

Abstract
Central memory CD4+ T cells provide a pool of lymph node-homing, antigen-experienced cells that are capable of responding rapidly after a secondary infection. We have previously described a population of central memory CD4+ T cells in Leishmania major infected mice that were capable of mediating immunity to a secondary infection. Here we show that the Leishmania-specific central memory CD4+ T cells require IL-12 in order to produce IFN-γ, demonstrating that this population needs additional signals to develop into Th1 cells. In contrast, effector cells isolated from immune mice produced IFN-γ in vitro or in vivo in the absence of IL-12. In addition, we found that when central memory CD4+ T cells were adoptively transferred into IL-12 deficient hosts many of the cells became IL-4 producers. These studies indicate that the central memory CD4+ T cell population generated during L. major infection is capable of developing into either Th1 or Th2 effectors. Thus, continued IL-12 production may be required to ensure the development of Th1 cells from this central memory T cell pool, a finding that has direct relevance to the design of vaccines dependent upon central memory CD4 T cells.
Keywords: Th1/Th2, T-cells, Parasite-protozoan, Memory, Cytokines
Introduction
Leishmania major is an intracellular protozoan parasite that causes considerable morbidity and mortality throughout the world. An IL-12-dependent CD4+ Th1 cell response is critical for the control of leishmaniasis and resolution of disease leads to life-long immunity (1, 2). Nevertheless, a vaccine for human leishmaniasis continues to prove elusive, a problem shared with several other diseases that require cell-mediated immune responses for protection. To a large extent, this is due to our limited understanding of memory CD4+ T cells. Two distinct populations of memory T cells have been described. The first, termed effector memory T cells, are similar to effector cells. They home to tissues and maintain the ability to rapidly produce cytokines upon re-stimulation. The other subset, termed central memory T cells (Tcm)3, act as a reservoir of antigen-specific T cells that can expand upon re-challenge and become effector T cells (3). Tcm cells express CD62L, which promotes their migration through the lymph nodes, a characteristic which enhances their ability to interact with dendritic cells presenting antigen (3). We found that CD4+ Tcm cells can confer immunity in mice that were re-challenged with L. major, and more importantly that they do not require the persistence of parasites to be maintained (4). Thus, CD4+ Tcm cells could be critical for the development of vaccines against a variety of pathogens, including Leishmania.
In addition to showing that Tcm cells can mediate immunity, we also found that IL-12 is required not only at the initiation of the infection to promote Th1 cell development, but also to maintain resistance to L. major in immune mice (5, 6). This could be because IL-12 enhances IFN-γ production by Th1 cells (7). Alternatively, if CD4+ Tcm cells have not yet differentiated into IFN-γ producing cells, they may require IL-12 to do so. Therefore, to distinguish between these possibilities, we analyzed both the IFN-γ responses of Th1 cells and the ability of Tcm cells to differentiate into Th1 cells in the absence of IL-12. These studies allow us to begin to draw conclusions regarding the nature of the Tcm cell population in L. major-infected mice. Although we have shown that L. major specific Tcm cells can become IFN-γ producers and promote resistance (4), it was unclear whether the Tcm cells were already predetermined to become Th1 cells. Therefore, to determine the level of plasticity in the L. major specific Tcm population, we made use of the ability of CD4+ T cells to differentiate into different effector cell subsets depending upon the cytokine environment. If the Tcm cell population isolated from an immune animal has developed directly from the effector pool then these cells should be fully differentiated, and should not require IL-12 to produce IFN-γ. However, if Tcm cells develop prior to effector cells than we might expect these cells to become IFN-γ producers only in the presence of IL-12. Here we show that the production of IFN-γ by L. major specific effector T cells is not dependent upon IL-12, and that there is no decrease in the frequency or amount of IFN-γ produced by T cells at the site of infection in the absence of IL-12. In contrast, the Tcm cell population generated in response to L. major infection required IL-12 to produce IFN-γ, and in the absence of IL-12 produced IL-4. These data show that the L. major specific CD4+ Tcm population is not fully differentiated since these cells maintain the ability to adopt different fates, and indicate that a major role for IL-12 in established infections may be to promote Th1 cell development from a pool of uncommitted Tcm cells.
Materials and Methods
Animals
C57BL/6J (B6 Thy1.2), C57BL/6J (B6 CD45.1) B6.PL-Thy1a/CyJ (B6 Thy1.1), and B6.129S1-IL-12a tm1Jm/J (B6 IL-12p35-/-) mice were obtained from the Jackson Laboratory (Bar Harbor, ME). B10.D2 ABLE mice (8) were a generous gift from the laboratory of Dr. Steven Reiner (University of Pennsylvania). Groups of two to five female mice were used at 4-8 wks of age. Animals were maintained in a specific pathogen-free environment and tested negative for pathogens in routine screening. All experiments were conducted following the guidelines of the University of Pennsylvania institutional animal care and use committee.
Parasites and Antigens
A clone of L. major (WHO/MHOM/IL/80/Friedlin) was used for these studies. Parasites were grown to stationary phase in Grace’s insect cell culture medium (Life Technologies, Grand Island, NY) with 20% FBS (HyClone, Logan, UT; 0.125 EU/ml) and 2 mM glutamine (Sigma-Aldrich, St. Louis, MO). Stationary-phase promastigotes were harvested and washed three times in PBS prior to use for infection. Mice were either infected by injecting 5 × 106 parasites suspended in 50 μl of PBS into the hind footpad or by injecting 1 × 106 parasites suspended in 10 μl of PBS into the ear dermis (9). LACK (Leishmania homologue of receptor for activated kinase) (LACK156-173) peptides were obtained from the Protein Chemistry Laboratory at the University of Pennsylvania. Freeze-thaw antigen (FTAg)3 was prepared from L. major stationary-phase promastigotes that were washed four times in PBS, re-suspended at 109/ml, and frozen (-80°C) and thawed rapidly (37°C) for five cycles. Mice were injected with 1-5 × 106 stationary-phase L. major parasites. Immune mice were greater than 12 weeks post-primary infection. Some animals were also treated with anti-IL-12 mAb (C17.8, 1 mg/mouse) on day -1 and day 3 post challenge.
Cell culture
Spleens and lymph nodes were mechanically dissociated and depleted of red blood cells using ammonium chloride potassium lysis buffer and re-suspended in complete tissue culture medium (DMEM supplemented with 10% heat-inactivated FBS, 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 25 mM HEPES, and 5×10-5 M 2-ME). Cells were stained with CFSE (Molecular Probes, Eugene, OR) as previously described (9), and cultured at 1-5 × 106 cells/ml in flat-bottom 24-well plates. Purified CD4+ T cells were cultured at 1:5 ratio in the presence of feeder cells consisting of splenocytes from Thy disparate naïve animals. Cells were cultured with L. major FTAg (5×106 parasites/ml), anti-IL-4 mAb (11B11, 50 μg/ml), and rIL-12 (1 ng/ml) for three days. For the rest and re-stimulation experiments cells were then cultured under Th1 conditions (LACK peptide (1 μg/ml), anti-IL-4 (10 μg/ml), rIL-12 (1 ng/ml)) for three days, followed by five days of rest in media and re-stimulated with a combination of LACK peptide, rIL-12, or anti-IL-12 (20 μg/ml) for 3 days.
In some experiments, lymphocytes were isolated from lesions for analysis as previously described (9). Briefly, the ventral and dorsal sheets of the infected ears were separated, deposited dermal side down in DMEM containing 100 U/ml penicillin, 100 μg/ml streptomycin, and liberase Cl enzyme blend (Roche, Indianapolis, IN). Ears were incubated for 30 min at 37°C. The sheets were dissociated in DMEM media using a medimachine (BD Bioscience, San Jose, CA) according to the manufacturer’s protocol. The tissue homogenates were filtered using a 50 μm filcon filters (BD Bioscience, San Jose, CA) and cell suspensions were then stained for flow cytometry.
CD4+ T cell purification and adoptive transfer
Naive or immune B6 Thy1.1 mice were depleted of CD8+ T cells by injection of 250 μg of H35 (Rat IgG2b) prior to being sacrificed (>95% effective). Cells were isolated from lymph nodes and spleens, and CD4+ T cells were purified using a T-cell purification column (R&D Systems, Minneapolis, MN) according to the manufacturer’s recommendations. In some experiments, CD4+ T cells were further separated based on expression of CD62L using MACs magnetic bead columns (>80% purity) (Miltenyi Biotec Inc., Auburn, CA) or CD44 expression by using a fluorescence activated FACS ARIA cells sorter (BD Biosciences, San Jose, CA). CD4+ T cells were then stained with CFSE and 5-8 million CFSE-labeled CD4+ T cells were transferred via the retro-orbital plexus into naive Thy1.2 B6 or Thy1.2 B6 IL-12p35-/- recipients (4). Mice were challenged 24 hrs later with L. major as described above. Donor lymphocytes were then isolated for analysis either from the draining lymph node (dLN)3 or the ear (10).
Flow cytometry and intracellular cytokine staining
Cells were analyzed by flow cytometry for surface markers and cytokines directly ex-vivo as previously described (4). Intra-nuclear T-bet staining (eBiosciences, San Diego, CA) was performed by fixation with 1% PFA in PBS, followed by permeabilization and staining in 0.1% Triton X-100 and 1% FBS in PBS. Data acquisition was on a FACSCalibur flow cytometer (BD Bioscience, San Jose, CA) and analysis was carried out using CellQuest Pro software (Version 5.1, BD Pharmingen) or FlowJo Software (Version 6.4.1, Tree Star Inc., Ashland, OR).
RT-PCR
Total RNA was extracted from homogenized ear tissues with RNeasy columns following the manufacturer’s instructions (Qiagen, Valencia, CA). The cDNA was generated using SuperScript II reagents (Invitrogen, Carlsbad, CA). Expression of IFN-γ relative to β-actin was determined by quantitative PCR using SYBR Green primers (Qiagen, Valencia, CA).
Results
IL-12 at the site of infection is not required for IFN-γ production
We have previously shown that IL-12 is necessary for the maintenance of immunity in mice that have resolved a primary infection with L. major (5, 6). It is possible that IL-12 is either acting to promote the development of Th1 cells from a pool of Tcm cells that are not terminally differentiated or it may be required to ensure maximal IFN-γ production from fully differentiated effector T cells. Consistent with previous studies (11), we found that in vitro-generated effector cells do not require IL-12 to maintain IFN-γ production (data not shown). However, while IL-12 may not be required for Th1 cells to produce IFN-γ, IL-12 can substantially increase the amount of IFN-γ produced by T cells by prolonging IFN-γ synthesis (7). Therefore, we wanted to determine if the absence of IL-12 would influence the ability of in vivo-generated CD4+ effector T cells to produce IFN-γ. We challenged naïve mice or mice that healed a primary infection (referred to here as immune mice) with L. major in the presence or absence of anti-IL-12/23p40 mAb. As expected, by 5 days there was a much higher frequency of CD4+ T cells producing IFN-γ at the site of infection in immune mice compared to naïve animals (Fig.1A). In naïve animals treated with neutralizing anti-IL-12/23p40 mAb, the frequency of IFN-γ producing CD4+ T cells decreased relative to untreated animals (25% to 14%), consistent with a role for IL-12 in promoting Th1 cell development from naïve T cells (12). In contrast, anti-IL-12/23p40 mAb treatment of immune mice did not result in a decreased frequency of IFN-γ producing cells at the site of infection.
Figure 1.
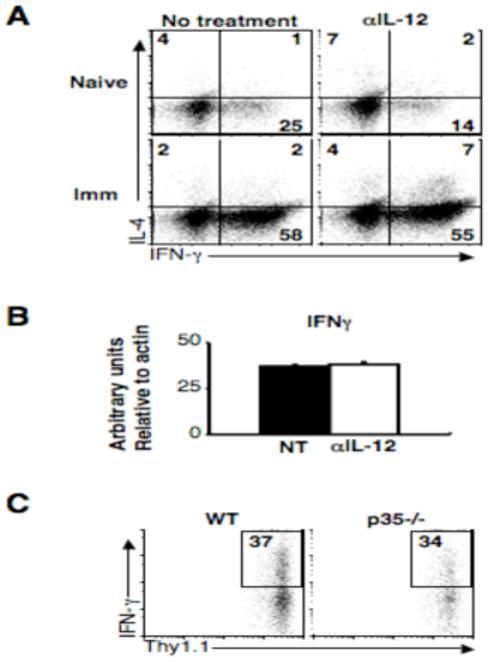
IL-12 at the site of infection is not required for IFN-γ production. (A) Naïve or immune C57BL/6 mice were infected and treated with 1mg αIL-12/23p40 mAb on day -1 and day 3, and on day 5 cells were isolated from infected ears. IL-4 and IFN-γ levels were determined by intracellular cytokine staining. Plots are gated on live CD4+ T cells and are representative of two experiments. Quadrant numbers are shown. (B) RNA was isolated from total ear dermis homogenate, reverse-transcribed and real-time quantitative PCR was performed with primers specific for IFN-γ. Graph shows arbitrary units relative to β-actin. (C) 20 ×106 CD4+ T cells from the spleens and lymph nodes of immune mice were transferred via the retro-orbital plexus into wild-type or IL-12p35-/- mice that were subsequently infected. After 5 days, cells were collected from infected ears and the percentage of IFN-γ+ cells is shown. Plots are gated on live Thy1.1+ donor cells and are representative of two experiments.
Interestingly, in the absence of IL-12, we consistently observed an increase in the frequency of CD4+ T cells producing both IFN-γ and IL-4 double producers in immune mice (Fig. 1A, upper), suggesting that perhaps a small portion of effector cells maintain a level of plasticity. However, despite the slight increase in IFN-γ/IL-4 double producers, the overall level of IFN-γ mRNA was unchanged at the site of infection in immune mice treated with anti-IL-12/23p40 mAb compared to untreated immune mice (Fig. 1B). In order to confirm this result we transferred CD4+ T cells from immune Thy1.1 mice into congenic wild-type or IL-12p35 deficient hosts and examined their ability to produce IFN-γ. The presence or absence of IL-12 did not affect the ability of effector cells at the site of infection to produce IFN-γ (Fig. 1C). Taken together, these data suggest that the majority of effectors cells generated following L. major infection are fully differentiated and do not require IL-12 to produce IFN-γ.
In vitro and in vivo generated Tcm cells require IL-12 in order to produce IFN-γ
We have shown thus far that IL-12 is not required to ensure IFN-γ production from fully differentiated effector T cells, although it is required for the maintenance of immunity to L. major (5, 6). We hypothesized that IL-12 may be required to promote the differentiation of Tcm cells into Th1 effectors. Previous studies have shown that following the removal of antigen and cytokines, in vitro generated effector cells rapidly transition into a population of long-lived, antigen-specific CD62Lhigh central memory and CD62Llow effector memory T cells (13, 14). In order to determine the requirement of in vitro-generated Tcm cells for IL-12 we sorted in vitro cultured CD4+ memory T cells into CD62Lhigh (central memory) and CD62Llow (effector/effector memory) populations and restimulated them in the presence or absence of IL-12. Upon re-stimulation, a small percentage of CD4+ CD62Lhigh cells produced IFN-γ and this was not affected by the addition of αIL-12 mAb (Fig. 2A). However, it was only when CD62Lhigh cells were restimulated in the presence of peptide and rIL-12 that the frequency of IFN-γ producing cells increased to a level similar to that of CD62Llow effector cells, suggesting that the CD62Lhigh central memory population generated in vitro requires IL-12 to become Th1 effectors. Here again, we observed a marked increase in both the frequency and mean fluorescence intensity of IFN-γ by CD62Llow cells upon re-stimulation. These results are similar to those obtained by analyzing T cells from the peak of a leishmanial infection, where CD62Lhigh T cells were also found to require IL-12 to produce IFN-γ (15, 16).
Figure 2.
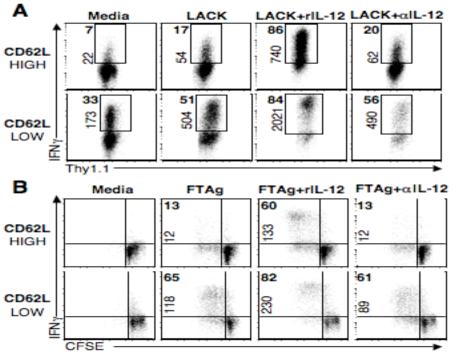
In vitro and in vivo generated Tcm cells require IL-12 in order to produce IFN-γ. (A) Splenocytes from B10.D2 LACK TCR transgenic ABLE-2 mice were stimulated in vitro for 3 days with LACK peptide, rIL-12, and αIL-4 mAb. Cells were then rested for five days, sorted based upon their CD62L expression and restimulated in the presence of feeder cells with LACK peptide with or without IL-12. Plots are gated on live CD4+ cells, numbers in bold represent the frequency of IFN-γ+ cells, numbers on axis represent MFI of IFN-γ+ cells. (B) CD4+ T cells from immune C57BL/6 mice were isolated and sorted into CD62Lhigh or CD62Llow groups, CFSE labeled, cultured at 1:5 ratio with splenocytes from naïve Thy disparate mice, and stimulated for four days with FTAg in the presence or absence of IL-12. Numbers represent percentage of CD4+ IFN-γ+ cells and plots are representative of two experiments.
In the in vitro system, TCR transgenic CD4+ T cells receive a strong stimulus resulting in the majority of the cells proliferating and producing IFN-γ (data not shown). This is in contrast to the heterogeneous effector T cell response that we observe following an L. major infection. Therefore, we next wanted to ask whether L. major specific CD62Lhigh CD4+ T cells isolated from an immune animal also require IL-12 in order to become IFN-γ producers. In order to address this question, CD62Lhigh and CD62Llow populations were sorted from L. major infected animals, labeled with CFSE to track proliferation, and restimulated with antigen in the presence or absence of IL-12. We found that CD62Llow cells stimulated with antigen and anti-IL-12/23p40 mAb produced similar levels of IFN-γ production as cells stimulated with antigen alone (Fig. 2B). This observation confirmed that while effector cells maintain the ability to respond to IL-12, as evidenced by the increased MFI and frequency of IFN-γ producers when cells were given rIL-12, they do not require IL-12 to produce IFN-γ. Similar to what we observed with in vitro-generated Tcm cells, CD62Lhigh cells sorted from infected animals proliferated in response to antigen, but were only able to become IFN-γ producers in the presence of IL-12. Thus, in vivo-generated Tcm cells isolated from an infected animal appear to require IL-12 in order to produce IFN-γ, suggesting that these cells are not developing from fully differentiated effector cells.
The CD62Lhigh compartment of an L. major infected animal contains a mixture of both Tcm and naïve cells. It is therefore possible that the naïve cells, and not the Tcm cells, require IL-12 to produce IFN-γ. In order to rule out this possibility, we sorted cells from immune mice based upon their expression of the activation marker CD44 into three groups, naïve (CD4+ CD62Lhigh CD44low), effector (CD4+ CD62Llow CD44high), and Tcm cells (CD4+ CD62Lhigh CD44high) and re-stimulated them in vitro. As expected, naïve cells were unable to produce IFN-γ upon antigen stimulation in the absence or presence of exogenous IL-12. In contrast, effector cells were able to produce IFN-γ in response to antigen and this response was further augmented by the addition of IL-12 (Fig. 3). However, only a small frequency (5%) of Tcm cells were able to produce IFN-γ when stimulated with antigen alone. They only acquired the ability to produce IFN-γ at high frequencies (17%) when cultured in the presence of rIL-12, suggesting that the L. major specific Tcm population is not fully differentiated and requires additional signals to promote IFN-γ production.
Figure 3.
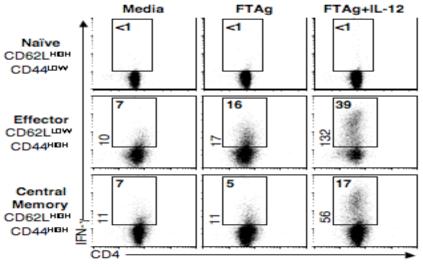
Central memory CD4+ T cells generated by Leishmania infection require IL-12 to become IFN-γ producers. CD4+ T cells from immune C57BL/6 mice were sorted into naïve (CD62Lhigh CD44low), effector (CD62Llow CD44high), or central memory (CD62Lhigh CD44high) and cultured at a 1:5 ratio with splenocytes from naïve Thy disparate animals, and stimulated with FTAg alone or in the presence of IL-12 and αIL-4. After 4 days cells were harvested and stained for CD4 and IFN-γ. Numbers represent percentage of CD4+ IFN-γ+ cells and plots are representative of two experiments.
Central memory CD4+ T cells do not express high levels of T-bet
It is well established that as CD4+ T cells differentiate, they up-regulate lineage specific transcription factors such as T-bet, which leads to the up-regulation of IFN-γ, and the suppression of Th2-associated cytokines (7, 17-19). Recently, T-bet has also been shown to regulate memory T cell potential (7, 20, 21). Decreases in T-bet expression levels have been linked to increases in Tcm cell numbers (20). Additionally, the levels of T-bet induced by the initial immune response have been shown to affect the number of memory precursors generated (21). These studies would suggest that Tcm cells are not fully differentiated. Although we have shown that L. major specific Tcm cells are capable of becoming IFN-γ producers (4), it remains unclear whether this population is terminally differentiated or maintains the ability to adopt different cells fates. If the Tcm cells generated during L. major infection are not fully differentiated, as our data so far would suggest, then these cells should express low levels of T-bet and these levels should increase as the cells proliferate, become activated, and down-regulate CD62L.
In order to test this, we transferred cells from naïve or immune animals into congenic recipients and challenged them with L. major. Two weeks following infection we examined the ability of these to cells to home to lymph nodes and proliferate. As we would expect, CD4+ T cells from immune animals proliferated at much higher frequencies than cells from naïve animals (Fig. 4A). We next wanted to determine the levels of T-bet expression in the responding populations. We gated on CFSEdim CD4+ T cells and analyzed expression of CD62L and T-bet. While a portion of CD4+ T cells from naïve animals had proliferated and down-regulated CD62L, only a very small frequency (3%) expressed T-bet (Fig. 4B). In contrast, CD4+ T cells from immune mice that had proliferated and down-regulated CD62L had a marked much higher frequencies of T-bet+ cells (27%), indicating that these cells were on their way to becoming Th1 effectors. However, there was minimal T-bet expression in proliferating CD62Lhigh T cells from either naïve or immune animals (Fig. 4B), suggesting that the majority of Tcm cells generated in response to L. major infection are not committed to a Th1 lineage. This result is consistent with the requirement for IL-12 to promote Th1 cell development from the Tcm cell population.
Figure 4.
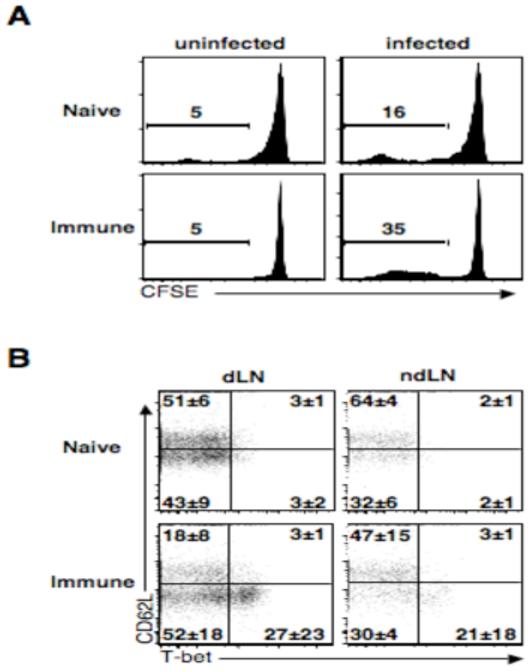
Central memory CD4+ T cells do not express high levels of T-bet. CD4+ T cells from immune mice were labeled with CFSE and transferred into wild-type mice that were subsequently infected and two weeks later donor cells from the draining and non-draining lymph nodes were isolated and analyzed by flow cytometry. (A) Plots are gated on live CD4+ donor T cells. Numbers represent percentage of cells that have diluted CFSE. Plots are indicative of three experiments (B) Plots are gated on proliferated live CD4+ donor T cells. Average quadrant numbers ± SD are shown.
In the absence of IL-12, central memory CD4+ T cells become IL-4 producers
If the Tcm cell population generated in response L. major infection is not derived from Th1 effector cells, then these cells should maintain the ability to develop into different effector cell subsets. To test the capacity of L. major specific Tcm cells to develop into Th1 or Th2 cells, T cells from an immune mouse were transferred into either a wild-type or an IL-12p35 deficient mouse. Adoptive transfers were done using polyclonal CD4+ T cells (4), therefore avoiding any confounding variables that might arise from non-physiologic frequencies of antigen specific T cells (22-24). Donor T cells proliferated extensively in the dLN, but only minimally in the non-dLNs, during the first 2 weeks of infection (Fig. 5A). We observed no significant differences in the ability of donor cells from immune mice to proliferate in the presence or absence of IL-12 (Fig. 5A). We also examined the ability of the immune T cells to become activated in the absence of IL-12 by measuring the down regulation of CD62L, and found no significant difference between proliferating donor T cells in wild-type or IL-12 deficient mice (data not shown).
Figure 5.
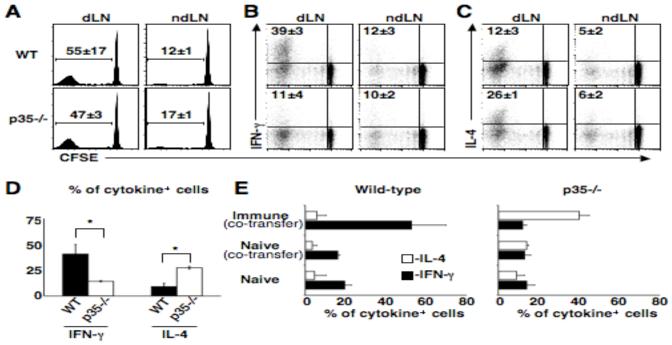
In the absence of IL-12, the Tcm cell population becomes IL-4 producers. CD4+ T cells from immune mice (Thy1.1) were CFSE labeled and transferred into wild-type or IL-12p35-/- mice that were subsequently infected and two weeks later donor cells were isolated and analyzed by flow cytometry. (A) Numbers represent average percentage of cells that had diluted CFSE ± SD. (B) Numbers represent average percentage of proliferated IFN-γ+ ± SD. (C) Numbers represent average percentage of proliferated IL-4+ ± SD. Plots are representative of three experiments, three mice per group (D) CD4+ CD62Lhigh T cells from immune mice (Thy1.1) were sorted and transferred into wild-type or IL-12p35-/- mice that were subsequently infected, two weeks later donor cells were isolated from the dLN and analyzed by flow cytometry. Graph represents average percent of proliferated cells that are cytokine positive ± SD. *=p < 0.05 by T-test. (E) CD4+ T cells from immune (Thy1.1, CD45.2) and/or naïve C57BL/6 mice (CD45.1) were transferred into wild-type or IL-12p35-/- mice (Thy1.2, CD45.2) that were subsequently infected and two weeks later donor cells from the dLNs were isolated and analyzed by flow cytometry. Numbers represent average percent of proliferated cells that are cytokine positive ± SD. Plots are representative of two experiments.
We previously demonstrated that Tcm cells from immune mice not only migrate to the dLN and proliferate, but that after several cell divisions develop the ability to produce IFN-γ (4). However, these same Tcm cells when transferred to IL-12 deficient hosts were unable to become IFN-γ producers (Fig. 5B). In addition, associated with the decrease in the generation of IFN-γ producing cells from the Tcm population was an increase in the frequency of IL-4+ cells (Fig. 5C). These results further suggest that the Tcm population generated in response to L. major is not fully differentiated. To confirm that the cells that were becoming IFN-γ and IL-4 producing cells were derived from the CD4+ Tcm pool, we purified CD62Lhigh CD4+ T cells from immune mice, and transferred these cells to either normal or IL-12 deficient hosts. Similar to our results with unfractionated immune T cells, we found that IL-12 was required for the development of a population of IFN-γ producing T cells and in its absence there was a marked increase in the number of CD4+ T cells producing IL-4 (Fig. 5D). These results confirm that the Leishmania specific Tcm population is not fully differentiated and remain capable of becoming either Th1 or Th2 effector cells.
Although our studies suggested that CD62Lhigh cells from immune mice have the flexibility to differentiate into either IFN-γ or IL-4 producing cells, the transferred population of cells contained both immune and naïve T cells, and the naïve T cells may contribute to the responses we observed. This did not seem likely, since when we transferred naïve T cells alone the proliferative response was limited. Nevertheless, we considered the possibility that the response of naïve T cells within the context of an immune T cell response might be enhanced. To address this issue, we tracked the response of naïve CD4+ T cells following adoptive transfer with or without immune T cells using allelic T cell markers to identify the two populations. Whether naïve T cells were transferred with or without immune T cells, they failed to substantially contribute to the cytokine response observed by the immune T cells (Fig. 5E). Taken together, these results suggest that the Tcm cell population generated during L. major infection is not fully differentiated and requires IL-12 in order become Th1 cells.
Discussion
L. major infection induces a pool of antigen-specific Tcm cells that are capable of mediating immunity (4). Tcm cells can be identified by their expression of CD62L, which facilitates entry of Tcm cells into lymph nodes. Here we show that the CD62Lhigh Tcm cells require IL-12 in order to produce IFN-γ, while CD62Llow effector T cells from immune mice produce IFN-γ in the presence or absence of IL-12. Furthermore, we found that Tcm cells retain the ability to develop into either Th1 or Th2 cells. Thus, when Tcm cells from immune animals were transferred into wild-type mice that were subsequently infected, many of these cells proliferated and began to produce IFN-γ. In contrast, when these same cells were transferred into IL-12p35-/- mice, few of the cells were able to produce IFN-γ after proliferation, but an increased frequency produced IL-4. These results demonstrate that in addition to the previously reported role of IL-12 in the differentiation of naïve CD4+ T cells into Th1 cells, IL-12 is also required to promote the differentiation of Tcm cells into Th1 effector cells.
IL-12 is essential for generating a protective Th1 immune response following infection with L. major, acting directly to promote the survival of T cells that are expressing the Th1 transcription factor T-bet, and indirectly by inhibiting the Th2 inducing transcription factor, GATA3 (7, 18, 25). The requirement for IL-12 at the initiation of the infection is demonstrated by the fact that normally susceptible BALB/c mice are rendered resistant to L. major by IL-12 administration (26-28). In addition, our previous studies demonstrate that IL-12 is also required to maintain immunity. Thus, when we treated B6 IL-12p35-/- mice infected with L. major with exogenous IL-12 they mounted a Th1 response, were able to control the primary infection, but were unable to maintain long-term immunity (5, 6). Similar results have been reported in Toxoplasma and Mycobacterium tuberculosis infections (29, 30). Our current findings suggest that one role for IL-12 in the maintenance of immunity is to promote the differentiation of Tcm cells into IFN-γ-producing Th1 cells. This is a process that may be continually required, since even during the peak of the leishmanial infection there are CD62Lhigh T cells that exhibit the capacity to differentiate into either Th1 or Th2 cells (15, 16).
In addition to promoting the differentiation of Tcm cells into Th1 cells, we cannot exclude the possibility that IL-12 is also playing other roles. For example, we considered the possibility that IL-12 might be acting on effector cells to increase their production of IFN-γ, since IL-12 has also been reported to act as a trans-activator of IFN-γ production (7). We found that in vitro addition of IL-12 increased the production of IFN-γ by Th1 effector T cells. However, when we treated mice in vivo with an IL-12 blocking antibody there was no change in the production of IFN-γ by T cells at the site of infection. Additionally, IL-12 has been reported to promote the expression of tissue homing chemokines, and can induce anti-apoptotic factors involved in T cell survival, such as Bcl-2 and Bcl-3 (31-35). Each of these might play some role in the ability of IL-12 to maintain immunity in L. major infected animals.
Tcm cells were initially described as a population of cells that could home to the lymph nodes, due to the expression of CD62L, and that had not yet gained the capacity to make effector cytokines (3). However, later studies showed convincingly that effector memory CD8+ T cells could re-express CD62L, suggesting that Tcm cells may develop from effector T cells (36). Consistent with this, we found that Trichuris muris infection induces a population of IL-4 competent memory CD4+ T cells that express CD62L (37). In contrast, we show here that Tcm cells generated by L. major infection can become either IFN-γ or IL-4 producing T cells, suggesting that these cells were not derived from effector T cells. These divergent results raise questions as to the true lineage of Tcm cells. The most straightforward interpretation of these results is that differences in the immune response determine the nature of the memory T cells generated. Factors such as the type of innate immune response induced, the replication rate of the pathogen, the site of pathogen replication, and whether CD4+ or CD8+ T cells dominate the response may all be critical. In the case of leishmaniasis, we and others have found that no effector memory develops (4, 38, 39), while other infections clearly generate effector memory cells (40-42). It may be that when no effector memory T cells are generated, there is no opportunity to develop CD62Lhigh memory T cells that are committed to producing IFN-γ. Defining the pathogen-associated factors responsible for these differences will be important in fully understanding how infections induce immunologic memory.
Currently there are no vaccines for human leishmaniasis, in spite of the fact that control of a primary infection leads to resistance to re-infection. This resistance is now known to be due in part to the presence of persistent parasites (38, 39). However, we have previously found that immunization of mice with an attenuated parasite led to the generation of protective Tcm cells that could be maintained in the absence of persistent parasites (4). Our findings here indicate that the majority of the Tcm population is not destined to become Th1 cells, which raises the issue of how to ensure that the CD4+ Tcm cells generated by a vaccine develop into Th1 cells upon re-challenge. One solution might be to promote the expansion of both CD4+ and CD8+ Tcm cells, since we have previously demonstrated that CD8+ T cells play a critical role in promoting Th1 cell development (43). An alternative approach would be to determine if some Tcm cells are pre-committed to developing into Th1 cells, and discern how to enhance their development (44, 45). In our studies, we did find a low frequency of Tcm cells that produced IFN-γ even in the absence of IL-12, and these might represent such pre-committed cells.
IL-12 is known to promote the IFN-γ production necessary to eliminate intracellular Leishmania parasites and is a critical cytokine in initiating immunity to leishmaniasis. Our results suggest that the continued presence of IL-12 is also required to ensure that non-committed Tcm cells differentiate into Th1 effector cells thereby highlighting the need for additional studies to better understand how IL-12 production is induced and regulated in leishmaniasis.
Acknowledgments
We thank members of the Department of Pathobiology for useful discussions and Dr. Leanne Johnson and Betsy Taylor in the preparation of the manuscript. We would also like to thank Dr. Charles Kao for his assistance with our intra-nuclear staining protocol.
Footnotes
Funding provided by NIH grant AI35914. Colby Zaph supported by the Irvington Institute Fellowship Program of the Cancer Research Institute.
- Tcm
- central memory T cell
- dLN
- draining lymph node
- FTAg
- freeze thaw antigen.
References
- 1.Scott P, Artis D, Uzonna J, Zaph C. The development of effector and memory T cells in cutaneous leishmaniasis: the implications for vaccine development. Immunol Rev. 2004;201:318–338. doi: 10.1111/j.0105-2896.2004.00198.x. [DOI] [PubMed] [Google Scholar]
- 2.Sacks D, Noben-Trauth N. The immunology of susceptibility and resistance to Leishmania major in mice. Nat Rev Immunol. 2002;2:845–858. doi: 10.1038/nri933. [DOI] [PubMed] [Google Scholar]
- 3.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 4.Zaph C, Uzonna J, Beverley SM, Scott P. Central memory T cells mediate long-term immunity to Leishmania major in the absence of persistent parasites. Nat Med. 2004;10:1104–1110. doi: 10.1038/nm1108. [DOI] [PubMed] [Google Scholar]
- 5.Park AY, Hondowicz B, Kopf M, Scott P. The role of IL-12 in maintaining resistance to Leishmania major. J Immunol. 2002;168:5771–5777. doi: 10.4049/jimmunol.168.11.5771. [DOI] [PubMed] [Google Scholar]
- 6.Park AY, Hondowicz BD, Scott P. IL-12 is required to maintain a Th1 response during Leishmania major infection. J Immunol. 2000;165:896–902. doi: 10.4049/jimmunol.165.2.896. [DOI] [PubMed] [Google Scholar]
- 7.Mullen AC, High FA, Hutchins AS, Lee HW, Villarino AV, Livingston DM, Kung AL, Cereb N, Yao TP, Yang SY, Reiner SL. Role of T-bet in commitment of TH1 cells before IL-12-dependent selection. Science. 2001;292:1907–1910. doi: 10.1126/science.1059835. [DOI] [PubMed] [Google Scholar]
- 8.Reiner SL, Fowell DJ, Moskowitz NH, Swier K, Brown DR, Brown CR, Turck CW, Scott PA, Killeen N, Locksley RM. Control of Leishmania major by a monoclonal alpha beta T cell repertoire. J Immunol. 1998;160:884–889. [PubMed] [Google Scholar]
- 9.Lyons AB, Parish CR. Determination of lymphocyte division by flow cytometry. J Immunol Methods. 1994;171:131–137. doi: 10.1016/0022-1759(94)90236-4. [DOI] [PubMed] [Google Scholar]
- 10.Belkaid Y, Jouin H, Milon G. A method to recover, enumerate and identify lymphomyeloid cells present in an inflammatory dermal site: a study in laboratory mice. J Immunol Methods. 1996;199:5–25. doi: 10.1016/s0022-1759(96)00117-2. [DOI] [PubMed] [Google Scholar]
- 11.Manetti R, Gerosa F, Giudizi MG, Biagiotti R, Parronchi P, Piccinni MP, Sampognaro S, Maggi E, Romagnani S, Trinchieri G, et al. Interleukin 12 induces stable priming for interferon gamma (IFN-gamma) production during differentiation of human T helper (Th) cells and transient IFN-gamma production in established Th2 cell clones. J Exp Med. 1994;179:1273–1283. doi: 10.1084/jem.179.4.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seder RA, Gazzinelli R, Sher A, Paul WE. Interleukin 12 acts directly on CD4+ T cells to enhance priming for interferon gamma production and diminishes interleukin 4 inhibition of such priming. Proc Natl Acad Sci U S A. 1993;90:10188–10192. doi: 10.1073/pnas.90.21.10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKinstry KK, Golech S, Lee WH, Huston G, Weng NP, Swain SL. Rapid default transition of CD4 T cell effectors to functional memory cells. J Exp Med. 2007;204:2199–2211. doi: 10.1084/jem.20070041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harbertson J, Biederman E, Bennett KE, Kondrack RM, Bradley LM. Withdrawal of stimulation may initiate the transition of effector to memory CD4 cells. J Immunol. 2002;168:1095–1102. doi: 10.4049/jimmunol.168.3.1095. [DOI] [PubMed] [Google Scholar]
- 15.Mocci S, Coffman RL. Induction of a Th2 population from a polarized Leishmania-specific Th1 population by in vitro culture with IL-4. J Immunol. 1995;154:3779–3787. [PubMed] [Google Scholar]
- 16.Mocci S, Coffman RL. The mechanism of in vitro T helper cell type 1 to T helper cell type 2 switching in highly polarized Leishmania major-specific T cell populations. J Immunol. 1997;158:1559–1564. [PubMed] [Google Scholar]
- 17.Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2:933–944. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]
- 18.Martins GA, Hutchins AS, Reiner SL. Transcriptional activators of helper T cell fate are required for establishment but not maintenance of signature cytokine expression. J Immunol. 2005;175:5981–5985. doi: 10.4049/jimmunol.175.9.5981. [DOI] [PubMed] [Google Scholar]
- 19.Mullen AC, Hutchins AS, High FA, Lee HW, Sykes KJ, Chodosh LA, Reiner SL. Hlx is induced by and genetically interacts with T-bet to promote heritable T(H)1 gene induction. Nat Immunol. 2002;3:652–658. doi: 10.1038/ni807. [DOI] [PubMed] [Google Scholar]
- 20.Intlekofer AM, Takemoto N, Kao C, Banerjee A, Schambach F, Northrop JK, Shen H, Wherry EJ, Reiner SL. Requirement for T-bet in the aberrant differentiation of unhelped memory CD8+ T cells. J Exp Med. 2007;204:2015–2021. doi: 10.1084/jem.20070841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. Inflammation Directs Memory Precursor and Short-Lived Effector CD8(+) T Cell Fates via the Graded Expression of T-bet Transcription Factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marzo AL, Klonowski KD, Le Bon A, Borrow P, Tough DF, Lefrancois L. Initial T cell frequency dictates memory CD8+ T cell lineage commitment. Nat Immunol. 2005;6:793–799. doi: 10.1038/ni1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Badovinac VP, Haring JS, Harty JT. Initial T cell receptor transgenic cell precursor frequency dictates critical aspects of the CD8(+) T cell response to infection. Immunity. 2007;26:827–841. doi: 10.1016/j.immuni.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hataye J, Moon JJ, Khoruts A, Reilly C, Jenkins MK. Naive and memory CD4+ T cell survival controlled by clonal abundance. Science. 2006;312:114–116. doi: 10.1126/science.1124228. [DOI] [PubMed] [Google Scholar]
- 25.Usui T, Preiss JC, Kanno Y, Yao ZJ, Bream JH, O’Shea JJ, Strober W. T-bet regulates Th1 responses through essential effects on GATA-3 function rather than on IFNG gene acetylation and transcription. J Exp Med. 2006;203:755–766. doi: 10.1084/jem.20052165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heinzel FP, Schoenhaut DS, Rerko RM, Rosser LE, Gately MK. Recombinant interleukin 12 cures mice infected with Leishmania major. J Exp Med. 1993;177:1505–1509. doi: 10.1084/jem.177.5.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sypek JP, Chung CL, Mayor SE, Subramanyam JM, Goldman SJ, Sieburth DS, Wolf SF, Schaub RG. Resolution of cutaneous leishmaniasis: interleukin 12 initiates a protective T helper type 1 immune response. J Exp Med. 1993;177:1797–1802. doi: 10.1084/jem.177.6.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nabors GS, Afonso LC, Farrell JP, Scott P. Switch from a type 2 to a type 1 T helper cell response and cure of established Leishmania major infection in mice is induced by combined therapy with interleukin 12 and Pentostam. Proc Natl Acad Sci U S A. 1995;92:3142–3146. doi: 10.1073/pnas.92.8.3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yap G, Pesin M, Sher A. Cutting edge: IL-12 is required for the maintenance of IFN-gamma production in T cells mediating chronic resistance to the intracellular pathogen, Toxoplasma gondii. J Immunol. 2000;165:628–631. doi: 10.4049/jimmunol.165.2.628. [DOI] [PubMed] [Google Scholar]
- 30.Feng CG, Jankovic D, Kullberg M, Cheever A, Scanga CA, Hieny S, Caspar P, Yap GS, Sher A. Maintenance of pulmonary Th1 effector function in chronic tuberculosis requires persistent IL-12 production. J Immunol. 2005;174:4185–4192. doi: 10.4049/jimmunol.174.7.4185. [DOI] [PubMed] [Google Scholar]
- 31.Myers KJ, Eppihimer MJ, Hall L, Wolitzky B. Interleukin-12-induced adhesion molecule expression in murine liver. Am J Pathol. 1998;152:457–468. [PMC free article] [PubMed] [Google Scholar]
- 32.Pearlman E, Lass JH, Bardenstein DS, Diaconu E, Hazlett FE, Jr., Albright J, Higgins AW, Kazura JW. IL-12 exacerbates helminth-mediated corneal pathology by augmenting inflammatory cell recruitment and chemokine expression. J Immunol. 1997;158:827–833. [PubMed] [Google Scholar]
- 33.Wang S, Fan Y, Han X, Yang J, Bilenki L, Yang X. IL-12-dependent vascular cell adhesion molecule-1 expression contributes to airway eosinophilic inflammation in a mouse model of asthma-like reaction. J Immunol. 2001;166:2741–2749. doi: 10.4049/jimmunol.166.4.2741. [DOI] [PubMed] [Google Scholar]
- 34.Yoo JK, Cho JH, Lee SW, Sung YC. IL-12 provides proliferation and survival signals to murine CD4+ T cells through phosphatidylinositol 3-kinase/Akt signaling pathway. J Immunol. 2002;169:3637–3643. doi: 10.4049/jimmunol.169.7.3637. [DOI] [PubMed] [Google Scholar]
- 35.Valenzuela JO, Hammerbeck CD, Mescher MF. Cutting edge: Bcl-3 up-regulation by signal 3 cytokine (IL-12) prolongs survival of antigen-activated CD8 T cells. J Immunol. 2005;174:600–604. doi: 10.4049/jimmunol.174.2.600. [DOI] [PubMed] [Google Scholar]
- 36.Wherry EJ, Teichgraber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, Ahmed R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol. 2003;4:225–234. doi: 10.1038/ni889. [DOI] [PubMed] [Google Scholar]
- 37.Zaph C, Rook KA, Goldschmidt M, Mohrs M, Scott P, Artis D. Persistence and function of central and effector memory CD4+ T cells following infection with a gastrointestinal helminth. J Immunol. 2006;177:511–518. doi: 10.4049/jimmunol.177.1.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uzonna JE, Wei G, Yurkowski D, Bretscher P. Immune elimination of Leishmania major in mice: implications for immune memory, vaccination, and reactivation disease. J Immunol. 2001;167:6967–6974. doi: 10.4049/jimmunol.167.12.6967. [DOI] [PubMed] [Google Scholar]
- 39.Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420:502–507. doi: 10.1038/nature01152. [DOI] [PubMed] [Google Scholar]
- 40.Swain SL, Agrewala JN, Brown DM, Jelley-Gibbs DM, Golech S, Huston G, Jones SC, Kamperschroer C, Lee WH, McKinstry KK, Roman E, Strutt T, Weng NP. CD4+ T-cell memory: generation and multi-faceted roles for CD4+ T cells in protective immunity to influenza. Immunol Rev. 2006;211:8–22. doi: 10.1111/j.0105-2896.2006.00388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hikono H, Kohlmeier JE, Ely KH, Scott I, Roberts AD, Blackman MA, Woodland DL. T-cell memory and recall responses to respiratory virus infections. Immunol Rev. 2006;211:119–132. doi: 10.1111/j.0105-2896.2006.00385.x. [DOI] [PubMed] [Google Scholar]
- 42.Lohning M, Hegazy AN, Pinschewer DD, Busse D, Lang KS, Hofer T, Radbruch A, Zinkernagel RM, Hengartner H. Long-lived virus-reactive memory T cells generated from purified cytokine-secreting T helper type 1 and type 2 effectors. J Exp Med. 2008;205:53–61. doi: 10.1084/jem.20071855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uzonna JE, Joyce KL, Scott P. Low dose Leishmania major promotes a transient T helper cell type 2 response that is down-regulated by interferon gamma-producing CD8+ T cells. J Exp Med. 2004;199:1559–1566. doi: 10.1084/jem.20040172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lozza L, Rivino L, Guarda G, Jarrossay D, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A, Geginat J. The strength of T cell stimulation determines IL-7 responsiveness, secondary expansion, and lineage commitment of primed human CD4(+)IL-7R(hi) T cells. Eur J Immunol. 2008;38:30–39. doi: 10.1002/eji.200737852. [DOI] [PubMed] [Google Scholar]
- 45.Rivino L, Messi M, Jarrossay D, Lanzavecchia A, Sallusto F, Geginat J. Chemokine receptor expression identifies Pre-T helper (Th)1, Pre-Th2, and nonpolarized cells among human CD4+ central memory T cells. J Exp Med. 2004;200:725–735. doi: 10.1084/jem.20040774. [DOI] [PMC free article] [PubMed] [Google Scholar]