

Abstract
Receptor protein tyrosine phosphatase T (PTPRT/PTPρ) is frequently mutated in human cancers including colon, lung, gastric and skin cancers. More than half of the identified tumor-derived mutations are located in the extracellular part of PTPρ. However, the functional significance of those extracellular domain mutations remains to be defined. Here we report that the extracellular domain of PTPρ mediates homophilic cell-cell aggregation. This homophilic interaction is very specific, as PTPρ does not interact with its closest homolog PTPμ in a cell aggregation assay. We further demonstrated that all 5 tumor-derived mutations located in the N-terminal MAM and Ig domains impair, to varying extents, their ability to from cell aggregates, indicating that those mutations are loss-of-function mutations. Our results suggest that PTPρ may play an important role in cell-cell adhesion and mutational inactivation of this phosphatase could promote tumor migration and metastasis.
Introduction
Tyrosine phosphorylation is coordinately controlled by protein tyrosine kinases and phosphatases and is a central feature of many signaling pathways involved in tumor development (1). While activating mutations in protein tyrosine kinases have been shown to play vital roles in tumorigenesis (1), the role of phosphatases is less well-defined. We recently identified PTPRT, also known as PTPρ, as the most frequently mutated PTP gene in colorectal cancers (CRCs) (2). PTPRT was also mutationally altered in lung, gastric cancers and melanomas (2). The spectrum of mutations, which included nonsense mutations and frameshifts, suggested that these mutations were inactivating (2). Biochemical analyses demonstrated that missense mutations in the catalytic domains of PTPρ diminished its phosphatase activity and overexpression of PTPρ inhibited CRC cell growth (2). Taken together, these studies strongly supported the notion that PTPρ normally acts as a tumor suppressor gene. This conclusion was also supported by a transposon-based somatic mutagenesis screen in mice, wherein PTPρ was isolated as a target gene from two different mouse transgenic sarcomas (3).
PTPRT (PTPρ) is a member of the type IIB receptor protein tyrosine phosphatase (RPTP) subfamily (4). Other members of this subfamily include PTPRM (PTPμ), PTPRK (PTPκ) and PCP2 (also called PTPλ, PTPψ, PTPRO-omicron, PTPπ, hPTP-J) (5). These 4 RPTPs share the same domain structure: an extracelluar domain, a juxtamembrane region and two phosphatase domains (6). The extracellular domains of type IIB RPTPs have high sequence identities (7, 8), all consist of a MAM (memprin/A5/PTPμ) domain, an Ig domain, and four fibronectin type III (FNIII) repeats (6). The MAM domain is suggested to play a role in protein dimerization (6). The Ig domain is a disulfide structure that is found in many cell surface proteins and has been shown to mediate homophilic and heterophilic interactions between cell adhesion molecules (6). The FNIII motif was originally identified in the extracellular matrix protein fibronectin and later found to be present in many immunoglobulin superfamily cell adhesion molecules (6). We have identified ~15 somatic mutations that are localized in the extracellular domain of PTPRT/PTPρ. How these mutations affect the functions of PTPρ remains to be determined.
Three close homologs of PTPρ, PTPμ, PTPκ and PTPλ (PCP-2), are known to mediate homophilic cell-cell adhesion (9– 12). Expression of the full-length PTPμ (9) (10) or a construct encoding the extracellular, transmembrane and 55 amino acids of the juxtamembrane domain of PTPμ in non-adhesive Sf9 insect cells induces cell aggregation (9). Furthermore, homophilic binding of PTPμ was demonstrated between PTPμ-coated fluorescent beads and cells that endogenously express PTPμ (9). The minimal region required for homophilic binding was mapped to the Ig domain of PTPμ (13). In addition, the Ig domain is required for proper cell surface localization (14). However, assays that test cell-cell aggregation demonstrate that the MAM and Ig domains as well as the first two fibronectin type III repeats are required for efficient cell-cell aggregation (15– 18). Similarly, PTPκ was also demonstrated to mediate homophilic cell-cell aggregation (11). Here we report that PTPRT (PTPρ), like its homologs, mediates homophilic cell-cell aggregation in Sf9 cells. Most importantly, the tumor-derived mutations located in the MAM and Ig domains are defective in this cell-cell adhesion function.
Results
The extracellular domain of PTPρ mediates cell-cell aggregation in Sf9 cells
To test whether PTPρ mediates homophilic cell-cell adhesion, we made the following PTPρ baculoviral constructs expressing: (1) full-length PTPρ; (2) the intracellular fragment of PTPρ (Intra); (3) the extracellular fragment of PTPρ with its transmembrane domain (Extra-TM); (4) the extracellular fragment of PTPρ with its transmembrane, juxtamembrane and wedge domains (Extra-JMD-W); (5) a chimera containing the extracellular and transmembrane domains of the EGF receptor and the intracellular PTP domains of PTPρ (Fig. 1A). All of those fragments were tagged with V5 epitope tags. Proteins of the expected size were expressed in Sf9 cells infected with those baculoviral constructs (Fig. 1B). All of these proteins, but the intracellular PTPρ, were expressed on the surface on Sf9 cells (Fig. 1C). To test whether these PTPρ recombinant proteins mediate cell-cell adhesion, a cell aggregation assay was performed following the procedure of Brady-Kalnay et al (9). Sf9 cells infected with a baculovirus encoding PTPμ, which was previously demonstrated to mediate cell-cell aggregation, were treated in parallel as a positive control (Fig. 2). As shown in Fig 2, the non-adhesive Sf9 cells expressing either the full-length PTPρ or PTPρ-JMD-W formed cell aggregates. On the contrary, Sf9 cells expressing either the intracellular fragments of PTPρ or the EGFR/PTPρ chimeras remained as a suspension of single cells (Fig 1C). These results indicate that the extracullar fragment of PTPρ play an important role in mediating cell-cell aggregation and the phosphatase activity of PTPρ is not required for this function. Surprisingly, although the construct that encodes the PTPρ extracellular fragment with the transmembrane domain sequences only (Extra-TM) was expressed on the cell surface (see Figure 7), this protein failed to mediate cell aggregation of Sf9 cells, suggesting that additional juxtamembrane sequences are required for either forming or stablizing the cell aggregates.
Figure 1. Expression of PTPρ constructs in Sf9 cells.

(A) Schematic diagram of PTPρ constructs. MAM domain (gray oval); Ig domain (pink circle); Juxtamembrane domain (yellow oval); Wedge domain (red circle); Phosphatase domain (green rectangles). (B) Western blot of lysates from Sf9 cells expressing the indicated PTPρ constructs. (C) Immunofluorescent staining of Sf9 cells expressing the indicated PTPρ proteins.
Figure 2. PTPρ mediates cell-cell aggregation.

Sf9 cells were infected with indicated baculoviral constructs for 48 hrs. Cell aggregation assay was performed under low shear conditions and representative phase pictures were captured with a microscope.
Figure 7. Cell Surface Protein expression of PTPμ and the PTPρ mutant proteins.
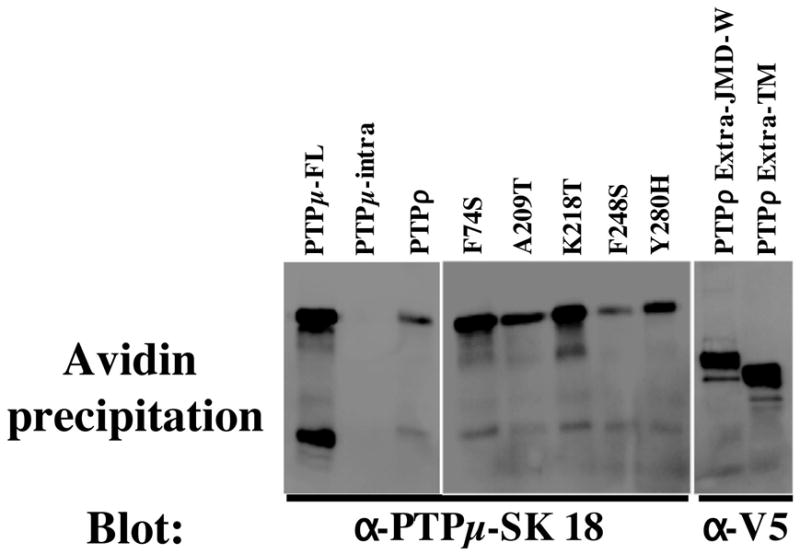
The cell surface proteins were labeled with Sulfo-NHS-SS-Biotin. The biotinylated proteins were isolated on NeutrAvidin Gel. The bound proteins were eluted, run on SDS-PAGE gels, transferred to nitrocellulose and immunoblotted with antibodies to PTPμ (SK18) or the V5 tag. The SK18 antibody recognizes the cytoplasmic domain of both PTPμ and PTPρ.
PTPρ mediates highly specific homophilic cell-cell aggregation
To test whether PTPρ mediates homophilic cell-cell aggregation, one set of Sf9 cells were infected with virus expressing enhanced green fluorescent protein (EGFP) and another set of Sf9 cells were co-infected with viruses expressing either the full-length PTPρ or red fluorescent protein (RFP). The GFP expressing cells were mixed with equal number cells expressing full-length PTPρ (co-expressing RFP) and the aggregation assays were performed. As shown in Fig 3A, the GFP labeled cells did not aggregate and did not contribute to the aggregates formed by cells expressing full-length PTPρ (RFP). These results strongly suggest that the aggregation of Sf9 cells expressing PTPρ is mediated through a homophilic interaction among PTPρ molecules in trans, but not through a heterophilic interaction between molecules expressed on the surface of Sf9 cells. Interestingly, the homophilic interaction of PTPρ is very specific. Although 63% of the amino acid sequences of the extracellular domain of PTPρ are identical with that of PTPRM (PTPμ), Sf9 cells expressing PTPρ (co-expressing RFP) did not aggregate with PTPμ expressing cells (co-expressing GFP) as shown in Fig. 3B.
Figure 3. PTPρ mediates homophilic cell-cell aggregation.

(A) Sf9 cells were infected with GFP while another flask of Sf9 cells was co-infected with PTPρ and RFP viruses. Sf9 cells from the two flasks were mixed at a 1:1 ratio and a cell aggregation assay was performed. (B) One flask of Sf9 cells was co-infected with PTPμ and GFP viruses, while another flask of Sf9 cells was co-infected with PTPρ and RFP viruses. Sf9 cells from the two flasks were mixed at 1:1 ratio and cell aggregation assay was performed. Images of cell aggregates were captured under phase, GFP and RFP fluorescent filters respectively.
The tumor-derived PTPRT/PTPρ mutations in the MAM and Ig domains are defective in cell-cell aggregation
Several studies demonstrated that the MAM and Ig domains as well as the first two FNIII repeats of PTPμ are the minimal essential regions required for its homophilic interaction (13, 15, 17, 18). We identified 5 tumor-specific mutations that are located in the two domains (2). Among these five mutations, the F74S mutation is located in the MAM domain and the other four mutations (A209T, K218T, F248S and Y280H) are located in the Ig domain (Fig. 4A). To test whether these mutations affect their ability to mediate cell-cell aggregation in Sf9 cells, we engineered the five mutations by site-directed mutagenesis using the full-length PTPρ baculoviral construct as the template. The five mutant proteins are expressed at the expected size and at similar levels to wild-type PTPρ on a western blot (Fig. 4B), indicating that these mutations do not affect their protein stability. None of these mutations significantly alter their cell surface expression, as all five mutant PTPρ proteins showed strong plasma membrane staining (Fig. 4C and Fig. 7). However, as shown in Fig. 5, all mutant PTPρ proteins formed smaller aggregates than that of the wild-type PTPρ protein. To quantify the cells aggregates, percentage of cells that are incorporate into > 5-cell clusters were counted (as described in the material and methods section). As shown in Fig. 5, Sf9 cells expressing all the mutants formed significantly less aggregates than that of cells expressing wild-type PTPρ. The deletion construct of PTPρ lacking the two phosphatase domains (PTPρ Extra-JMD-W) and the K218T mutant exhibited similar levels of aggregation that were statistically less than wild-type PTPρ (p < 0.01). Taken together, our data demonstrated that the five tumor-derived mutations located the MAM and Ig domains of PTPρ are deficient in cell-cell adhesion. Therefore, these mutations exhibit varying degrees of loss-of-function in cell adhesion.
Figure 4. Expression of tumor-derived PTPρ mutants in Sf9 cells.

(A) Schematic diagram of PTPρ mutations located in the MAM and Ig domain. (B) Western blot of lysates from Sf9 cells expressing with indicated PTPρ mutations. (C) Immunofluorescent staining of Sf9 cells expressing the indicated PTPρ mutant proteins.
Figure 5. Tumor-derived mutations of PTPρ are defective in cell-cell aggregation.

Cell aggregation assay of Sf9 cells expressing either wild-type or the indicated mutation of PTPρ was performed under low shear conditions. Representative phase pictures were captured with a microscope.
Mechanistically, the defect of the PTPρ mutations in cell-cell adhesion could be resulted from either impaired ability in homophilic binding or reduced cell surface expression. Although immunofluorescent staining indicated that those mutations do not alter cell surface expression qualitatively, it is imperative to quantify cell surface expression of mutant PTPρ proteins. As described in detail in the method section, baculovirus-infected Sf9 cell surface proteins were labeled with Sulfo-NHS-SS-Biotin and isolated on immobilized NeutrAvidin Gel. Western blots were performed with antibodies to PTPμ (SK18) or the V5 tag. The SK18 antibody recognizes the cytoplasmic domain of both PTPμ and PTPρ. The results shown in Fig. 7 demonstrate that full length PTPμ and the wild-type, mutant and deleted PTPρ proteins were expressed at the cell surface. As a control for a non-cell surface protein, the cytoplasmic intracellular domain of PTPμ (PTPμ-intra) was expressed that is detected by SK18 (9). PTPμ intra does not contain an extracellular domain and therefore is not biotinylated nor precipitated by the NeutrAvidin gel (Fig. 7). Quantitatively, the levels of cell surface expression of the 5 tumor-derived PTPρ mutations were similar to that of wild-type protein. If the cell surface expression level (CSE value) of wild-type PTPρ is arbitrarily assigned as 1, the CSE values of F74S, A209T, K218T, F248S and Y280H mutations were 2.3, 1.1, 1.6, 0.6 and 0.8 respectively. Interestingly, some PTPρ mutants that contain the extracellular domain but are unable to mediate cell-cell aggregation are expressed at higher levels on the Sf9 cell surface than wild-type PTPρ. These results suggest that defect of PTPρ mutation in cell-cell adhesion is mainly due to reduced homophilic binding affinity of the mutant proteins. Interestingly, the cell surface expression levels PTPρ Extra-TM (CSE value = 27) and PTPρ Extra-JMD-W (CSE value = 20) were much higher than that of full-length protein for unknown reasons.
Discussion
We demonstrated that PTPRT/PTPρ meditates homophilic cell-cell aggregation and five tumor-derived mutations located in the MAM and Ig domains impair this function. Our results strongly suggest that those PTPρ loss-of-function mutations are selected for during the process of tumor development; therefore, these mutations should play a causal role in tumorigenesis. We chose to examine whether the MAM and Ig domain mutations are deficient in cell aggregation, because previous studies indicated that these two domains of PTPμ, a close homolog of PTPρ, are required for its cell-cell aggregation function (13, 15, 17). Recently, the crystal structure of the N-terminal MAM and Ig domains of PTPμ was determined at 2.7 Å resolution (17, 18). Five tumor-derived mutations of PTPρ are located in the two domains and all of them can be mapped to equivalent residues in the MAM and Ig domain structure of PTPμ. Based on the crystal structure, the authors predicted that 4 of them (F74S, A209T, F248S and Y280H) would cause significant perturbation of the structure of PTPRT, because these mutations dramatically alter the character of buried side-chains (17). Moreover, the fifth mutation K218T is located on the surface of the Ig domain and the authors predicted that this mutation would directly alter protein-protein interactions (17). Our data indicated that the K218T mutation affects the size of aggregates and only a small effect on the percentage of cells incorporated into the aggregates, suggesting that the K218T mutant may decrease the binding affinity of PTPρ. Interestingly, the other four mutations significantly reduce the percentage of aggregation, therefore, these data suggest that the F74S, A209T, F248S and Y280H mutations dramatically alter the domain structure and lead to perturbation of the homophilic interaction of PTPρ. Our previous studies showed that the tumor-derived phosphatase domain mutations of PTPρ diminish its phosphatase activity and overexpression of PTPρ suppresses the growth of colorectal cancer cells, indicating that PTPρ normally act as a tumor suppressor (2). The loss-of-function nature of these five extracellular mutations provides further supportive evidence to this notion.
The finding that PTPρ mediates homophilic cell aggregation of non-adhesive insect Sf9 cells suggests that this protein could be involved in cell-cell adhesion in human epithelial cells where it is endogenously expressed. The type IIB RPTP subfamily consists of PTPμ, PTPκ, PTPρ and PCP-2. It has been shown that PTPμ, PTPκ and PTPλ (PCP-2), mediate homophilic cell-cell adhesion (9–12). Interestingly, the homophilic interaction of these RPTPs seems to be highly specific, because we and others showed that neither PTPρ nor PTPκ (15) interact with PTPμ. It is worth noting that the juxtamembrane domain of PTPρ may be necessary for its cell adhesion function. As shown in Figure 2, Sf9 cells expressing the extracellular and juxtamembrane domains of PTPρ, but not cells expressing the extracellular and transmembrane portion of the protein (PTPρ Extra-TM), formed cell aggregates, although both proteins were expressed on the cell surface (Fig. 7). Expression of a construct encoding the extracellular, transmembrane and only 55 amino acids of the juxtamembrane domain of PTPμ in non-adhesive Sf9 insect cells induced cell aggregation (9). It is possible that cis interactions between the membrane proximal juxtamembrane domains are required for initiating, stabilizing or maintaining cell-cell adhesion. Interestingly, there are two PTPRT mutations (K788E and R790I) located in the juxtamembrane domain that are found in lung cancers (1). Moreover, Besco et al. showed that PTPρ interacts with cell adhesion molecules including E-cadherin, α, β, and γ-catenin, and α;-actinin (19). PTPμ also binds the cadherin-catenin complex (7, 20–22). Loss of cell-cell adhesion, such as genetic and epigenetic inactivation of cell adhesion molecule E-cadherin, is thought to be a prerequisite for carcinoma invasion and metastasis (23). Therefore, mutational inactivation of the cell adhesion function PTPρ may also play a critical role in tumor migration and metastasis.
Materials and Methods
Cell culture
Insect Sf9 cells (BD Bioscience) were maintained at 27°C in TNM-FH insect medium (BD Bioscience).
Plasmid and Baculovirus Construction
To tag our protein of interest with a V5 tag, we first constructed a pVL1393-V5-His vector. A 450bp fragment containing the V5 and 6xHis epitope was digested with Bam HI and Pme I restriction enzymes from pcDNA3.1 and then cloned into the pVL1393 vector cleavage by Bam HI and Bgl II (blunt ended). The full-length PTPRT/PTPρ was inserted and in-frame fused with the V5 tag in the pVL1393-V5-His vector. PCR were used to generate the following constructs: PTPρ Intra (663 amino acids, base pair 2401–4392), PTPρ Extra-TM (787 amino acids, base pair 1–2361) and PTPρ Extra-JMD-W (916 amino acids, base pair 1–2748). A chimera containing the extracellular and transmembrane domains of the EGF receptor (673 amino acids, base pair 170–2190) and the intracellular PTP domains of PTPRT (663 amino acids, base pair 2401–4392) was generated by in-frame fusion of the EGF receptor fragment with the PTPρ Intra construct. All constructs were sequenced to ensure that no mutations were introduced by PCR. These constructs were co-transfected with the BaculoGold™ Linearized Baculovirus DNA (BD biosciences) according to manufacturer instructions to generate recombinant viruses. The PTPμ baculovirus has been described (9).
Sf9 Cell Aggregation Assays
The cell aggregation assays were performed as previously described (9) with minor modifications. Sf9 cells were harvested two days post-infection. The cell suspensions were added to glass scintillation vials and incubated at 25°C at 90 rpm in a gyratory shaker for 30 minutes. For a given condition, half of the cell suspension was poured into a 100mm Petri dish either prior to (0 time point) or after 30 minutes of aggregation. Images were captured with a Nikon inverted fluorescence microscope (TE-200). To quantify cell aggregates, 10 pictures of randomly selected fields were taken for each time point and condition in that experiment. The area of each object (single cells and aggregates) was determined by the Metamorph software (Molecular Devices) using a threshold setting of 160 and appropriate size filters that allowed the counting of cells and aggregates but not debris. The percentage of aggregation for an experiment was calculated as the average area at 30 minutes minus the average area at the zero time point divided by the average area at the 30 minute time point {N30-N0/N30}. At least three independent experiments were performed for each condition.
Western blotting
Sf9 Cells were harvested two days post-infection and lysed in RIPA buffer (50 mM Tris·HCl, pH 8.0, 0.5% Triton X-100, 0.25% sodium deoxycholate, 150 mM sodium chloride, 1 mM EDTA) with complete protease inhibitor cocktail. Western blots were performed as described (24) with a mouse anti-V5 antibody (Invitrogen) or SK18 (9).
PTPρ Immunofluorescence
Sf9 cells were fixed with 4% paraformaldehyde for 30 minutes at room temperature and permeabilized with 0.2% Triton X 100 (diluted in PBS). Cells were blocked with Image-iT™ FX signal enhancer (Invitrogen) for 30 minutes and then stained with mouse anti-V5 antibody (Invitrogen) followed by an Alexa 488-conjugated secondary antibody (Invitrogen). Images were captured with a Nikon fluorescence microscope.
Site-directed mutagenesis
The F74S, A209T, K218T, F248S and Y280H mutations were generated by fusion PCR (25) using the full-length PTPRT pVL1393-V5-His vector as the template. The primers GCAGTGCCCACAGGATCTTCCATGATGGTGAACAGCTCT and GAAGATCCTGTGGGCACTGC were used as the mutagenic primers for the F74S mutant; the primers GGTGAATGTGGGGCAGAATACCACATTTCAGTGCATTGC and TATTCTGCCCCACATTCACC were used as the mutagenic primers for the A209T mutant; the primers CGTGGTCTCAGCATGACAAG and CTTGTCATGCTGAGACCACGTCCCACCAGCAATGCACTG were used as the mutagenic primers for the K218T mutant; the primers GTGGTCAACCACAGGCGCTCCTCAGCCACAGTCAGTGTG and GAGCGCCTGTGGTTGACCAC were used as the mutagenic primers for the F248S mutant; the primers CACGCGGAGCTGATCGTGAA and TTCACGATCAGCTCCGCGTGGTTGGACACACCAGACCCA were used as the mutagenic primers for the Y280H mutant.
Quantification of cell surface expression of PTPρ and PTPμ proteins
The amount of PTPμ and the PTPρ proteins expressed at the Sf9 cell surface was determined using the Cell Surface Protein Isolation Kit [catalog # 89881] (Pierce, Rockford, IL). The kit provides the proprietary reagents referred to below including wash buffer, lysis buffer and the column. Sf9 cells were grown to 80% confluence in T25 flasks and infected with baculovirus as described above. 48 hours post-infection, the media was removed and 5ml of 0.25mg/ml Sulfo-NHS-SS-Biotin in 1X PBS was added to each flask. The flasks were placed on a rocking platform for 30min at 4°C. 250μl of Quenching Solution was added to each flask to quench the reaction. The cells were mechanically removed from the flask and centrifuged at 5000rpm for 3min, washed and centrifuged again. The cells were lysed by incubated on ice for 30min with lysis buffer, vortexed and centrifuged at 10, 000rpm for 2min. 250μl of immobilized NeutrAvidin Gel slurry was added to the column. The column was washed 3 times. The cell lysate was added to the column and incubated for 60 min at RT with rocking. The column was centrifuged for 1min at 1000rpm and washed 3 times. 200μl of sample buffer containing a final concentration of 55mM DTT was added to the gel and incubated for 60min on platform rocker at RT. The column was centrifuged for 2min at 1000rpm and the flow through was collected. The flow through was run on 6% SDS-PAGE gels, transferred to nitrocellulose. Total cell surface protein levels were normalized by immunoblotting with Strepavidin-HRP (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). The normalized cell surface protein was run on 6% SDS-PAGE gels, transferred to nitrocellulose and immunoblotted with antibodies to PTPμ (SK18) or the V5 tag followed by secondary antibody conjugated to HRP. The HRP signal was detected using a Fluor-S MAX MultiImager (Bio-Rad Laboratories Inc., Hercules, CA). HRP signal was quantitated by densitometry using the Quantity One Software from Bio-Rad. The densitometry in the results represents a comparison of the quantitation of the V5 signal for all PTPρ proteins with the number for wild-type PTPρ which we assigned a value of 1. We define this value as the cell surface expression value (CSE value).
Figure 6. Quantification of the cell-cell aggregation assays.

The percentage of aggregated Sf9 cells for each indicated protein was calculated from three independent experiments. Student t.tests were performed between wild-type PTPρ and each of the mutants (asterisk indicated that the P < 0.01).
Acknowledgments
We thank Drs. Sanford Markowitz and David Sedwick for helpful discussions, Sara Lou for preparation of the figures and graphs. This research was supported by grants from the National Institutes of Health Grant R01-CA127590, U54CA116867 (Z.W), R01-EY12251 and RO1-NS051520 (S.B.K). Additional support was obtained from the Concern Foundation, the V foundation (Z.W) and the Visual Sciences Research Center Core Grant PO-EY11373 from the National Eye Institute (S.B.K).
References
- 1.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411(6835):355–65. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 2.Wang Z, Shen D, Parsons DW, et al. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science. 2004;304(5674):1164–6. doi: 10.1126/science.1096096. [DOI] [PubMed] [Google Scholar]
- 3.Collier LS, Carlson CM, Ravimohan S, Dupuy AJ, Largaespada DA. Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature. 2005;436(7048):272–6. doi: 10.1038/nature03681. [DOI] [PubMed] [Google Scholar]
- 4.McAndrew PE, Frostholm A, White RA, Rotter A, Burghes AH. Identification and characterization of RPTP rho, a novel RPTP mu/kappa-like receptor protein tyrosine phosphatase whose expression is restricted to the central nervous system. Brain Res Mol Brain Res. 1998;56(1–2):9–21. doi: 10.1016/s0169-328x(98)00014-x. [DOI] [PubMed] [Google Scholar]
- 5.Ensslen-Craig SE, Brady-Kalnay SM. Receptor protein tyrosine phosphatases regulate neural development and axon guidance. Dev Biol. 2004;275(1):12–22. doi: 10.1016/j.ydbio.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 6.Brady-Kalnay SM. Protein tyrosine phosphatases. In: Beckerle M, editor. Cell Adhesion: Frontiers in Molecular Biology. Oxford University Press; Oxford, UK: pp. 217–258. [Google Scholar]
- 7.Brady-Kalnay SM, Mourton T, Nixon JP, et al. Dynamic interaction of PTPmu with multiple cadherins in vivo. J Cell Biol. 1998;141(1):287–96. doi: 10.1083/jcb.141.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Besco J, Popesco MC, Davuluri RV, Frostholm A, Rotter A. Genomic structure and alternative splicing of murine R2B receptor protein tyrosine phosphatases (PTPkappa, mu, rho and PCP-2) BMC Genomics. 2004;5(1):14. doi: 10.1186/1471-2164-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brady-Kalnay SM, Flint AJ, Tonks NK. Homophilic binding of PTP mu, a receptor-type protein tyrosine phosphatase, can mediate cell-cell aggregation. J Cell Biol. 1993;122(4):961–72. doi: 10.1083/jcb.122.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gebbink MF, Zondag GC, Wubbolts RW, Beijersbergen RL, van Etten I, Moolenaar WH. Cell-cell adhesion mediated by a receptor-like protein tyrosine phosphatase. J Biol Chem. 1993;268(22):16101–4. [PubMed] [Google Scholar]
- 11.Sap J, Jiang YP, Friedlander D, Grumet M, Schlessinger J. Receptor tyrosine phosphatase R-PTP-kappa mediates homophilic binding. Mol Cell Biol. 1994;14(1):1–9. doi: 10.1128/mcb.14.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng J, Wu K, Armanini M, O'Rourke N, Dowbenko D, Lasky LA. A novel protein-tyrosine phosphatase related to the homotypically adhering kappa and mu receptors. J Biol Chem. 1997;272(11):7264–77. doi: 10.1074/jbc.272.11.7264. [DOI] [PubMed] [Google Scholar]
- 13.Brady-Kalnay SM, Tonks NK. Identification of the homophilic binding site of the receptor protein tyrosine phosphatase PTP mu. J Biol Chem. 1994;269(45):28472–7. [PubMed] [Google Scholar]
- 14.Del Vecchio RL, Tonks NK. The conserved immunoglobulin domain controls the subcellular localization of the homophilic adhesion receptor protein-tyrosine phosphatase mu. J Biol Chem. 2005;280(2):1603–12. doi: 10.1074/jbc.M410181200. [DOI] [PubMed] [Google Scholar]
- 15.Zondag GC, Koningstein GM, Jiang YP, Sap J, Moolenaar WH, Gebbink MF. Homophilic interactions mediated by receptor tyrosine phosphatases mu and kappa. A critical role for the novel extracellular MAM domain. J Biol Chem. 1995;270(24):14247–50. doi: 10.1074/jbc.270.24.14247. [DOI] [PubMed] [Google Scholar]
- 16.Cismasiu VB, Denes SA, Reilander H, Michel H, Szedlacsek SE. The MAM (meprin/A5-protein/PTPmu) domain is a homophilic binding site promoting the lateral dimerization of receptor-like protein-tyrosine phosphatase mu. J Biol Chem. 2004;279(26):26922–31. doi: 10.1074/jbc.M313115200. [DOI] [PubMed] [Google Scholar]
- 17.Aricescu AR, Hon WC, Siebold C, Lu W, van der Merwe PA, Jones EY. Molecular analysis of receptor protein tyrosine phosphatase mu-mediated cell adhesion. Embo J. 2006 doi: 10.1038/sj.emboj.7600974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aricescu AR, Siebold C, Choudhuri K, et al. Structure of a Tyrosine Phosphatase Adhesive Interaction Reveals a Spacer-Clamp Mechanism. Science. 2007;317(5842):1217–20. doi: 10.1126/science.1144646. [DOI] [PubMed] [Google Scholar]
- 19.Besco JA, Hooft van Huijsduijnen R, Frostholm A, Rotter A. Intracellular substrates of brain-enriched receptor protein tyrosine phosphatase rho (RPTPrho/PTPRT) Brain Res. 2006;1116(1):50–7. doi: 10.1016/j.brainres.2006.07.122. [DOI] [PubMed] [Google Scholar]
- 20.Brady-Kalnay SM, Rimm DL, Tonks NK. Receptor protein tyrosine phosphatase PTPmu associates with cadherins and catenins in vivo. J Cell Biol. 1995;130(4):977–86. doi: 10.1083/jcb.130.4.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hiscox S, Jiang WG. Association of PTPmu with catenins in cancer cells: a possible role for E-cadherin. Int J Oncol. 1998;13(5):1077–80. doi: 10.3892/ijo.13.5.1077. [DOI] [PubMed] [Google Scholar]
- 22.Zondag GC, Reynolds AB, Moolenaar WH. Receptor protein-tyrosine phosphatase RPTPmu binds to and dephosphorylates the catenin p120(ctn) J Biol Chem. 2000;275(15):11264–9. doi: 10.1074/jbc.275.15.11264. [DOI] [PubMed] [Google Scholar]
- 23.Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer. 2004;4(2):118–32. doi: 10.1038/nrc1276. [DOI] [PubMed] [Google Scholar]
- 24.Zhang X, Guo A, Yu J, et al. Identification of STAT3 as a substrate of receptor protein tyrosine phosphatase T. PNAS. 2007;104(10):4060–4. doi: 10.1073/pnas.0611665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Higuchi R, Krummel B, Saiki RK. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 1988;16(15):7351–67. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]