

Abstract
Patients with chronic kidney disease (CKD) are at significantly higher risk of death from sepsis, although the mechanism by which CKD increases mortality has not been investigated. We established a mouse two-stage model of pre-existing renal disease with subsequent sepsis by combining folic acid (FA) injection and sub-lethal cecal ligation and puncture (CLP) surgery. Mice were injected with FA then made septic (FA-CLP) or were injected with vehicle then made septic (Veh-CLP). FA-CLP mice had significantly higher mortality than Veh-CLP mice. Sepsis increased serum creatinine in the FA-CLP but not in the Veh-CLP group. FA-CLP mice had more severe septic shock and significantly increased vascular permeability, plasma vascular endothelial growth factor (VEGF), bacteremia, serum IL-10 and splenocyte apoptosis compared to Veh-CLP. To evaluate the contribution of vascular and immunological dysfunction, we treated FA-CLP mice with soluble Flt-1 and chloroquine. Mice treated with combination therapy showed a significant improvement in kidney injury, hemodynamics, and survival. In conclusion, the sequential FA-CLP model mimics human sepsis that is frequently complicated with pre-existing conditions including CKD. This animal model would be useful to evaluate preventative and therapeutic strategies under conditions more typical of human sepsis.
Introduction
Sepsis is the leading cause of death in critically ill patients and the incidence of sepsis is increasing.1, 2 Sepsis causes acute kidney injury (AKI) and patients with both sepsis and AKI show an especially high mortality rate.3 Chronic kidney disease (CKD) is found in approximately 30% of AKI patients in ICU4 and severe sepsis occurring in patients with underlying chronic diseases (co-morbidities) including CKD, liver disease, and diabetes has an extremely high mortality rate.1, 5 These findings suggest that clinical sepsis and sepsis-induced AKI are dramatically influenced by underlying diseases, which may explain why simple animal models of sepsis do not mimic human sepsis, and do not predict human response to therapeutics.6 Developing a new animal model that allow us to investigate the mechanism by which pre-existing CKD increases the mortality of sepsis might help discovery efforts to improve the mortality of sepsis, because the prevalence of CKD is increasing worldwide.
In the present study, we developed a new two-stage mouse model that mimics sepsis in patients with pre-existing renal dysfunction. This “two-hit” animal model consists of folic acid (FA)-induced renal injury followed by a sub-lethal cecal ligation and puncture (CLP) model of sepsis. Folic acid injection induces renal injury, as documented by an increase of serum creatinine, about 60% decrease in glomerular filtration rate (GFR) and a remarkable interstitial fibrosis within two weeks.7 Two weeks after FA injection, we induced sepsis by using a clinically relevant CLP sepsis model which we have established;8 animals are treated with fluid resuscitation and antibiotics similar to septic patients in an ICU. We then used this new two stage FA-CLP sepsis model to investigate the pathophysiological mechanisms through which CKD increases sepsis mortality. We used two probes that are known to modulate vascular (sFlt-1) and inflammatory (chloroquine) dysfunction in sepsis. Since the two-hit model is sufficiently complex and models the propensity of pre-existing disease to dramatically increase the risk of sepsis, we evaluated the effect of combination therapy in this model that better mimics human sepsis.
Results
FA-CLP mice showed higher mortality after sepsis with amplified acute kidney injury
The protocol of the two-stage FA-CLP model is shown in Figure 1. Blood urea nitrogen (BUN) and serum creatinine (Cr) of FA group at 48 hr after injection were substantially higher than vehicle (Veh) group. Although BUN and serum Cr decreased at two weeks after injection, there was a significant and persistent elevation of both BUN and Cr in the FA group. The glomerular filtration rate at two weeks after injection was 62% lower in the FA group than in the Veh group (Table 1).
Figure 1.

Two-stage mouse model of FA-induced renal injury and subsequent sepsis with CLP surgery. Schema of FA-CLP animal model protocol is shown.
Table 1. Renal function after folic acid injection.
| BUN | serum Cr | BUN | serum Cr | GFR | |
|---|---|---|---|---|---|
| 48 hr | 48 hr | 2 wk | 2 wk | 2 wk | |
| (mg/dl) | (mg/dl) | (mg/dl) | (mg/dl) | (μl/min) | |
| FA | 204.9 ± 14.2* | 1.32 ± 0.14* | 50.3 ± 3.2* | 0.28 ± 0.03* | 162.0 ± 20.2* |
| Veh | 18.5 ± 1.3 | 0.10 ± 0.01 | 24.5 ± 2.0 | 0.11 ± 0.01 | 425.5 ± 14.9 |
Data are means ± SEM; n = 14∼17 in BUN and serum Cr and n =5 in GFR measurement.
p < 0.05 versus Veh group.
Sepsis induced by sub-lethal CLP surgery showed a significantly higher mortality in the FA-CLP group compared with the Veh-CLP group [FA-CLP 93%, Veh-CLP 18% at 96 hr; p<0.05] (Figure 2A). In the Veh-CLP group, BUN but not serum Cr showed a modest increase following sub-lethal CLP surgery. In contrast, BUN and serum Cr in the FA-CLP group were both increased significantly at 18 hr after CLP surgery compared to 0 hr (Figure 2B and 2C). Renal morphologic evaluation was performed at 0 and 18 hr after CLP surgery. In accordance with previous reports,7, 9 FA induced patchy interstitial fibrotic lesions two weeks after injection, although the cortical tubules in non-fibrotic areas were grossly normal (Figure 2D and 2E). We found no histological evidence of damage to other organs (data not shown). We have reported previously that sepsis induced by CLP caused renal tubular damage mainly consisting of tubular vacuolization.8, 10, 11 In the present study, tubular vacuolization was found in the cortex of Veh-CLP group and non-fibrotic cortical area of FA-CLP (Figure 2F and 2G). The number of vacuolized tubules in these areas was higher in the FA-CLP group than in the Veh-CLP group (Figure 2H).
Figure 2. Survival and kidney injury of FA-CLP model.
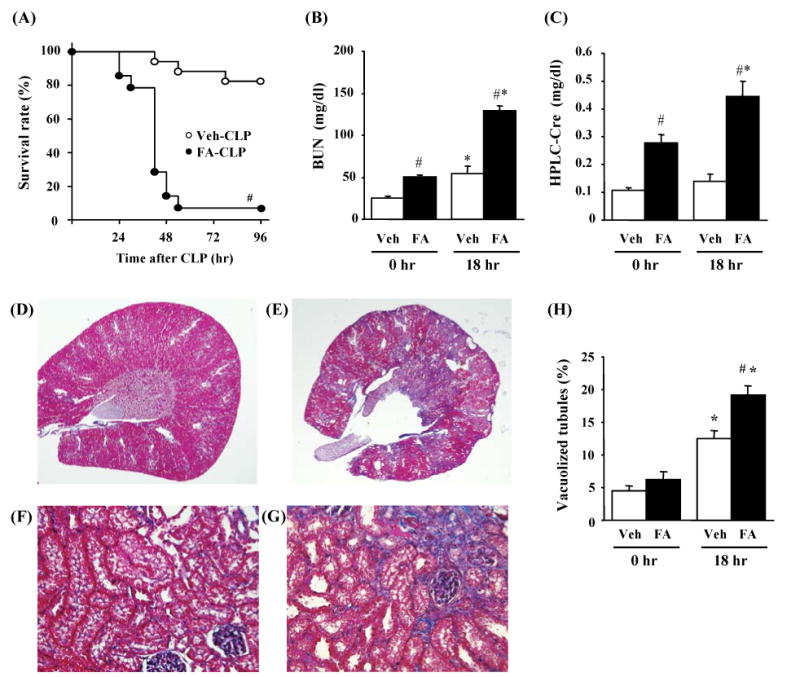
(A) Survival analysis of FA-CLP (n = 14) and Veh-CLP (n = 17). (B-C) Renal function of Veh-CLP and FA-CLP mice. BUN and serum Cr were measured 0 and 18 hr after CLP (n = 14 in FA-CLP, n = 17 in Veh-CLP). (D-G) Representive renal histology of Veh-CLP (D, F) and FA-CLP (E, G) at 18 hr are shown with Masson trichrome staining. Original magnification: X20 in (D, E) and X400 in (F, G). (H) Percentage of vacuolized tubules (n = 3 at 0hr, n = 5 at 18 hr per group). #, P < 0.05 versus Veh-CLP. *, P < 0.05 versus CLP 0 hr.
Severe septic shock and hyperkalemia in FA-CLP mice
Blood pressure (BP) and heart rate (HR) were measured in conscious animals by radiotelemetery. Pre-sepsis BP was slightly, but not significantly, higher in FA-injected mice than vehicle injected mice. Sepsis induced by sublethal CLP surgery caused mild decreases of BP and HR in Veh-CLP mice. On the other hand, severe hypotension and decreased HR were found in FA-CLP mice (Figure 3A and 3B).
Figure 3. Blood pressure and heart rate.

(A-B) Telemetric recordings of mean arterial pressure (MAP) and heart rate (HR) in Veh-CLP (open circle) and FA-CLP (closed circle) (n = 5 per group). #, P < 0.05 versus Veh-CLP.
Serum potassium levels of FA-CLP mice at 0 hr (i.e., before CLP) were similar to Veh-CLP mice. Sepsis induced hyperkalemia along with AKI in FA-CLP mice. Bilateral nephrectomy (BNx) induced higher serum potassium levels than FA-CLP mice 18 hr after surgery (Supplementary Figure 1). However, bilateral nephrectomized mice started to die later than FA-CLP mice [Time to death: FA-CLP 41.5 ± 5.8 hr (n = 10), BNx 51.3 ± 3.7 hr (n = 11), p < 0.05], although total survival rate were not significantly different between these two groups. This suggests that hyperkalemia is unlikely to be the primary cause of death in FA-CLP mice.
FA-CLP mice showed higher vascular permeability, plasma VEGF
Vascular permeability was examined with Evans blue dye which binds to circulating albumin. Renal vascular permeability in FA-CLP mice at 0 hr was increased compared with Veh-CLP (p < 0.05). There was no difference in peritoneum or lung vascular permeability before CLP. FA-CLP mice showed a significantly higher vascular permeability compared with Veh-CLP mice in kidney (6 hr), peritoneum (6, 18 hr) and lung (6 hr) (Figure 4A-C). The plasma level of VEGF, a growth factor known to enhance vascular permeability,12 in FA-CLP mice was higher than in Veh-CLP mice before CLP. Sepsis significantly increased plasma VEGF in FA-CLP compared with Veh-CLP mice (6, 18 hr). Moreover, bilateral nephrectomy dramatically increased plasma VEGF 18 hr after nephrectomy (Figure 4D). These results suggest that the kidney plays an important role in handling circulating VEGF. Recombinant sFlt-1 peptide can bind to circulating VEGF and has been reported to improve the survival of mouse sepsis models including CLP.13, 14 We found that treatment with sFlt-1 peptide significantly reduced peritoneal, kidney, and lung vascular permeability in FA-CLP mice, best seen at 6 hr after CLP (Figure 4A-C).
Figure 4. Vascular permeability and plasma VEGF levels.

Evans blue dye leakage in peritoneum (A), kidney (B), and lung (C) was measured at 0, 6 and 18 hr after CLP (n = 4∼8 per group). (D) Plasma VEGF levels were measured at 0, 6 and 18 hr after CLP by ELISA (n = 5∼6 per group). #, P < 0.05 versus Veh-CLP. *, P < 0.05 versus FA-CLP.
Bacterial count, splenocyte apoptosis and serum IL-10 increased in FA-CLP mice
Sepsis induces a state of altered host defenses, which was assessed via measurement of bacterial counts from blood and peritoneal cavity, splenocyte apoptosis, and serum IL-10 levels. FA-CLP mice showed higher bacterial counts both in blood and peritoneal cavity compared with Veh-CLP (Figure 5A and 5B). Splenocyte apoptosis evaluated by activated caspase-3 immunohistochemisty was not increased by FA injection alone. After CLP surgery, the number of activated caspase-3 cells was higher in FA-CLP compared with Veh-CLP mice at 18 hr after CLP surgery (Figure 5C). Serum IL-10 levels were not different between FA-CLP and Veh-CLP mice two weeks after FA injection. After induction of sepsis, FA-CLP mice showed significantly higher serum IL-10 levels compared with Veh-CLP mice (Figure 5D). Chloroquine (CQ) is reported to improve mortality of several CLP models via improving splenocyte function,15 splenocyte apoptosis and serum IL-10.16 In the current FA-CLP model, we found that CQ treatment also significantly attenuated the bacterial count in blood, splenocyte apoptosis and serum IL-10 levels. In contrast, CQ did not significantly decrease peritoneal fluid bacterial counts (Figure 5A-D).
Figure 5. Bacterial counts in blood and peritoneal fluid, splenic apoptosis, and serum IL-10 levels.

Bacterial counts in blood (A) and peritoneal cavity (B) were evaluated at 18 hr after CLP (n = 8 per group). (C) Splenic apoptosis was evaluated by activated caspase-3 immunohistochemistry (n = 4 per group). (D) Serum IL-10 levels were measured at 0 and 18 hr after CLP by ELISA (n = 5∼6 per group). #, P < 0.05 versus Veh-CLP. *, P < 0.05 versus FA-CLP.
Liver damage and lung pathological damage in FA-CLP mice
There was no statistically significant difference of AST and ALT between FA-CLP and Veh-CLP mice, although both groups showed increases of liver enzymes after CLP surgery (Figure 6A and 6B). Folic acid injection did not cause any lung damage (data not shown). Although lung vascular leakage in FA-CLP mice at 6 hr was significantly increased, there was no detectable histological change [i.e., increase of interstitial cellularity and/or extension of cellular infiltrates into the alveolar space] in either FA-CLP or Veh-CLP at 6 and 18 hr after CLP (Figure 6C and 6D).
Figure 6. Liver damage and lung pathology.
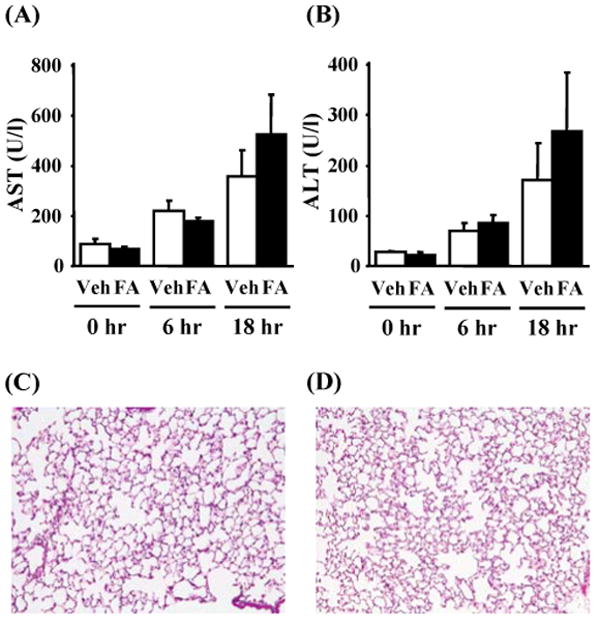
(A-B) Serum aspartate transaminase (AST) and alanine transaminase (ALT) were measured at 0, 6 and 18 hr after CLP (n = 5∼6 per group). (C-D) Lung histology in Veh-CLP and FA-CLP at 18 hr with HE staining. Original magnification: X200.
Combination treatment with soluble Flt-1 and chloroquine improved survival of FA-CLP mice
Since sFlt-1 peptide and CQ appear to affect sepsis by different mechanisms, we treated FA-CLP mice with sFlt-1 and CQ, alone or in combination. Each treatment alone tended to decrease BUN and serum Cr levels 18 hr after CLP surgery, but only the combination treatment showed a statistically significant protective effect (Figure 7A and 7B). Hypotension and bradycardia after sepsis induction were improved only in sFlt-1 and combination treatment groups (Figure 7C and 7D), whereas CQ partially improved blood pressure without improving heart rate at all. In the survival analysis, sFlt-1 and CQ treatment decreased mortality compared with control group individually [sFlt-1 40%, CQ 29%, control 12% at 96 hr], however only the combination treatment group showed a statistically significant improvement of survival compared with the control group [survival rate: 60% at 96 hr, p < 0.05 versus FA-CLP] (Figure 7E).
Figure 7. Combination treatment of sFlt-1 and chloroquine.

(A-B) Renal function of combination treatment. BUN and serum Cr were measured 18 hr after CLP in control group (n = 18), sFlt-1 treatment group, CQ treatment group, and combination treatment group (n = 6∼7 per treatment group). (C-D) Telemetric recordings of mean arterial pressure (MAP) and heart rate (HR) (n = 5∼6 per group) and (E) Survival analysis of combination treatment (n = 15∼17 per group). #, P < 0.05 versus control.
Discussion
We have developed a new two-stage mouse model of folic acid-induced renal injury and subsequent sepsis with cecal ligation and puncture surgery. This model replicated the clinical finding that pre-existing CKD amplifies sepsis-induced AKI, induces severe septic shock, and worsens outcome in sepsis.1, 4 We demonstrated the participation of several different pathophysiological mechanisms (i.e., increased capillary permeability, decreased bacteria clearance, and splenocyte apoptosis), and the benefit of combination treatment with soluble Flt-1 and chloroquine, compared to treatment with the individual agents.
Animal studies typically examine sepsis and related organ failure in otherwise healthy animals, despite the inability of simple sepsis models to predict human drug response6 and numerous epidemiological studies of human sepsis that show the importance of pre-existing co-morbidity.1, 5 Angus et al. reported that 55 percent of 200,000 patients with severe sepsis had underlying co-morbidity.1 As CKD patients have been recently recognized as being at high risk for cardiovascular death and mortality from all-causes,17, 18 patients with CKD also have an increased risk of morbidity and mortality of sepsis.1, 19-22 In one large multicenter study involving thirty thousand critically ill patients, 50% of AKI was associated with septic shock and 30% of AKI had pre-existing renal dysfunction.4 A new animal model that mimics the complexity of human sepsis is required. To simulate CKD, we employed a mouse folic acid injection model. This model causes acute tubular damage with increases of BUN and serum Cr peaking two days after injection and mild renal dysfunction with remarkable interstitial fibrosis were subsequently found at two weeks after. Folic acid injection did not cause other organ damage (i.e., liver and lung), possibly because the rodent folate receptors are highly expressed in the kidney23 and/or the kidney damage is induced by chemical precipitation of folic acid in the renal tubules. Although the FA-induced renal injury model does not show any glomerular lesions nor does it progress to end-stage renal disease, it does transiently mimic human CKD in terms of decreased glomerular filtration rate (Table 1) and histologic evidence of kidney damage (Figure 2E and F) as defined in the K/DOQI guideline.24
Our study is the first “two-stage” sepsis animal model that consists of prior kidney injury followed by subsequent sepsis. Previous “two-hit” animal models of sepsis generally include two closely adjacent “hits” that mimic prior surgical procedures, acute hemorrhagic shock or burns followed in rapid succession by sepsis induced by CLP, endotoxin or bacteria injections.25-27 These two-hit models generate substantially more severe sepsis than induced by sepsis alone. This heightened susceptibility was also found in the current two-stage acute-on-chronic disease animal model that showed a higher mortality in animals with reduced renal function (FA-CLP) compared with normal renal function (Veh-CLP). FA-CLP mice had substantially increased acute kidney injury and severity of septic shock, but not liver enzyme elevations or lung inflammatory changes compared to Veh-CLP mice. It is well-known that severe shock and sepsis-induced AKI are the most important predicting factors for sepsis outcomes.28, 29 Our data indicate that pre-existing renal dysfunction worsens sepsis by amplifying additional renal damage and promoting septic shock.
VEGF plays a critical role in promoting endothelial survival and maintaining the microvasculature.30 On the other hand, high levels of VEGF can cause vascular leakage by destruction of vascular barrier function12 and are associated with heightened severity of human sepsis.31, 32 Interestingly, plasma VEGF levels were already increased in FA-CLP mice just before CLP and bilateral nephrectomy caused a large increase of plasma VEGF. Increased plasma VEGF levels have been reported in predialysis or hemodialyized patients and subtotal nephrectomized rats.33-35 VEGF production in fibrotic kidney in mouse FA-induced renal injury model was decreased.9 These data suggest that the kidney plays an important role in removing VEGF from the systemic circulation. Other factors induced by renal dysfunction might enhance systemic VEGF production and/or suppress VEGF degradation. Impaired handling of VEGF in FA-CLP mice and CKD patients could contribute to the high mortality from sepsis. In the present study, we found a protective effect of sFlt-1 in our FA-CLP model that was associated with a decrease in vascular permeability and prevention of severe hypotension. Recently, two different groups reported that VEGF plays an important pathophysiologic role in sepsis. Injection of recombinant human VEGF worsened the survival of lipopolysaccharide-injected mice.14 Soluble Flt-1 (VEGF receptor-1) peptide injection improved the survival in a lipopolysaccaride(LPS) injection model and a simple CLP model.13 soluble Flt-1 treatment had multiple effects including attenuation of increased vascular permeability, depression of cardiac function,13 and enhanced pro-inflammatory cytokine productions.14 Soluble Flt-1 modulates sepsis by several different mechanisms and further investigations are required to clarify the precise mechanisms of action of sFlt-1 in this FA-CLP model.
Infectious complications in CKD patients are important causes of their morbidity and mortality. CKD patients are at significant risk of hospitalization for sepsis.19, 22 It is reported that mortality associated with systemic bacteremia is significantly higher in patients with pre-existing CKD (serum creatinine above 3 mg/dl).21 Uremia is associated with alterations in host defense systems and increases the risk of bacterial infections through a number of possible mechanisms such as impaired neutrophil activation and deficient cell immunity.36, 37 In the present study, bacterial counts in blood and peritoneal fluid after CLP-induced sepsis were significantly higher in FA-CLP mice than Veh-CLP mice. We also found significantly higher levels of anti-inflammatory cytokine IL-10 in FA-CLP mice. It is reported that IL-10 in septic patient serum could deactivate human monocytes38 and impaired antigen presentation of human monocytes induced by lipopolysaccharide (LPS) is dependent on IL-10.39 In human sepsis, apoptosis was detected dominantly in lymphocytes40 and several strategies to decrease immune cell apoptosis has been reported to improve the survival from sepsis by CLP.41-43 FA-CLP mice showed more splenocyte apoptosis after sepsis compared with Veh-CLP mice. It is reported that chloroquine improved survival of CLP-induced sepsis following hemorrhagic shock while increasing splenocyte proliferation and IL-2 production.15 We also found that chloroquine improved sepsis via decreasing splenocyte apoptosis and serum IL-10 levels.16 In the present study, chloroquine treatment improved bacteremia, decreased IL-10 levels, and splenocyte apoptosis in FA-CLP mice.
Numerous basic research studies have investigated the multiple pathophysiological mechanisms of sepsis. However, clinical trials targeting specific pathways have not been successful, with the possible exception of activated protein C, although this is controversial.44 There are several possible reasons.6 First, some of these drug targets (e.g. tumor necrosis factor-α) were selected on the basis of results from animal models that do not replicate human sepsis. We established our CLP model to replicate human sepsis and sepsis-induced AKI by administrating fluid and broad-spectrum antibiotics.8 In addition, we introduced a pre-existing co-morbidity to mimic the common observation that human sepsis occurs more commonly in patients with underlying chronic diseases.1 Second, several therapies were extremely narrowly focused (e.g., target a single pro-inflammatory cytokine). As discussed above, our FA-CLP model is sufficiently complex since it includes both pre-existing renal dysfunction and polymicrobial sepsis. Therefore, we treated FA-CLP mice with a combination of agents that act on apparently different pathways; soluble Flt-1 for vascular dysfunction and chloroquine for altered host defense. The survival advantage from the combination treatment was statistically significant, whereas the individual treatments were not. The highest survival rate and improved systemic hemodynamics as measured by blood pressure and heart rate in the combination treatment group suggests that multiple therapeutic interventions may be required for the treatment of sepsis complicated with co-morbidity. We did not measure the therapeutic window for the combination therapy; this would need to be tested to determine whether this strategy might be considered to pre-empt or treat patients with established sepsis and/or sepsis-AKI.
In conclusion, we developed a new clinically relevant murine two-stage model of sepsis in the setting of pre-existing renal dysfunction. This model replicated the clinical findings of higher mortality of sepsis in CKD patients. We also found that combination therapy of soluble Flt-1 and chloroquine, which block vascular and immunological dysfunction, respectably showed the best survival rate. Our results strongly suggest that combination of complementary therapeutic approaches might be needed to treat human sepsis.
Methods
Folic acid injection and subsequent cecal ligation and puncture (FA-CLP) model
All animal experiments were conducted in accordance with an animal study protocol approved by the NIDDK animal care and use committee. Eight week old male CD-1 mice (Charles River Laboratories, Wilmington, MA) were used. Mice were administered FA (Sigma-Aldrich, St. Louis, MO) intraperitoneally at a dose of 250 mg/kg in vehicle (0.2 ml of 0.3 mM NaHCO3) or given vehicle alone. Two days later, 60 – 70% of the mice developed acute kidney injury (defined as BUN > 100 mg/dl) as previously.45, 46 In all experiments, we only used animals with sufficient acute renal damage (BUN > 100 mg/dl) at 48 hr after FA injection.
Cecal ligation and puncture (CLP) surgery was performed on FA-treated (FA-CLP) and vehicle-treated mice (Veh-CLP) at two weeks after injection. Under isoflurane anesthesia, a 4-0 silk ligature was placed 8 mm from the cecal tip. The length of 8 mm was shown to cause sub-lethal sepsis in normal CD-1 mouse.47 The cecum was punctured twice with a 21-gauge needle and gently squeezed to express a 1 mm column of fecal material. 1 ml of prewarmed normal saline (NS) was injected intraperitoneally. Treatment with fluid and antibiotic was started at 6 hr after surgery with subcutaneous injection of imipenem/cilastatin (14 mg/kg) in 1 ml of NS. Animals were killed at 6 and 18 hr after surgery for collecting specimens. In the survival study, treatment was continued every 12 hr with imipenem/cilastatin (7 mg/kg) in 1 ml of NS.
Bilateral nephrectomy
Under isoflurane anesthesia, the kidneys were exposed from flank, dissected and removed after the pedicles were ligated using 4-0 silk sutures. The wounds were closed in two layers using 6-0 nylon sutures and surgical staples. 1 ml of prewarmed normal saline (NS) was injected intraperitoneally. Animals received antibiotics and fluid treatment at 6 hr as described above and were killed 18 hr after surgery or observed for survival analysis.
Measurement of GFR in conscious mice
GFR was measured by FITC-inulin clearance.48 Blood samples were collected from the tail vein at 3, 7, 10, 15, 35, 55 and 75 min after single FITC-inulin (3.7 μl/g body wt) injection. Fluorescence was determined by a Nanodrop-ND-3300 fluorescence spectrometer (Nanodrop Technologies, Wilmington, DE). GFR was calculated using a two-compartment model of two-phase exponential decay.
Treatment of soluble Flt-1 and chloroquine
Recombinant human soluble Flt-1 domain D1-3 (Cell Sciences, Canton, MA) at the dose of 1 μg per mouse or an equal volume of NS was injected intravenously every 3 hr (four doses), beginning 1 hr after CLP.13 Chloroquine (CQ, Sigma-Aldrich Inc.) at the dose of 50 mg/kg or an equal volume of saline was administered by oral gavage at 3 hr before CLP surgery. When testing combination treatment, mice were randomly assigned to the following groups: sFlt-1 and CQ, sFlt-1 and vehicle (p.o.), CQ and vehicle (i.v.), or vehicle only (p.o. and i.v.).
Measurement of blood pressure and heart rate
The mean blood pressure and heart rate were measured by radiotelemetry.49 A telemeter catheter was implanted in the left carotid artery and advanced to the aortic arch. The attached telemetry transmitter (model TA11PA-C10, Data Sciences International, St. Paul. MN) was placed in a subcutaneous pocket on the left flank 3-5 days before CLP surgery. Blood pressure and heart rate data were analyzed from 9 hr before CLP surgery for 27 hrs.
Measurement of blood chemistry, vascular endothelial growth factor and IL-10
BUN and serum Cr was measured by a modified method of the Berthelot reaction with Urea Nitrogen (BUN) Colorimetric Reagent (Teco Diagnositcs, Anaheim, CA) and HPLC method 50 respectively. Serum potassium, asparate aminotransferase (AST) and alanine aminotransferase (ALT) were measured using an autoanalyzer (Hitachi 917, Boehringer Mannheim, Indianapolis, IN). Plasma VEGF and serum interleukin 10 (IL-10) were measured by ELISA (R&D Systems, Minneapolis, MN).
Vascular permeability assay with Evans blue dye
Mice were injected intravenously with 20 mg/kg Evans blue dye (Sigma-Aldrich). Thirty minutes after injection, peritoneal fluid was collected with 1.5 ml NS lavage. Mice were perfused with PBS through the right ventricle until blood was visably eliminated. The kidneys and lungs were weighed, snap-frozen in liquid nitrogen, and stored at −80°C. Peritoneal fluid was centrifuged for 10 min at 3,000 × g. The kidneys and lungs were homogenized in 1mL formamide and incubated 55°C for 18 hr and centrifuged at 10,000 × g for 30 min. The amount of Evans blue dye in the supernatants was analyzed by measuring absorbance at 620 nm and 740 nm as described previously.51 Results were expressed as concentration of Evans blue dye in peritoneal fluid lavage and micrograms of Evans blue dye per gm of kidney or lung (wet weight).
Bacterial count in blood and peritoneal cavity
The peritoneal cavity was lavaged with 1.5 ml sterile saline and blood was collected by cardiac puncture 18 hr after CLP surgery. Serial dilutions of lood or peritoneal fluid were plated onto tryptic soy agar (Remel, Lenexa KS) and colony counting after 24 hr incubation at 37°C. Bacterial counts were log normalized; samples that did not have detectable bacteria were assigned a value of 0.5 colony forming unit (CFU).
Morphologic evaluation of kidney and lung
4 μm kidney and lung specimens fixed in 10% formalin and embedded in paraffin were stained with Masson's trichrome and hematoxylin and eosin staining respectively. Renal tubular damage caused by CLP-induced sepsis was assessed by counting vacuolized tubules at 400X magnification using >100 randomly selected tubules from each animal.10, 11
Immunohistochemical analysis of activated caspase-3 in spleen
Immunohistochemical staining of 4 μm paraffin sections was performed with anti-activated caspase-3 antibody (Cell Signaling Technology, Beverly, MA) as described previously.52 The number of positive stained cells was determined from the mean of five randomly selected non-overlapping 200X fields in each section.
Statistical analysis
Results are expressed as mean ± SEM. Differences between groups were analyzed by Student's t-test or Mann-Whitney rank sum test. Differences among groups in the combination treatment experiments were confirmed by one-way ANOVA followed by Dunnett's test for individual comparison. Survival analyses were compared by a log-rank test with a multiple comparison correction. These calculations were done using SigmaStat ver 3.10 (Systat Software Inc, Richmond, CA). The null hypothesis was rejected when P < 0.05.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, NIDDK.
Sources of support
Intramural Research Program of the NIH, NIDDK
Footnotes
Disclosure: K.D. is a Japan Society for the Promotion of Science (JSPS) Postdoctoral Fellow for Research Abroad.
To verify these findings, we set up a second chronic kidney disease (CKD) model utilizing a modified 5/6 nephrectomy ‘remnant kidney’ model. CKD was confirmed by robust proteinuria, increases in serum creatinine and BUN, and histological evidence of chronic glomerular changes and interstitial fibrosis at 4 weeks. Subsequent sublethal CLP produced significant increases in BUN, AST, ALT at 18h in the CKD animals compared to non-CKD animals; the creatinine was increased but not significantly (as in the folic acid/sublethal model). There was also a trend toward higher mortality within 18h (Leelahavanichkul et al., unpublished observations). Therefore the worsening of sepsis by pre-existing kidney disease is not limited to the folic acid model.
References
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, et al. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Martin GS, Mannino DM, Eaton S, et al. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 3.Russell JA, Singer J, Bernard GR, et al. Changing pattern of organ dysfunction in early human sepsis is related to mortality. Crit Care Med. 2000;28:3405–3411. doi: 10.1097/00003246-200010000-00005. [DOI] [PubMed] [Google Scholar]
- 4.Uchino S, Kellum JA, Bellomo R, et al. Acute renal failure in critically ill patients: a multinational, multicenter study. Jama. 2005;294:813–818. doi: 10.1001/jama.294.7.813. [DOI] [PubMed] [Google Scholar]
- 5.Guidet B, Aegerter P, Gauzit R, et al. Incidence and impact of organ dysfunctions associated with sepsis. Chest. 2005;127:942–951. doi: 10.1378/chest.127.3.942. [DOI] [PubMed] [Google Scholar]
- 6.Riedemann NC, Guo RF, Ward PA. The enigma of sepsis. J Clin Invest. 2003;112:460–467. doi: 10.1172/JCI19523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doi K, Okamoto K, Negishi K, et al. Attenuation of folic Acid-induced renal inflammatory injury in platelet-activating factor receptor-deficient mice. Am J Pathol. 2006;168:1413–1424. doi: 10.2353/ajpath.2006.050634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miyaji T, Hu X, Yuen PS, et al. Ethyl pyruvate decreases sepsis-induced acute renal failure and multiple organ damage in aged mice. Kidney Int. 2003;64:1620–1631. doi: 10.1046/j.1523-1755.2003.00268.x. [DOI] [PubMed] [Google Scholar]
- 9.Yuan HT, Li XZ, Pitera JE, et al. Peritubular capillary loss after mouse acute nephrotoxicity correlates with down-regulation of vascular endothelial growth factor-A and hypoxia-inducible factor-1 alpha. Am J Pathol. 2003;163:2289–2301. doi: 10.1016/s0002-9440(10)63586-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yasuda H, Yuen PS, Hu X, et al. Simvastatin improves sepsis-induced mortality and acute kidney injury via renal vascular effects. Kidney Int. 2006;69:1535–1542. doi: 10.1038/sj.ki.5000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dear JW, Kobayashi H, Jo SK, et al. Dendrimer-enhanced MRI as a diagnostic and prognostic biomarker of sepsis-induced acute renal failure in aged mice. Kidney Int. 2005;67:2159–2167. doi: 10.1111/j.1523-1755.2005.00321.x. [DOI] [PubMed] [Google Scholar]
- 12.Weis SM, Cheresh DA. Pathophysiological consequences of VEGF-induced vascular permeability. Nature. 2005;437:497–504. doi: 10.1038/nature03987. [DOI] [PubMed] [Google Scholar]
- 13.Yano K, Liaw PC, Mullington JM, et al. Vascular endothelial growth factor is an important determinant of sepsis morbidity and mortality. J Exp Med. 2006;203:1447–1458. doi: 10.1084/jem.20060375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsao PN, Chan FT, Wei SC, et al. Soluble vascular endothelial growth factor receptor-1 protects mice in sepsis. Crit Care Med. 2007;35:1955–1960. doi: 10.1097/01.CCM.0000275273.56547.B8. [DOI] [PubMed] [Google Scholar]
- 15.Ertel W, Morrison MH, Ayala A, et al. Chloroquine attenuates hemorrhagic shock-induced immunosuppression and decreases susceptibility to sepsis. Arch Surg. 1992;127:70–75. 75–76. doi: 10.1001/archsurg.1992.01420010084012. discussion. [DOI] [PubMed] [Google Scholar]
- 16.Yasuda H, Leelahavanichkul A, Tsunoda S, et al. Chloroquine and inhibition of Toll-like receptor 9 protect from sepsis-induced acute kidney injury. Am J Physiol Renal Physiol. 2008 doi: 10.1152/ajprenal.00461.2007. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sarnak MJ, Levey AS, Schoolwerth AC, et al. Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation. 2003;108:2154–2169. doi: 10.1161/01.CIR.0000095676.90936.80. [DOI] [PubMed] [Google Scholar]
- 18.Tonelli M, Wiebe N, Culleton B, et al. Chronic kidney disease and mortality risk: a systematic review. J Am Soc Nephrol. 2006;17:2034–2047. doi: 10.1681/ASN.2005101085. [DOI] [PubMed] [Google Scholar]
- 19.Thamer M, Ray NF, Fehrenbach SN, et al. Relative risk and economic consequences of inpatient care among patients with renal failure. J Am Soc Nephrol. 1996;7:751–762. doi: 10.1681/ASN.V75751. [DOI] [PubMed] [Google Scholar]
- 20.Sarnak MJ, Jaber BL. Mortality caused by sepsis in patients with end-stage renal disease compared with the general population. Kidney Int. 2000;58:1758–1764. doi: 10.1111/j.1523-1755.2000.00337.x. [DOI] [PubMed] [Google Scholar]
- 21.Shmuely H, Pitlik S, Drucker M, et al. Prediction of mortality in patients with bacteremia: the importance of pre-existing renal insufficiency. Ren Fail. 2000;22:99–108. doi: 10.1081/jdi-100100856. [DOI] [PubMed] [Google Scholar]
- 22.Naqvi SB, Collins AJ. Infectious complications in chronic kidney disease. Adv Chronic Kidney Dis. 2006;13:199–204. doi: 10.1053/j.ackd.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 23.Parker N, Turk MJ, Westrick E, et al. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal Biochem. 2005;338:284–293. doi: 10.1016/j.ab.2004.12.026. [DOI] [PubMed] [Google Scholar]
- 24.Foundation NK. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis. 2002;39:S1–266. [PubMed] [Google Scholar]
- 25.Garrison RN, Spain DA, Wilson MA, et al. Microvascular changes explain the “two-hit” theory of multiple organ failure. Ann Surg. 1998;227:851–860. doi: 10.1097/00000658-199806000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clancy KD, Lorenz K, Dries D, et al. Chlorpromazine modulates cytokine expression in the liver and lung after burn injury and endotoxemia. J Trauma. 2000;48:215–222. 222–213. doi: 10.1097/00005373-200002000-00003. discussion. [DOI] [PubMed] [Google Scholar]
- 27.Bauhofer A, Lorenz W, Kohlert F, et al. Granulocyte colony-stimulating factor prophylaxis improves survival and inflammation in a two-hit model of hemorrhage and sepsis. Crit Care Med. 2006;34:778–784. doi: 10.1097/01.ccm.0000201900.01000.6b. [DOI] [PubMed] [Google Scholar]
- 28.Russell JA. Management of sepsis. N Engl J Med. 2006;355:1699–1713. doi: 10.1056/NEJMra043632. [DOI] [PubMed] [Google Scholar]
- 29.Schrier RW, Wang W. Acute renal failure and sepsis. N Engl J Med. 2004;351:159–169. doi: 10.1056/NEJMra032401. [DOI] [PubMed] [Google Scholar]
- 30.Kang DH, Johnson RJ. Vascular endothelial growth factor: a new player in the pathogenesis of renal fibrosis. Curr Opin Nephrol Hypertens. 2003;12:43–49. doi: 10.1097/00041552-200301000-00008. [DOI] [PubMed] [Google Scholar]
- 31.Pickkers P, Sprong T, Eijk L, et al. Vascular endothelial growth factor is increased during the first 48 hours of human septic shock and correlates with vascular permeability. Shock. 2005;24:508–512. doi: 10.1097/01.shk.0000190827.36406.6e. [DOI] [PubMed] [Google Scholar]
- 32.van der Flier M, van Leeuwen HJ, van Kessel KP, et al. Plasma vascular endothelial growth factor in severe sepsis. Shock. 2005;23:35–38. doi: 10.1097/01.shk.0000150728.91155.41. [DOI] [PubMed] [Google Scholar]
- 33.Harper SJ, Downs L, Tomson CR, et al. Elevated plasma vascular endothelial growth factor levels in non-diabetic predialysis uraemia. Nephron. 2002;90:341–343. doi: 10.1159/000049071. [DOI] [PubMed] [Google Scholar]
- 34.Jacobi J, Porst M, Cordasic N, et al. Subtotal nephrectomy impairs ischemia-induced angiogenesis and hindlimb re-perfusion in rats. Kidney Int. 2006;69:2013–2021. doi: 10.1038/sj.ki.5000448. [DOI] [PubMed] [Google Scholar]
- 35.Pawlak K, Pawlak D, Mysliwiec M. Possible association between circulating vascular endothelial growth factor and oxidative stress markers in hemodialysis patients. Med Sci Monit. 2006;12:CR181–185. [PubMed] [Google Scholar]
- 36.Girndt M, Sester U, Sester M, et al. Impaired cellular immune function in patients with end-stage renal failure. Nephrol Dial Transplant. 1999;14:2807–2810. doi: 10.1093/ndt/14.12.2807. [DOI] [PubMed] [Google Scholar]
- 37.Jaber BL. Bacterial infections in hemodialysis patients: pathogenesis and prevention. Kidney Int. 2005;67:2508–2519. doi: 10.1111/j.1523-1755.2005.00364.x. [DOI] [PubMed] [Google Scholar]
- 38.Brandtzaeg P, Osnes L, Ovstebo R, et al. Net inflammatory capacity of human septic shock plasma evaluated by a monocyte-based target cell assay: identification of interleukin-10 as a major functional deactivator of human monocytes. J Exp Med. 1996;184:51–60. doi: 10.1084/jem.184.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wolk K, Docke WD, von Baehr V, et al. Impaired antigen presentation by human monocytes during endotoxin tolerance. Blood. 2000;96:218–223. [PubMed] [Google Scholar]
- 40.Hotchkiss RS, Swanson PE, Freeman BD, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27:1230–1251. doi: 10.1097/00003246-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 41.Oberholzer C, Tschoeke SK, Moldawer LL, et al. Local thymic caspase-9 inhibition improves survival during polymicrobial sepsis in mice. J Mol Med. 2006:1–7. doi: 10.1007/s00109-005-0017-1. [DOI] [PubMed] [Google Scholar]
- 42.Weaver JG, Rouse MS, Steckelberg JM, et al. Improved survival in experimental sepsis with an orally administered inhibitor of apoptosis. Faseb J. 2004;18:1185–1191. doi: 10.1096/fj.03-1230com. [DOI] [PubMed] [Google Scholar]
- 43.Wesche-Soldato DE, Chung CS, Lomas-Neira J, et al. In vivo delivery of caspase-8 or Fas siRNA improves the survival of septic mice. Blood. 2005;106:2295–2301. doi: 10.1182/blood-2004-10-4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eichacker PQ, Natanson C, Danner RL. Surviving sepsis--practice guidelines, marketing campaigns, and Eli Lilly. N Engl J Med. 2006;355:1640–1642. doi: 10.1056/NEJMp068197. [DOI] [PubMed] [Google Scholar]
- 45.Bosch RJ, Woolf AS, Fine LG. Gene transfer into the mammalian kidney: direct retrovirus-transduction of regenerating tubular epithelial cells. Exp Nephrol. 1993;1:49–54. [PubMed] [Google Scholar]
- 46.Surendran K, McCaul SP, Simon TC. A role for Wnt-4 in renal fibrosis. Am J Physiol Renal Physiol. 2002;282:F431–441. doi: 10.1152/ajprenal.0009.2001. [DOI] [PubMed] [Google Scholar]
- 47.Doi K, Hu X, Yuen PS, et al. AP214, an analogue of alpha-melanocyte-stimulating hormone, ameliorates sepsis-induced acute kidney injury and mortality. Kidney Int. 2008 doi: 10.1038/ki.2008.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen L, Kim SM, Oppermann M, et al. Regulation of renin in mice with Cre recombinase-mediated deletion of G protein Gsalpha in juxtaglomerular cells. Am J Physiol Renal Physiol. 2007;292:F27–37. doi: 10.1152/ajprenal.00193.2006. [DOI] [PubMed] [Google Scholar]
- 49.Kim SM, Chen L, Mizel D, et al. Low plasma renin and reduced renin secretory responses to acute stimuli in conscious COX-2-deficient mice. Am J Physiol Renal Physiol. 2007;292:F415–422. doi: 10.1152/ajprenal.00317.2006. [DOI] [PubMed] [Google Scholar]
- 50.Yuen PS, Dunn SR, Miyaji T, et al. A simplified method for HPLC determination of creatinine in mouse serum. Am J Physiol Renal Physiol. 2004;286:F1116–1119. doi: 10.1152/ajprenal.00366.2003. [DOI] [PubMed] [Google Scholar]
- 51.Standiford TJ, Kunkel SL, Lukacs NW, et al. Macrophage inflammatory protein-1 alpha mediates lung leukocyte recruitment, lung capillary leak, and early mortality in murine endotoxemia. J Immunol. 1995;155:1515–1524. [PubMed] [Google Scholar]
- 52.Dear JW, Yasuda H, Hu X, et al. Sepsis-induced organ failure is mediated by different pathways in the kidney and liver: acute renal failure is dependent on MyD88 but not renal cell apoptosis. Kidney Int. 2006;69:832–836. doi: 10.1038/sj.ki.5000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.