It is unlikely that nuclear splicing intermediates of the prototype AR gene contributed to the detected AR variant signals, because nuclear RNA contributed less than 5% of the signal when compared to cytoplasmic RNA on a per cell basis (Supplemental Figure 3). A subset of hormone naïve PCa expressed AR variants at levels comparable to those in HRPC specimens (Figure 2B). This elevated AR-V7 expression was associated with worse clinical outcome (log rank p=0.012), as defined by PSA recurrence following surgical treatment (Figure 2C), in 66 RRP cases for which long-term clinical follow-up data were available. In this same sample set (n=66), higher prototype AR mRNA levels did not predict PSA failure (Supplemental Figure 4A). Similarly, higher ratio of V7/AR did not predict PSA failure (Supplemental Figure 4B) though there appeared to be a trend. AR-V1 expression was not associated with this clinical outcome (log rank p=0.498) (not shown). It is unknown why AR-V1 and AR-V7, while both overexpressed in HRPC specimens, differed in their association with PSA recurrence. It is worth noting that our preliminary analysis predicted that AR-V1 variant specific sequences (Figure 1A) lack the basic amino acids characteristic of the bipartite nuclear localizing sequence (15), and therefore may not be a fully functional nuclear receptor (not shown).
Figure 2. Quantification of AR variant transcripts in clinical specimens.

A. Representative gel images of amplified AR variant transcripts detected using primer sets designed for real-time RT-PCR assays. An identical forward primer, P6/P7/P8(F) in exon 3 was paired with different reverse primers P6(R), P7(R), and P8(R) (Supplemental Table II), to amplify the AR-V1, AR-V7 and prototype AR transcripts, respectively. SF3A3 was used as a reference gene transcript (Methods and Materials). Normal (RRP): normal prostate tissues from radical retropubic prostatectomy (RRP) specimens; PCa (RRP): Hormone Naïve PCa from RRP specimens; HRPC (TURP): HRPC samples from transurethral resection of prostate (TURP); HRPC (autopsy): metastatic HRPC samples from autopsies (Supplemental Table I)
B. Quantitative results of AR-V7 in 124 clinical prostate specimens by real time PCR. Normalized expression values (in log2 scale) for AR-V7 derived from comparative threshold analysis were shown in 4 groups of clinical specimens. Normal (n=17): normal prostate tissues from radical retropubic prostatectomy (RRP) specimens; Hormone Naïve PCa (n=82): PCa samples from RRP specimens; HRPC (TURP) (n=4): HRPC samples from transurethral resection of prostate (TURP); HRPC (autopsy) (n=21): metastatic HRPC samples from autopsies (Supplemental Table I)
C. Kaplan-Meier plot comparing progression free survival in patients with less than median AR-V7 expression (n = 38) to those with greater than median AR-V7 expression (n = 28). The survival curves were compared using the Log-rank test. Follow-up years were marked on the X axis. Censored subjects were marked with vertical ticks in blue. Note that the PSA recurrence status was annotated in years, not months.
AR-V7 is translated and constitutively active
Transcript variants harboring PTC may be subjected to nonsense-mediated decay (16). Indeed, although similar transcript variants have been previously characterized for other steroid hormone receptor family members (17), no corresponding protein product has been reliably demonstrated. Using the unique peptide sequence encoded by AR CE3, we generated polyclonal antibodies specifically against AR-V7. The antibodies recognized a single band of expected size (80KD) in VCaP and CWR22Rv1 cells (Figure 3A), which expressed highest levels of AR-V7 mRNA (Supplemental Figure I). Similarly, AR-V7 protein was detected in protein extracts from these two cell lines that were enriched for AR proteins by immunoprecipitation (IP) with an antibody against the AR NTD (Figure 3B). In addition, the antibody detected the AR-V7 antigen in two clinical HRPC specimens, using both whole tissue lysates and IP concentrated extracts (Figure 3C). Moreover, using IP concentrated protein extracts, we detected AR-V7 protein expression in 10 of 14 human prostate cancer xenografts, 12 of which were derived from HRPC patients (18), but in only 1 of the 9 hormone naïve radical prostatectomy specimens (Supplemental Figure 5). In CWR22Rv1 cells, siRNA mediated knockdown of AR-V7 expression or depletion of AR-V7 using anti-AR-V7 both resulted in significant reduction of the commonly observed ~80KD protein band but did not affect prototype AR expression (Supplemental Figure 6), suggesting nearly equivalent AR-V7 and prototype AR protein levels in this cell line. While the prototype AR responded to the treatment of androgen by localizing to the nucleus, a large fraction of endogenous AR-V7 was localized in the nucleus in the absence of androgen and the proportion of nuclear AR-V7 did not change upon androgen stimulation (Figure 3D). The putative functional role of AR-V7 was investigated using exogenously transfected AR-V7 in AR negative PC-3 cells. AR-V7 localized to the nucleus (Figure 4A) and induced PSA reporter gene expression in an androgen-independent manner (Figure 4B). Furthermore, in androgen responsive LNCaP cells AR-V7 induced canonical androgen responsive genes such as KLK3, KLK2, NKX3-1, FKBP5, and TMPRSS2 in the absence of androgens, as demonstrated by global gene expression analysis following transfection of the exogenous AR-V7 cDNA in LNCaP cells (Figure 4C).
Figure 3. AR-V7 protein detection and analysis using a variant specific antibody.

A. Detection of AR-V7 protein product in cell lines expressing high levels of AR-V7 transcript (Supplemental Figure 1). Following immunoblot analysis for AR-V7 (upper panel), the same membrane was stripped and subjected to immunoblot analysis with anti-AR(N20) antibody (middle panel) to detect the prototype AR. Loading of total protein was monitored by Ponceau S staining of the PVDF membrane (Lower panel)
B. Detection of AR-V7 protein following enrichment of all NTD-containing AR proteins by immunoprecipitation (IP) using the anti-AR(441) antibody. Note that following enrichment, AR-V7 was detected in cell lines expressing highest levels of AR-V7 mRNA, VCaP and CWR22Rv1 cells, but not in LNCaP cells, which expressed low levels of AR-V7 (Supplemental Figure 1). Control: mouse IgG. anti-AR: anti-AR(441) monoclonal antibody
C. Detection of AR-V7 protein in HRPC. Western blot analysis were performed to detect AR-V7 in whole tissue lysates and enriched AR protein extracts derived from 4 hormone naïve human PCa tissue (RRP5, 6, 7 and 8) and two hormone refractory human PCa tissues (TURP1 and 2). Protein loading was monitored by Ponceau S staining of the PVDF membrane (Middle panel). IP with the anti-AR(441) antibody was performed to enrich the AR proteins and immunoblotted (IB) with anti-AR(N20) to detect the prototype AR, and AR-V7 by the antiAR-V7 antibody (Lower panel)
D. Biochemical analysis of cellular localization of AR-V7 protein. VCaP and CWR22Rv1 cells were grown in phenol red-free RPMI 1640 containing charcoal stripped serum (CSS) with or without 10nM R1881. The cytosolic fraction (C) and nuclear fraction (N) of lysates with equivalent number of cells were isolated and subjected to immunoblot analysis of AR-V7, prototype AR by anti-AR(N20) antibody, and β-actin.
Figure 4. Constitutive function of AR-V7.
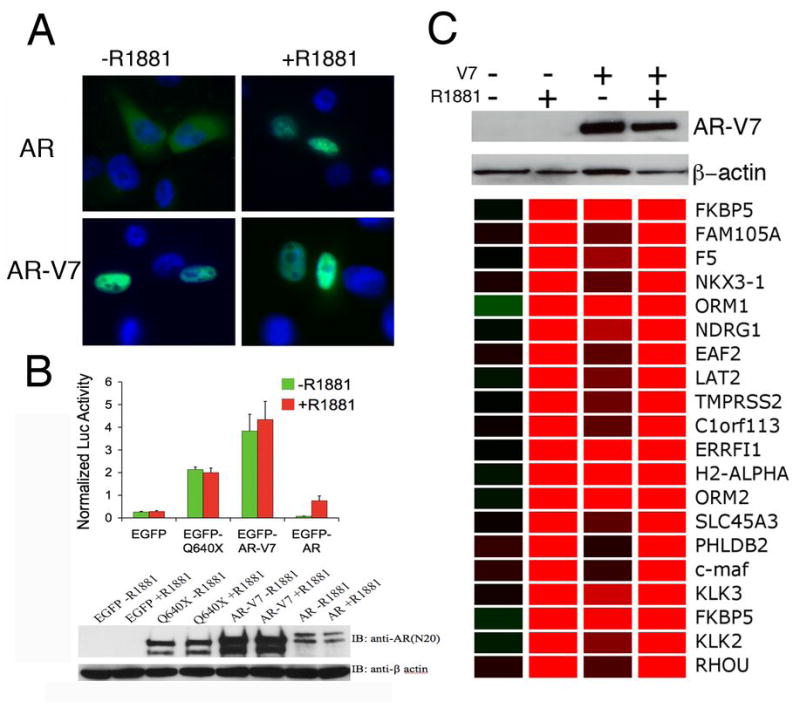
A. Constitutive nuclear localization of transfected AR-V7 in the absence of androgen. PC-3 cells were transfected with pEGFP-AR and pEGFP-AR-V7 to express the prototype AR or AR-V7, and examined for the localization of GFP-tagged AR proteins in the absence or presence of 5 nM R1881
B. AR-V7 constitutively activates an AR luciferase reporter. PC-3 cells were transfected with vector control (EGFP), a LBD truncated AR mutant (EGFP-Q640X), AR-V7 (EGFP-AR-V7), and prototype AR (EGFP-AR), and subjected to luciferase assays and western blot analysis following culturing in the absence or presence of R1881
C. Androgen-independent induction of AR responsive genes by AR-V7 in LNCaP cells. LNCaP cells were transfected with pcDNA-AR-V7 to express the untagged AR-V7 protein or the control pcDNA vector, and cultured with or without 10nM R1881 before being harvested for western blot analysis or RNA extraction for expression microarray analysis. The genes shown were the top 20 ranked genes by fold induction following R1881 treatment in pcDNA empty vector transfected LNCaP cells. Expression ratios of the test sample versus the common reference (pcDNA empty vector transfected LNCaP without R1881) were represented by red (Greater than 1) and green colors (less than 1).
Hormonal therapy for advanced PCa is most commonly achieved by orchiectomy, systemic administration of LHRH agonists (e.g., leuprolide), and/or anti-androgens (e.g., bicalutamide). There are significant drawbacks associated with all existing androgen manipulation approaches. First, a variable period of clinical regression is followed by progression to HRPC, a lethal manifestation of the disease that is resistant to further therapies (4). Second, there are debilitating consequences from these treatments that must be considered when deciding whether and when to commence hormone therapy (2). Furthermore, sufficient levels of local androgens continue to be present in patients treated with combined androgen blockade (19). In spite of these challenges, hormone therapies remain the mainstay of treatment for patients with advanced PCa primarily due to the often dramatic clinical responses. The discovery of multiple LBD truncated AR variants that mediate androgen-independent AR functions in HRPC and a subset of advanced, but hormone naïve PCa adds another level of detail to the complex molecular mechanisms underlying the development of HRPC, and may suggest new diagnostic and therapeutic approaches targeting this lethal disease. Indeed, these findings reinforce arguments for specific targeting of the AR N-terminal domain to achieve complete abrogation of AR signaling (20). Our quantitative mRNA data suggested that AR-V7 is a low abundance variant relative to the prototype AR in the vast majority of clinical specimens including HRPC (Supplemental Figure 7). The relative contribution of the prototype AR and the less abundant, yet androgen-independent AR variants to the development of HRPC is currently unknown and will require detailed investigation. Nevertheless, the detection of such variants in proper target tissues or cells, upon further refinement of the detection methods, may predict or monitor hormone therapy efficacy and could potentially help guide the decision making process regarding the type and timing of therapies given to patients with advanced PCa.
Supplementary Material
Acknowledgments
Financial Support: The David H. Koch Foundation.
We would like to thank Dr. Patrick Walsh, Ms. Sally Isaacs, and Ms. Kathleen Wiley for assistance with the clinical data, Dr. Angelo De Marzo and Ms. Helen Fedor for assisting with tissue preparation, Drs. Robert Getzenberg and John Isaacs for critical reading of the manuscript, Dr. Donald Coffey for insightful discussions, and Mr. Wasim Chowdhury for technical assistance. This study is supported by the David H. Koch Foundation. The Brady Urological Research Institute Prostate Specimen Repository is supported by the Prostate SPORE grant (NIH/NCI P50CA58236). J.L. is a Phyllis and Brian L. Harvey Scholar, awarded by the Patrick C. Walsh Prostate Cancer Research Fund.
References
- 1.Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25:276–308. doi: 10.1210/er.2002-0032. [DOI] [PubMed] [Google Scholar]
- 2.Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol. 2002;20:3001–15. doi: 10.1200/JCO.2002.10.018. [DOI] [PubMed] [Google Scholar]
- 3.Shang Y, Myers M, Brown M. Formation of the androgen receptor transcription complex. Mol Cell. 2002;9:601–10. doi: 10.1016/s1097-2765(02)00471-9. [DOI] [PubMed] [Google Scholar]
- 4.Armstrong AJ, Carducci MA. New drugs in prostate cancer. Curr Opin Urol. 2006;16:138–45. doi: 10.1097/01.mou.0000193390.69845.bb. [DOI] [PubMed] [Google Scholar]
- 5.Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253–61. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- 6.Agoulnik IU, Weigel NL. Androgen receptor action in hormone-dependent and recurrent prostate cancer. J Cell Biochem. 2006;99:362–72. doi: 10.1002/jcb.20811. [DOI] [PubMed] [Google Scholar]
- 7.Céraline J, Cruchant MD, Erdmann E, Erbs P, Kurtz JE, Duclos B, et al. Constitutive activation of the androgen receptor by a point mutation in the hinge region: a new mechanism for androgen-independent growth in prostate cancer. Int J Cancer. 2004;108:152. doi: 10.1002/ijc.11404. [DOI] [PubMed] [Google Scholar]
- 8.Libertini SJ, Tepper CG, Rodriguez V, Asmuth DM, Kung HJ, Mudryj M. Evidence for calpain-mediated androgen receptor cleavage as a mechanism for androgen independence. Cancer Res. 2007;67:9001–5. doi: 10.1158/0008-5472.CAN-07-1072. [DOI] [PubMed] [Google Scholar]
- 9.Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–77. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo J, Duggan DJ, Chen Y, Sauvageot J, Ewing CM, Bittner ML, et al. Human prostate cancer and benign prostatic hyperplasia: molecular dissection by gene expression profiling. Cancer Res. 2001;61:4683–8. [PubMed] [Google Scholar]
- 11.Suzuki H, Freije D, Nusskern DR, Okami K, Cairns P, Sidransky D, et al. Interfocal heterogeneity of PTEN/MMAC1 gene alterations in multiple metastatic prostate cancer tissues. Cancer Res. 1998;58:204–9. [PubMed] [Google Scholar]
- 12.Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, et al. Delineation of prognostic biomarkers in prostate cancer. Nature. 2001;412:822–6. doi: 10.1038/35090585. [DOI] [PubMed] [Google Scholar]
- 13.Tepper CG, Boucher DL, Ryan PE, Ma AH, Xia L, Lee LF, et al. Characterization of a novel androgen receptor mutation in a relapsed CWR22 prostate cancer xenograft and cell line. Cancer Res. 2002;62:6606–14. [PubMed] [Google Scholar]
- 14.Quigley CA, Evans BA, Simental JA, Marschke KB, Sar M, Lubahn DB, et al. Complete androgen insensitivity due to deletion of exon C of the androgen receptor gene highlights the functional importance of the second zinc finger of the androgen receptor in vivo. Mol Endocrinol. 1992;6:1103–12. doi: 10.1210/mend.6.7.1508223. [DOI] [PubMed] [Google Scholar]
- 15.Zhou ZX, Sar M, Simental JA, Lane MV, Wilson EM. A ligand-dependent bipartite nuclear targeting signal in the human androgen receptor. Requirement for the DNA-binding domain and modulation by NH2-terminal and carboxyl-terminal sequences. J Biol Chem. 1994;269:13115–23. [PubMed] [Google Scholar]
- 16.Pan Q, Saltzman AL, Kim YK, Misquitta C, Shai O, Maquat LE, et al. Quantitative microarray profiling provides evidence against widespread coupling of alternative splicing with nonsense-mediated mRNA decay to control gene expression. Genes Dev. 2006;20:153–8. doi: 10.1101/gad.1382806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirata S, Shoda T, Kato J, Hoshi K. Isoform/variant mRNAs for sex steroid hormone receptors in humans. Trends Endocrinol Metab. 2003;14:124–9. doi: 10.1016/s1043-2760(03)00028-6. [DOI] [PubMed] [Google Scholar]
- 18.Saramäki OR, Porkka KP, Vessella RL, Visakorpi T. Genetic aberrations in prostate cancer by microarray analysis. Int J Cancer. 2006;119(6):1322–9. doi: 10.1002/ijc.21976. [DOI] [PubMed] [Google Scholar]
- 19.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447–54. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dehm SM, Tindall DJ. Androgen receptor structural and functional elements: role and regulation in prostate cancer. Mol Endocrinol. 2007;21:2855–63. doi: 10.1210/me.2007-0223. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.