

Summary
Enteropathogenic E. coli (EPEC) have been previously shown to alter sodium hydrogen exchange (NHE3) activity in human intestinal epithelial cells. To further characterize these observations, PS120 fibroblasts transfected with NHE3 were studied. EPEC E2348/69 infection decreased NHE3 activity in PS120 fibroblasts. The effect on NHE3 was enhanced when PS120 cells were co-transfected with the scaffolding/regulatory proteins NHERF1 or NHERF2 or EBP50 and E3KARP, respectively. The decrease in NHE3 activity was dependent on an intact type III secretion system, although intimate attachment mediated by Tir was not required. Despite its ability to bind to NHERF proteins, the EPEC effector Map had no impact on the regulation of NHE activity. Instead, EspF was found to be responsible for decreased NHE3 activity. However, neither EspF induced apoptosis nor the interaction of EspF with sorting nexin-9, an endocytic protein, were involved.
Introduction
Enteropathogenic Escherichia coli (EPEC) are members of a family of diarrheal pathogens best known for the formation of actin pedestals through the use of a type III secretion system (TTSS) for delivery of the translocated intimin receptor (Tir) and its subsequent binding to intimin on the bacterial surface. EPEC infections are primarily of concern in developing countries and cause severe dehydration and often death in infants. A number of factors contribute to EPEC-associated diarrhea including disruption of epithelial barrier function, increased expression of galanin-1 receptor, and the production of 5′-AMP a pro-secretagogue by neutrophils (McNamara et al., 2001; Hecht et al., 1999; Madara et al., 1993). However, the onset of diarrhea in EPEC infections occurs within a few hours in humans and prior to the earliest studied occurrence of the above mentioned events in animal models, suggesting additional unidentified causes.
Changes in ion secretion/absorption are the underlying causes of diarrheal diseases. A classic example involves secretion of Cl− via CFTR in response to cholera toxin leading to the efflux of water into the intestinal lumen. While EPEC does not alter electrogenic Cl− secretion, changes in electroneutral ion exchange processes have been recently shown (Dean et al., 2006; Hecht et al., 2004; Hecht and Koutsouris, 1999). Although these pathways are not electrogenic and, therefore, cannot be detected through changes in short circuit current Isc, they are equally important for intestinal absorptive/secretory functions. Sodium hydrogen exchanger (NHE) activity is differentially regulated in response to EPEC infection with an increase in NHE1 and NHE2 activity accompanying a decrease in NHE3 activity (Hecht et al., 2004). In addition, Cl−/OH− exchange, butyrate uptake, and SGLT1 functions are impaired (Gill et al., 2007; Borthakur et al., 2006; Dean et al., 2006). NHE3 is the primary absorptive route for Na+ entering the intestinal epithelium from the lumen and is often suppressed by diarrheal pathogens in addition to alterations in Cl− secretion (Zachos et al., 2005; Hayashi et al., 2004; Kandasamy et al., 1995). Both NHE2 and NHE3 activity are suppressed by cholera toxin in a cAMP-dependent manner (Subramanya et al., 2007). Since water osmotically moves towards areas of higher salt concentration, changes in either Na+ and Cl− absorption or secretion lead to diarrhea. NHE3 is the primary mediator of Na+ absorption in the intestine and the loss of NHE3, but not NHE2, activity in knockout mice has been shown to cause diarrhea (Zachos et al., 2005; Gawenis et al., 2002).
Our previous studies pertaining to the effects of EPEC infection on NHEs were carried out in the human colonic epithelial cell lines Caco-2, T-84 and HT-29 (Hecht et al., 2004). Of these, only Caco-2 cells express NHE3 and do so in conjunction with NHE2, another apical isoform. The dramatic increase in NHE2 activity in response to EPEC infection and the requirement for non-commercially available inhibitors makes it exceedingly difficult to reliably study NHE3 activity in these cells. Therefore, we used PS120 fibroblasts, a derivative of the hamster cell line CCL39 that was specifically selected for the absence of NHE activity and transfected to stably express NHE3 in order to examine the effect of EPEC infection on NHE3 in a “clean” model system (Yun et al., 1997; Tse et al., 1993; Franchi et al., 1986). The PS120 fibroblasts and the NHE-transfected variants have been analyzed in great detail and shown to mimic native NHE activity although co-factors, known as NHERFs or NHE regulatory factors, are required for response to some signal transduction pathways (Cha et al., 2005; Yun et al., 1997). In addition PS120 fibroblasts do not express microvilli, allowing the effects of EPEC on the absorptive surface and changes in NHE activity to be separated. We employed PS120 fibroblasts transfected with NHE3 alone or in combination with NHERF1 or NHERF2 (Yun et al., 1997; Tse et al., 1993) to specifically examine the effect of EPEC on this important intestinal transporter.
NHERF proteins are members of the PDZ family of proteins named after a conserved motif found in PSD-95, Dlg and ZO-1 (Kennedy, 1995). Both NHERF1 and NHERF2 contain two PDZ domains in addition to an ezrin/radixin/moesin (ERM) binding motif (Reczek et al., 1997). The primary function of PDZ proteins is to anchor membrane-associated proteins to the actin cytoskeleton through an indirect linkage to actin via an ERM protein. NHERF proteins are also thought to promote clustering of NHE3 and association with other transporters such as DRA (Lamprecht et al., 2002). PDZ motifs typically recognize a C-terminal motif consisting of (S/T)-X-(L/V). However, the PDZ binding motif in NHE3 is atypical and is not C-terminal but instead consists of a β-hairpin motif located internally (Lamprecht and Seidler, 2006).
EPEC contain a pathogenicity island known as the locus of enterocyte effacement (LEE) which encodes the components of a type III secretion system and a number of secreted effector molecules (Daniell et al., 2001; Kaper et al., 1997). The effector molecules encoded by the LEE include EspB, EspF, EspH, EspG, EspG2, Tir, Map and SepZ. In addition, a number of non-LEE-encoded (Nle) effector proteins have been recently described (Gruenheid et al., 2004). The effector molecule EspB is part of the pore-forming complex which results in delivery of bacterial effectors but it is also secreted into the host cell cytosol (Taylor et al., 1998). EspF is primarily known for its role in disrupting epithelial barrier integrity both in tissue culture and animal models (Shifflett et al., 2005; McNamara et al., 2001). Tir is the primary effector responsible for accumulation of actin and pedestal formation although EspH and Map have been shown to have effects on pedestal length (Kenny et al., 2002). Map is also known to interact with NHERF1 (EBP50) via a TRL motif located in the C-terminus and mimics the function of the small G-protein Cdc42 (Alto et al., 2006; Simpson et al., 2006). The direct interaction between NHERF1 and Map make it an interesting candidate in terms of NHE regulation. The effector molecule EspG and its homolog disrupt microtubules which could inhibit NHE3 recycling as was recently shown for the Cl−/OH− exchanger DRA (Gill et al., 2007; Tomson et al., 2005; Sabolic et al., 2002). SepZ/EspZ has only recently been identified as a TTSS secreted effector but its function is not known (Kanack et al., 2005).
In this study we use the transfected PS120 fibroblast model to examine in more detail the EPEC-induced decrease in NHE3 activity. The role of the NHERF scaffolding proteins and of individual TTSS effector molecules are explored.
Experimental procedures
Bacterial strains
Enteropathogenic E. coli strains used in this study are described in Table 1.
Table 1.
Bacterial strains
| Strain name | Mutation | Phenotype | Reference |
|---|---|---|---|
| E2348/69 | Wt | wt | |
| UMD874 | ΔespF-orf32 | EspF mutant | (McNamara and Donnenberg, 1998) |
| UMD874 pBPM19 | ΔespF+pespF | EspF complement | (McNamara and Donnenberg, 1998) |
| SE1114 | espG (Km) | EspG mutant | (Elliott et al., 2001) |
| JAC719 | Δtir | Tir deletion | (Crawford and Kaper, 2002) |
| orf19 | Δmap | Map deletion | (Kenny and Jepson, 2000) |
| EPECΔmap+pmap | Δmap+pmap | Map complement | (Alto et al., 2006) |
| MapΔTRL | Δmap+pmap TRL | PDZ ligand motif | (Alto et al., 2006) |
| SE651orf18 | espH (Km) | EspH mutant | James Kaper |
| CVD452 | escN (Km) | TTSS (ATPase) | (Jarvis et al., 1995) |
| HB101 | Wt | Non-pathogenic E. coli | |
| SE1207 | ΔespG/G2 | EspG/G2 double mutant | James Kaper |
| GH290 | espF+ | IPTG inducible espF in UMD874 | (Alto et al., 2007) |
| GH393 | espF-D3 | GH290 with Arg75,122,169 → Asp | (Alto et al., 2007) |
| GH406 | L16E | GH290 with Leu16→Glu |
Infection and Acid Loading of Cells
Bacteria were grown overnight in Luria Bertani broth (LB) and then 300 μl were subcultured into 10 ml of serum free media consisting of 50% DMEM and 50% Ham’s F-12 supplemented with 14 mM sodium bicarbonate, 15 mM HEPES and 5% mannose.
Cultures were grown approximately 3 hours until reaching mid-log phase, OD600 of 0.4. Bacteria were pelleted and resuspended at equivalent density in fresh serum free media. PS120 fibroblasts or Caco-2 cells were placed in serum free media overnight prior to infection. A volume of 200 μl was used per well to infect a 24 well plate, with 6 wells per sample, half of which are necessary for EIPA inhibition. Cells were typically infected for 90 minutes then rinsed in phosphate buffered saline (PBS). This was followed by 30 minutes in an acid load solution consisting of 50 mM NH4Cl, 70 mM choline chloride, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 5 mM glucose and 15 mM MOPS (pH 7.0). The acid load process is carried out under neutral pH but lowers pHi upon removal of this buffer resulting in increase in Na-dependent pHi recovery. This NHE activity is generated by an allosteric response to protons and does not interfere with other regulatory mechanisms typically resulting from NHE3 trafficking. After acid loading, the cells are washed in 120 mM choline chloride and 15 mM Tris-HEPES, pH 7.5 prior to sodium uptake.
Sodium Uptake Studies
Sodium uptake was carried out for 5 minutes in a solution of 3mM NaCl, 110 mM choline chloride, 1 mM MgCl2, 2 mM CaCl2, 20 mM Tris-HEPES (pH 7.4) and 1 μCi/ml of 22Na+, with or without 50 μM EIPA (Hecht et al., 2004). EIPA specifically inhibits NHE activity, and is routinely used to separate NHE activity from other forms of Na+ uptake. Alternatively, 10μM S3226 (generously donated by Aventis) was utilized to specifically inhibit NHE3 activity in Caco-2 cells. For each assay condition, triplicate wells were assayed for Na+ uptake and another set of triplicate wells were assayed for uptake in the presence of the inhibitor EIPA. Subtraction of the EIPA inhibited values from uninhibited values gives the portion of Na+ uptake that is specific to NHEs. Only NHE3 is expressed in the cells used for these studies. NHE activity is quantified as the EIPA or S3226-inhibited portion of sodium uptake (CPM) × [NaCl]/(CPM input) × (volume) × (mg protein), giving the final value of NHE activity expressed in nmol/mg protein/5 min.
Cell culture
PS120 fibroblasts stably transfected with NHE3V with a carboxy terminal VSVG tag were a generous gift from Dr. Chris Yun. Cells either contained NHE3V alone or in conjunction with NHERF1/EBP50 or E3KARP/NHERF2 (Yun et al., 1998; Yun et al., 1997; Levine et al., 1993). PS120 cells were split twice per week and acid selected to maintain NHE activity every 2 weeks (Levine et al., 1993; Franchi et al., 1986). Cells were grown in DMEM supplemented with 15 mM HEPES, 26 mM sodium bicarbonate, 50 U/ml penicillin, 50 μg/ml streptomycin and 10% fetal bovine serum. After trypsin treatment, a 75 cm2 flask of PS120 cells was resuspended in 10 ml of media and seeded at a 1:24 dilution into 24 well plates, which were used upon confluence, within 3–5 days of plating. Those cells containing NHERF1 or NHERF2 were kept under selective pressure with the use of 300 μg/ml hygromycin both during culture and plating (Yun et al., 1998). All cells were placed in serum and antibiotic free media the night before infection. Caco-2 cells were grown as previously described (Hecht et al., 2004).
Statistical analysis
Data were analyzed by Student’s t test and single outliers were tested using the Q-test. A single outlier was removed from Figure 1 at the timepoint 1.5 hours which was 20 σ from the mean and four data points were removed from Figure 6B, one from each condition with a range of deviation between 4 and 20 σ from the mean. All points removed were major outliers and met Qc values sufficient for exclusion. Data were significantly different by Student’s t test with or without outlier inclusion. Data were considered significant if P < 0.05.
Figure 1.
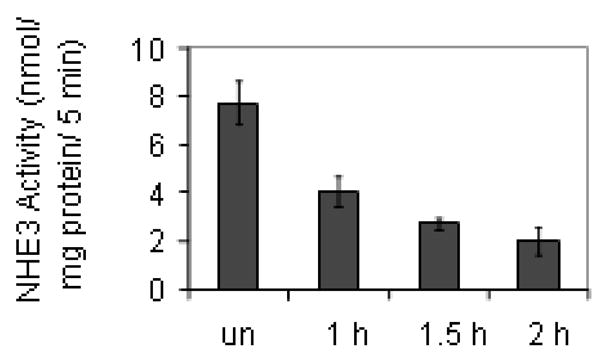
EPEC infection causes a decrease in NHE3 activity over time. PS120 cells transfected with NHE3 and NHERF2 were infected with EPEC for 30, 60 or 90 minutes followed by a PBS rinse and a 30 minute acid load. The stated times reflect both infection time and the following acid load. EPEC infection progressively decreased NHE3 activity. Values at all time points were significantly decreased compared to uninfected cells by Student’s t test P < 0.01. Values at 2 hours were significantly decreased compared to those at 1 hour (P < 0.05) while the values at 1.5 hours were not significantly different from those at either 1 or 2 hours. Data represent an n of 4 performed in triplicate. un = uninfected
Figure 6.

The effector protein EspF causes the decrease in NHE3 activity seen with EPEC infection. Cells were infected with an espF mutant or wt EPEC for 1.5 hours followed by 30 minutes of acid load prior to sodium uptake. EspF mutants are comparable to uninfected controls rather than wt EPEC suggesting that EspF mediates the decrease in NHE3 activity seen in infected cells. B. Cells were infected with a ΔespF mutant carrying a vector control or the complement pespF. Complementation was able to restore the ability of EPEC to cause a decrease in NHE activity while the vector control was comparable to uninfected. Data represent an n of 4, performed in triplicate where * denotes P < 0.001 by Student’s t test. C. These data were verified in Caco-2 human intestinal epithelial cell line which are polarized and express NHE3 on the apical surface. As in the PS120 model, the reduction in NHE3 activity was abrogated with an ΔespF mutant and restored through complementation with pespF. NHE3 activity was distinguished from NHE2 through the use of 10oM S3226. Data represent an n of 10, performed in triplicate where * indicates P < 0.001 by Student’s t test.
Results
EPEC decreases NHE3 activity in transfected PS120 fibroblasts
We initially characterized the effect of wt EPEC on NHE3 activity in a PS120 line co-transfected with NHE3 and NHERF2. This line was chosen as it shows regulatory responses that are akin to those seen in cultured intestinal epithelial cells. For example, NHERF2 co-expression with NHE3 allows for an expected down-regulation of NHE3 in response to Ca2+, cAMP and cGMP while NHERF1 only allows for regulation in response to cAMP (Cha et al., 2005; Lee-Kwon et al., 2003; Zizak et al., 1999). PS120 cells expressing NHE3 and NHERF2 were infected with EPEC for 30, 60, or 90 minutes then rinsed in PBS and treated with an acid load solution for 30 minutes. As seen in Figure 1, EPEC caused a significant decrease (48%) in NHE3 activity at 1 hour, the earliest time point studied. In addition, NHE3 activity continued to decrease over time reaching 64% at 1.5 hours and 74% at 2 hours. Therefore, we opted to use a 2 hour infection for the remainder of the experiments presented here. These results are comparable to the 51% decrease seen in NHE3 activity in Caco-2 cells (Hecht et al., 2004).
Wt EPEC but not non-pathogenic bacteria decrease NHE3 activity in PS120 cells
Bacterial infection causes changes in the media that could interfere with NHE activity. There is a very minor change in the pH over time (an acidification of 0.2 pH units for EPEC and 0.4 units for HB101). In addition, the presence of numerous bacteria could alter the osmotic load of the cells, thus affecting NHE activity. Therefore, we used the non-pathogenic bacterial cloning strain HB101 as a control. HB101 had no impact on NHE3 activity levels in PS120 cells expressing both NHERF2 and NHE3 despite minor acidification of the media (Figure 2A). This is consistent with our previous report that non-pathogenic bacteria do not change NHE activity in Caco-2 cells (Hecht et al., 2004).
Figure 2.
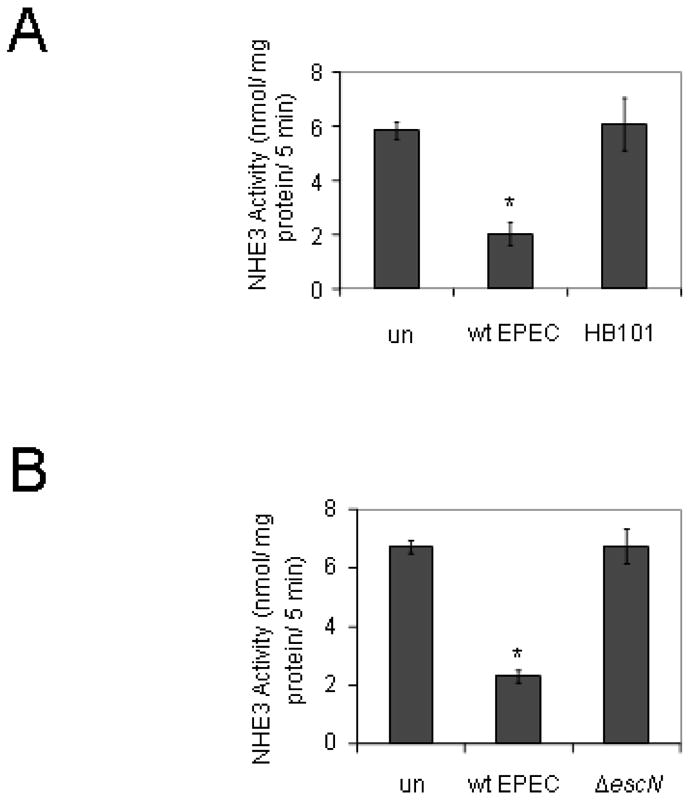
Non-pathogenic bacteria or those without a functional TTSS do not decrease NHE3 activity. A. PS120 cells transfected with NHE3 and NHERF2 were infected with either wild-type EPEC or the non-pathogenic E. coli HB101 for 90 minutes followed by a 30 minute acid load for a total infection time of 2 hours. NHE3 activity levels were comparable in uninfected controls and cells infected with HB101. B. Cells were infected with either wt EPEC or the TTSS defective mutant escN for a total of two hours and then assayed for NHE3 activity. The activity of NHE3 in cells infected with wt EPEC was significantly decreased while that of cells infected with the escN mutant was not significantly different from uninfected cells. * = P < 0.001 by Student’s t test compared to uninfected controls. Data are from an n of 4 or 8 experiments with triplicate wells for figures A and B respectively. un = uninfected
A functional TTSS is required for the reduction in NHE3 activity
Our previous work showed that mutation of escN, the gene encoding the ATPase that drives type III secretion, prevented EPEC from altering NHE activity. Similar findings were seen in PS120 cells expressing both NHERF2 and NHE3; that is, the escN mutant failed to alter NHE3 activity as compared to uninfected cells (Figure 2B).
NHE scaffolding proteins enhance the down-regulation of NHE activity
PS120 cells express a small amount of the scaffolding protein NHERF1 but not NHERF2 (Donowitz et al., 2005). Transfection of NHERF1 or NHERF2 in addition to NHE3 in PS120 cells has been shown to enhance the regulation of NHE3 by various signal transduction cascades elicited by including cAMP and Ca2+ (Lee-Kwon et al., 2003; Yun et al., 1997). Therefore, we determined whether scaffolding molecules contributed to the regulation of NHE3 activity by EPEC. While EPEC infection caused a 36% decrease in the NHE activity of PS120 cells transfected with NHE3 alone, the addition of NHERF1 or NHERF2 caused an additional decrease in NHE activity (Figure 3). Co-transfection with NHERF1 caused a 56% reduction in NHE activity during EPEC infection, while co-transfection of NHERF2 caused a 65% reduction, almost double that seen in cells transfected with NHE3 alone (Figure 3). These data may reflect the enhanced ability of NHE3 to enter recycling endosomes in the presence of NHERF proteins due to better cytoskeletal linkage.
Figure 3.

A. The regulatory proteins NHERF1 and NHERF2 enhance the EPEC-mediated decrease in NHE3 activity. Wt EPEC was used to infect PS120 cells which were transfected with either NHE3 alone or NHE3 in conjunction with either the scaffolding protein NHERF1 or NHERF2. Gray bars are uninfected while black bars are infected with wt EPEC. Co-transfection of NHE3 and either NHERF1 or NHERF2 significantly decreased NHE activity in comparison to cells expressing NHE3 alone. Cells were infected for 1.5 hours followed by a 30 minute acid load. EPEC infected cells showed significantly lower NHE3 activity than uninfected controls, P < 0.001 in all three cell lines by Student’s t test (*,**,***). In addition fibroblasts transfected with NHERF1(**) had lower NHE activity than those with NHE3 only after EPEC infection (P = 0.001) and those transfected with NHE3 and NHERF2 had lower activity after EPEC infection than either those with NHE3 and NHERF1 (P = 0.02) or those with NHE3 only (P < 0.001). Data represent an average of 9 experiments performed in triplicate shown with SEM. B. In contrast to EPEC, HB101 (black bars) a non pathogenic E. coli decreased NHE3 activity in PS120 cells expressing NHE3 only but not in those cells with the scaffolding protein NHERF1. Uninfected cells are shown in gray for comparison. n = 4, with triplicate wells per n. * indicates significance between control and infected by Student’s t test.
We tested the role of scaffolding proteins using the non-pathogenic E. coli HB101 in addition to EPEC. In the cell line PS120 transfected with both NHE3 and NHERF2, HB101 was similar to uninfected control (Figure 2A). However, there was variable response within the three cell lines tested. In cells expressing NHE3 only, there was a significant decrease in NHE3 activity (Figure 3B). In contrast, cells expressing NHE3 and NHERF1 behaved comparably to those expressing NHERF2 where the non-pathogen HB101 had no impact (Figure 3B). Taken in context of what is known about the regulation in this model system, these data suggest that only those cells expressing the scaffolding proteins NHERF1 or NHERF2 are appropriately regulated in response to infection.
Map mutants do not impair EPEC effects on NHE3
It has been recently shown that the EPEC effector protein Map interacts with NHERF1 (Alto et al., 2006; Simpson et al., 2006). Given the dependence of NHE3 response to EPEC infection on NHERF1/NHERF2, it seemed likely that an EPEC effector known to interact with NHERF1 would be involved in EPEC-mediated down-regulation of NHE3 activity. However, as seen in Figure 4A, neither deletion of map nor overexpression of map in the Δmap + pmap complement had any impact on the suppression of NHE3 activity seen with EPEC infection. This result was somewhat surprising given the known regulatory role of NHERF1 in NHE3 function. Therefore, we tested an additional map mutant. MapΔtrl is a plasmid-borne map construct missing the final three amino acids which form a consensus PDZ ligand and are necessary for interaction with NHERF1 (Alto et al., 2006). The Map trl mutant also caused a reduction in NHE3 activity equivalent to that of wt EPEC suggesting that Map interactions with NHERF1 are not involved in the down-regulation of NHE3 activity by EPEC (Figure 4B).
Figure 4.
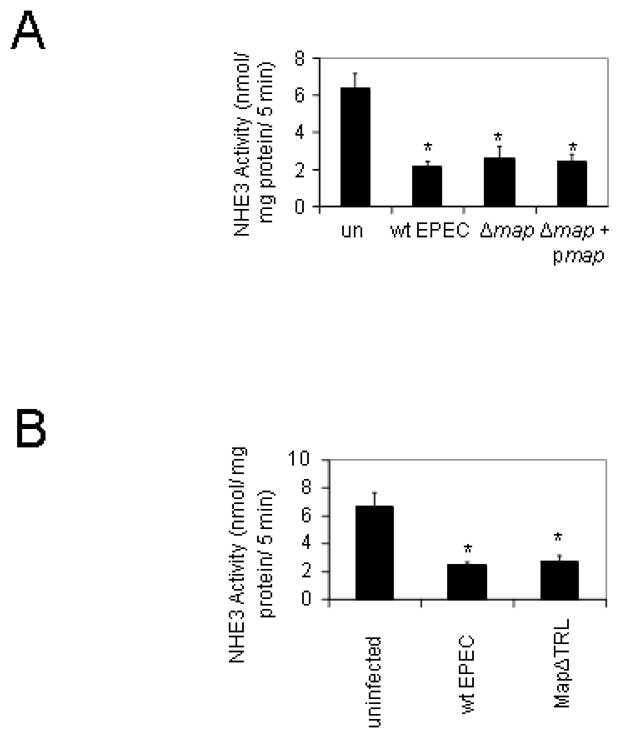
Map plays no role in the EPEC-induced decrease in NHE3 activity. Map is an EPEC effector protein that has recently been identified as a NHERF interacting protein which also has Cdc42-like activity (Alto et al., 2006). A. PS120 cells transfected with both NHERF2 and NHE3 were infected with wt EPEC, the Δmap mutant, or the complement Δmap + pmap for a total of 2 hours. There were no significant differences between the mutant, its complement and wt EPEC. B. Cells were infected with either wt EPEC or the strain MapΔTRL. MapΔTRL is a chromosomal map deletion complemented with a plasmid containing a 3′ deletion of the region encoding the last three amino acids of map. The TRL motif is part of a PDZ ligand domain in Map which is known to bind to NHERF proteins; however, deletion of the TRL motif failed to ablate the EPEC-mediated decrease in NHE3 activity suggesting that NHERF interaction with Map is not necessary for changing NHE3 activity. Data are the mean of 4 experiments performed in triplicate, * denotes P≤0.001.
Tir, EspG and EspH do not alter NHE3 activity
The EPEC effector protein Tir is known to play an important role in intimate attachment. Tir is a bacterial protein that is injected into the host cytosol and later becomes phosphorylated and associated with the cell membrane where it recruits actin in order to form a pedestal. Membrane-associated Tir then binds to intimin which is located on the bacterial surface. Intimate attachment is known to be an important factor in EPEC infection; however, EPEC also possesses additional adhesins such as the bundle forming pili which promote attachment without pedestal formation. Deletion of tir did not modify EPEC’s ability to diminish NHE activity (Figure 5A); therefore, we tested additional EPEC effector mutants. EspG and its homolog EspG2 cause disruption of microtubules, which are involved in vesicle trafficking; however, mutation of espG and espG2, did not block the decrease in NHE activity seen after EPEC infection (Figure 5B). Another effector, EspH has subtle effects on actin pedestal formation with mutants displaying shortened pedestals. Similar to Tir and EspG/G2, EspH was found to have no role in altering NHE activity (Figure 5B).
Figure 5.
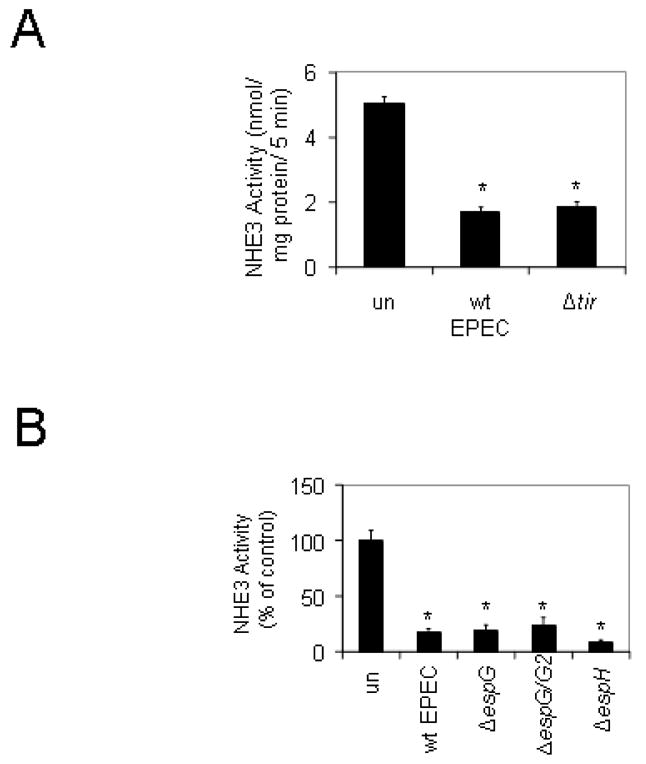
Intimate attachment is not required for EPEC’s ability to decrease NHE3 activity. A. The loss of Tir, an EPEC effector involved in intimate adherence, did not alter the ability of EPEC to alter NHE activity. Cells were infected with wt EPEC or a Δtir mutant for a total of 2 hours then NHE3 activity was measured. B. The effector EspG and its homolog EspG2 which cause microtubule disruption do not cause the decrease in NHE activity seen with EPEC infection. There was no significant difference between NHE3 activity in cells infected with an espH deletion compared to that of wt EPEC. Data represent an n of 3 with triplicate wells where * denotes P < 0.001.
EspF decreases NHE activity
EspF is primarily known for its role in disruption of barrier function and mediating a decrease in transepithelial resistance (McNamara et al., 2001); however a number of additional functions of EspF including a role in cell death have been described. The NHE activity of PS120 cells co-transfected with NHE3 and NHERF2 was essentially identical for uninfected cells and those infected with a ΔespF mutant in contrast to the decreased activity seen with wt EPEC infection (Figure 6A). The ΔespF mutant is also missing a short downstream region although espF represents the far 3′ terminus of the LEE pathogenicity island and this downstream region most likely is a remnant of translocation. Nonetheless, we tested an espF complement, and ΔespF + pespF not only restored the decrease in NHE3 activity to levels comparable to wt EPEC, but actually caused further suppression (Figure 6B). When compared to wt EPEC, there was an additional 50% drop in NHE activity in the presence of the complement, most likely due to overexpression of EspF. A pACYC184 vector control did not alter the phenotype of the espF mutant (Figure 6B).
Our initial data suggest that the NHE3/NHERF2 co-transfected PS120 fibroblast model is essentially equivalent to cultured human intestinal cell lines with regard to NHE3 activity. However, we wished to verify that this was also true in the case of the ΔespF mutant. Shown in Figure 6C, the ΔespF mutant had no effect on NHE3 activity in Caco-2 intestinal epithelial cells while the complemented strain ΔespF+pespF induced a 70% decrease.
Cell death is not the cause of reduced NHE3 activity
EspF is known to be associated with low levels of cell death. While the precise mechanism has not been determined. This cell death process is induced by mitochondrial targeting of EspF via a motif which is destroyed by the mutation Leu16→Glu or L16E (Nagai et al., 2005). Thus, this mutant can be used to rule out contributions to the loss of NHE3 activity by induction of cell death by EspF. However, at two hours post-infection when we perform these assays, the difference in cell death between wt EPEC and an EspF mutant is minimal (~5%), and cannot account for the massive reduction in NHE3 activity that we have seen. Therefore, given the possibility that mitochondrial targeting is playing some other role, we tested the espF mutant L16E. As seen in Figure 7A, complementation of our ΔespF mutant with an L16E construct restored the activity of EspF and reduced NHE3 levels significantly. These data suggest that neither mitochondrial targeting nor the associated cell death is responsible for altered NHE3 activity.
Figure 7.
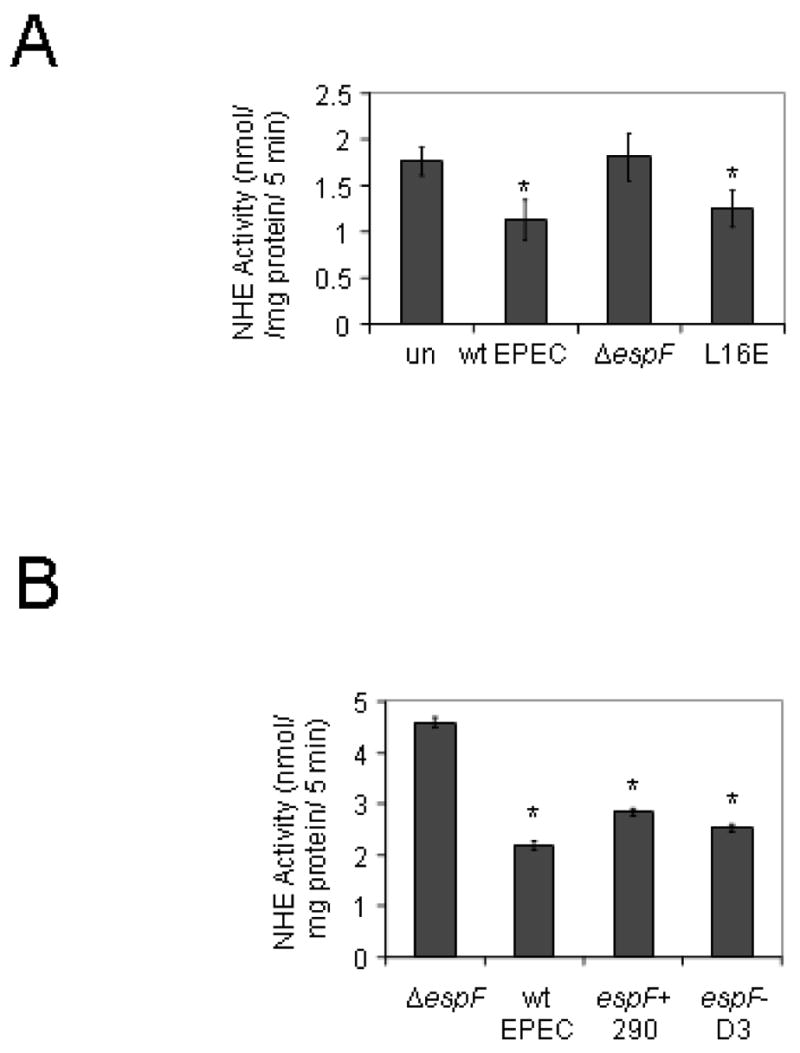
A. The espF point mutant L16E (Leu16→Glu) prevents targeting of EspF to mitochondria and the associated induction of cell death pathways. Loss of mitochondrial targeting does not impair the ability of EspF to reduce NHE3 activity. B. NHE3 is recycled from the surface to subapical membrane domains. EspF is known to interact with SNX9 which is involved in vesicle trafficking via its proline rich repeat domain. A triple point mutant which replaces Arg75,122,169 with Asp also known as EspF-D3 was used to test the role of SNX9 in alteration of NHE3 activity. n = 4, performed in triplicate. * = significant from uninfected by Student’s t test.
Modulation of vesicle trafficking through SNX9 does not alter NHE3 activity
Recycling of NHE3 from the apical cell surface to subapical membrane vesicles is the most common regulatory mechanism for NHE3. EspF has been recently been shown to bind sorting nexin 9 (SNX9) a bar-domain protein which is involved in pinching vesicles from the membrane (Alto et al., 2007). The binding site is found in the three proline rich regions at the carboxy terminus of EspF. The EspF-D3 mutant has three site specific mutations, Arg75,122,169 → Asp, which prevent the interaction of EspF with SNX9 (Alto et al., 2007). As can be seen in Figure 7B, disruption of the interaction between EspF and SNX9 does not prevent the reduction in NHE3 activity.
Discussion
EPEC infection modifies a number of transport processes including sodium/hydrogen exchange, chloride/bicarbonate exchange, sodium glucose co-transport and butyrate uptake (Gill et al., 2007; Borthakur et al., 2006; Dean et al., 2006; Hodges et al., 2006). While there are global disturbances in transport of ions and other critical substrates, the bacterial effectors triggering each of these processes are distinct. Alterations in Cl−/HCO3− exchange are caused by microtubule disruption mediated by EspG and EspG2 (Gill et al., 2007), while the effectors involved in increasing NHE2 activity and decreasing butyrate uptake have not been identified despite testing all of the LEE encoded effector proteins (Borthakur et al., 2006; Hodges et al., 2006). SGLT1 suppression appears to depend on a combination of proteins including EspF, Map, Tir and Intimin (Dean et al., 2006). In the current studies, we determined that changes in NHE3 activity are specific to the effector protein EspF. EspF may play a partial role in the decrease in SLGT1 activity, but there is no impact on Cl−/HCO−3 exchange, NHE2 activity, or butyrate uptake (Gill et al., 2007; Borthakur et al., 2006; Dean et al., 2006; Hodges et al., 2006). Also of note, there are temporal changes in the response of the transport proteins to EPEC infection. While NHE2 activity and butyrate uptake changes appear most intense at the earliest time points and then taper off, the decrease in NHE3 activity progressively increases over time.
Because of the specificity of each effector in modifying only select ion exchange processes, EPEC can alter one branch in what is usually a tightly interdependent cross-regulation between ion exchangers. Apical sodium hydrogen exchange is typically coordinately regulated where signal transduction pathways that affect NHE2 also have an impact on NHE3 activity although there can be divergent outcomes. For example, in our previous work we found that changes in NHE2 activity were due to activation of PKC α and ε (Hodges et al., 2006). PKC α is known to decrease NHE3 activity in addition to increasing NHE2 activity and we fully expected that the process involved in altering NHE2 activity would be directly related to the changes seen with NHE3 activity (Hodges et al., 2006; Lee-Kwon et al., 2003). However, the changes in NHE3 activity are solely attributable to EspF, while an espF mutant was found to be equivalent to wt EPEC in increasing NHE2 activity (Hodges et al., 2006). In addition, NHE3 activity is typically coupled to the apical Cl−/HCO−3 exchanger down-regulated in adenoma (DRA) via the interaction with NHERF proteins (Lamprecht et al., 2002). EspF mutants are equivalent to wt EPEC in their effect on Cl−/HCO−3 exchange and EspG has no impact on NHE3 activity (Gill et al., 2007). Thus, typically coordinated pathways are being independently modified by distinct bacterial effectors leading to an uncoupling of the host response. This provides EPEC with an exquisite level of control over host processes which appear to be regulated in a temporal manner. Initial infection causes a rapid absorption of sodium in a PKC-dependent manner (Hodges et al., 2006; Hecht et al., 2004). This initial absorptive state would likely assist during the early colonization phase of attachment before actin pedestals are formed. After intimate attachment of EPEC, a switch toward fluid imbalance, mediated in part by impaired Na+ and Cl− absorption due to a combined reduction in NHE3, Cl−/HCO−3, and SGLT1 activity would help in dispersing the pathogen, without dislodging already adherent bacteria.
Despite its ability to bind and interact with NHERF1, the effector protein Map did not play a role in regulating NHE3 activity (Alto et al., 2006; Simpson et al., 2006). However, the PDZ proteins NHERF1 and NHERF2 clearly enhanced the down-regulation of NHE3. NHE3 activity is typically regulated through endocytosis of the protein into subapical membrane vesicles which can subsequently recycle protein back to the apical membrane (Zachos et al., 2005). This recycling is dependent on an intact actin cytoskeleton and linkage of NHE3 to actin is mediated both through NHERF linkage to ezrin as well as an independent binding to ezrin (Cha et al., 2006; Donowitz et al., 2005). Point mutations in this secondary ezrin binding domain have been shown to impair NHE3 exocytosis leading to reduced surface expression (Cha et al., 2006). Map may not interfere with NHE3 function because of the functional redundancy provided by this second actin binding method involving direct interaction with ezrin, independent from NHERFs. This lack of an effect is not likely to be due to the overexpression of NHERFs as we saw similar results in PS120 cells transfected with NHE3 only (data not shown). These cells are known to express only low levels of NHERF1 (Donowitz et al., 2005).
EspF is well known for its role in modulating epithelial barrier function and redistributing tight junction components; however, the mechanism by which this occurs has not been defined (Muza-Moons et al., 2004; McNamara et al., 2001). In addition, EspF can be targeted to mitochondria leading to the activation of cell death pathways, (Nougayrede and Donnenberg, 2004). There is also direct binding to the intermediate filament cytokeratin 18 and association with 14-3-3 (Viswanathan et al., 2004). Cytokeratins are not known to play a role in NHE3 activity nor is the protein 14-3-3. While altered NHE activity is associated with changes in barrier function, the changes in NHE3 occur prior to loss of barrier function and a decrease in NHE3 is actually thought to enhance barrier function suggesting that these two phenotypes are not linked (Turner et al., 2000). Considering another known phenotype of EspF, perhaps targeting of mitochondria and initiation of caspase cleavage was involved with changes in NHE3. EspF is thought to induce cell death by causing membrane depolarization of mitochondria (Nougayrede and Donnenberg, 2004). Changes in mitochondrial membrane potential have not been linked to changes in NHE3 activity; however there is cross-regulation of mitochondrial death pathways and NHE1 activity. However, our data with the L16E mutant suggest that mitochondrial targeting and cell death are not responsible for the reduction in NHE3 activity (Figure 7A). The most recently identified role for EspF is alteration of vesicle trafficking through interaction with SNX9. Given that NHE3 is regulated by vesicle recycling, this interaction with SNX9 seemed a likely candidate for alteration of NHE3 activity; however, our data with the EspF-D3 mutant suggest that this is not the case (Figure 7B). EspF is clearly a multi-functional protein and its role in barrier disruption has been shown to play an important role in both tissue culture and animal models (Shifflett et al., 2005; McNamara et al., 2001). Coupled with the ability to modify NHE3 activity it is likely that EspF will prove important in both early and late processes of infection.
EPEC infection is most severe in infants and young children under the age of five although the vast majority of infections occur in children under the age of 1 year (Fagundes-Neto and Scaletsky, 2000). There are certainly differences in the immune system of children and adults but there has been no clear rationale for the striking ability of EPEC to infect children. Interestingly, the relative levels of NHE3 protein change dramatically during the developmental process. While there are no studies of human intestine, a number of studies have been carried out in rats. The relative contributions of NHE2 and NHE3 to brush border membrane vesicle NHE activity were determined between 2 and 6 weeks of age. At all times the activity of NHE3 was greater than NHE2 and this increased from a 59% share of brush border NHE activity at 2–3 weeks in age to 92% of NHE activity by 6 weeks (Collins et al., 1997). Overall, sodium uptake and the NHE3 specific portion were approximately 4-fold greater in 6 week old rats (post-weaning) than in adult (120 day old) rats (Collins et al., 1997). Thus in rats there appears to be a specific developmental period where NHE activity is enhanced and NHE3 is the dominant isoform. If these findings correspond to human development, the relative contribution of NHE3 to sodium absorption during early developmental stages could provide a unique susceptibility in infants. All in all, the role of EspF in EPEC-mediated disease seems highly significant since it is responsible for several mechanisms that actively promote disease including barrier disruption, apoptosis and a decrease in NHE3 activity.
References
- Alto NM, Shao F, Lazar CS, Brost RL, Chua G, Mattoo S, et al. Identification of a bacterial type III effector family with G protein mimicry functions. Cell. 2006;124:133–145. doi: 10.1016/j.cell.2005.10.031. [DOI] [PubMed] [Google Scholar]
- Alto NM, Weflen AW, Rardin MJ, Yarar D, Lazar CS, Tonikian R, et al. The type III effector EspF coordinates membrane trafficking by the spatiotemporal activation of two eukaryotic signaling pathways. J Cell Biol. 2007;178:1265–1278. doi: 10.1083/jcb.200705021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borthakur A, Gill RK, Hodges K, Ramaswamy K, Hecht G, Dudeja PK. Enteropathogenic Escherichia coli inhibits butyrate uptake in Caco-2 cells by altering the apical membrane MCT1 level. Am J Physiol Gastrointest Liver Physiol. 2006;290:G30–35. doi: 10.1152/ajpgi.00302.2005. [DOI] [PubMed] [Google Scholar]
- Cha B, Tse M, Yun C, Kovbasnjuk O, Mohan S, Hubbard A, et al. The NHE3 juxtamembrane cytoplasmic domain directly binds ezrin: dual role in NHE3 trafficking and mobility in the brush border. Mol Biol Cell. 2006;17:2661–2673. doi: 10.1091/mbc.E05-09-0843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha B, Kim JH, Hut H, Hogema BM, Nadarja J, Zizak M, et al. cGMP inhibition of Na+/H+ antiporter 3 (NHE3) requires PDZ domain adapter NHERF2, a broad specificity protein kinase G-anchoring protein. J Biol Chem. 2005;280:16642–16650. doi: 10.1074/jbc.M500505200. [DOI] [PubMed] [Google Scholar]
- Collins JF, Xu H, Kiela PR, Zeng J, Ghishan FK. Functional and molecular characterization of NHE3 expression during ontogeny in rat jejunal epithelium. Am J Physiol. 1997;273:C1937–1946. doi: 10.1152/ajpcell.1997.273.6.C1937. [DOI] [PubMed] [Google Scholar]
- Crawford JA, Kaper JB. The N-terminus of enteropathogenic Escherichia coli (EPEC) Tir mediates transport across bacterial and eukaryotic cell membranes. Mol Microbiol. 2002;46:855–868. doi: 10.1046/j.1365-2958.2002.03214.x. [DOI] [PubMed] [Google Scholar]
- Daniell SJ, Takahashi N, Wilson R, Friedberg D, Rosenshine I, Booy FP, et al. The filamentous type III secretion translocon of enteropathogeni Escherichia coli. Cell Microbiol. 2001;3:865–871. doi: 10.1046/j.1462-5822.2001.00168.x. [DOI] [PubMed] [Google Scholar]
- Dean P, Maresca M, Schuller S, Phillips AD, Kenny B. Potent diarrheagenic mechanism mediated by the cooperative action of three enteropathogenic Escherichia coli-injected effector proteins. Proc Natl Acad Sci U S A. 2006;103:1876–1881. doi: 10.1073/pnas.0509451103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donowitz M, Cha B, Zachos NC, Brett CL, Sharma A, Tse CM, Li X. NHERF family and NHE3 regulation. J Physiol. 2005;567:3–11. doi: 10.1113/jphysiol.2005.090399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott SJ, Krejany EO, Mellies JL, Robins-Browne RM, Sasakawa C, Kaper JB. EspG, a novel type III system-secreted protein from enteropathogenic Escherichia coli with similarities to VirA of Shigella flexneri. Infect Immun. 2001;69:4027–4033. doi: 10.1128/IAI.69.6.4027-4033.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagundes-Neto U, Scaletsky IC. The gut at war: the consequences of enteropathogenic Escherichia coli infection as a factor of diarrhea and malnutrition. Sao Paulo Med J. 2000;118:21–29. doi: 10.1590/S1516-31802000000100006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchi A, Cragoe E, Jr, Pouyssegur J. Isolation and properties of fibroblast mutants overexpressing an altered Na+/H+ antiporter. J Biol Chem. 1986;261:14614–14620. [PubMed] [Google Scholar]
- Gawenis LR, Stien X, Shull GE, Schultheis PJ, Woo AL, Walker NM, Clarke LL. Intestinal NaCl transport in NHE2 and NHE3 knockout mice. Am J Physiol Gastrointest Liver Physiol. 2002;282:G776–784. doi: 10.1152/ajpgi.00297.2001. [DOI] [PubMed] [Google Scholar]
- Gill RK, Borthakur A, Hodges K, Turner JR, Clayburgh DR, Saksena S, et al. Mechanism underlying inhibition of intestinal apical Cl/OH exchange following infection with enteropathogenic E. coli. J Clin Invest. 2007;117:428–437. doi: 10.1172/JCI29625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenheid S, Sekirov I, Thomas NA, Deng W, O’Donnell P, Goode D, et al. Identification and characterization of NleA, a non-LEE-encoded type III translocated virulence factor of enterohaemorrhagic Escherichia coli O157:H7. Mol Microbiol. 2004;51:1233–1249. doi: 10.1046/j.1365-2958.2003.03911.x. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Szaszi K, Coady-Osberg N, Furuya W, Bretscher AP, Orlowski J, Grinstein S. Inhibition and redistribution of NHE3, the apical Na+/H+ exchanger, by Clostridium difficile toxin B. J Gen Physiol. 2004;123:491–504. doi: 10.1085/jgp.200308979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht G, Koutsouris A. Enteropathogenic E. coli attenuates secretagogue-induced net intestinal ion transport but not Cl- secretion. Am J Physiol. 1999;276:G781–788. doi: 10.1152/ajpgi.1999.276.3.G781. [DOI] [PubMed] [Google Scholar]
- Hecht G, Marrero JA, Danilkovich A, Matkowskyj KA, Savkovic SD, Koutsouris A, Benya RV. Pathogenic Escherichia coli increase Cl-secretion from intestinal epithelia by upregulating galanin-1 receptor expression. J Clin Invest. 1999;104:253–262. doi: 10.1172/JCI6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht G, Hodges K, Gill RK, Kear F, Tyagi S, Malakooti J, et al. Differential regulation of Na+/H+ exchange isoform activities by enteropathogenic E. coli in human intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2004;287:G370–378. doi: 10.1152/ajpgi.00432.2003. [DOI] [PubMed] [Google Scholar]
- Hodges K, Gill R, Ramaswamy K, Dudeja PK, Hecht G. Rapid activation of Na+/H+ exchange by EPEC is PKC mediated. Am J Physiol Gastrointest Liver Physiol. 2006;291:G959–968. doi: 10.1152/ajpgi.00274.2005. [DOI] [PubMed] [Google Scholar]
- Jarvis KG, Giron JA, Jerse AE, McDaniel TK, Donnenberg MS, Kaper JB. Enteropathogenic Escherichia coli contains a putative type III secretion system necessary for the export of proteins involved in attaching and effacing lesion formation. Proc Natl Acad Sci U S A. 1995;92:7996–8000. doi: 10.1073/pnas.92.17.7996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanack KJ, Crawford JA, Tatsuno I, Karmali MA, Kaper JB. SepZ/EspZ is secreted and translocated into HeLa cells by the enteropathogenic Escherichia coli type III secretion system. Infect Immun. 2005;73:4327–4337. doi: 10.1128/IAI.73.7.4327-4337.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandasamy RA, Yu FH, Harris R, Boucher A, Hanrahan JW, Orlowski J. Plasma membrane Na+/H+ exchanger isoforms (NHE-1, -2, and -3) are differentially responsive to second messenger agonists of the protein kinase A and C pathways. J Biol Chem. 1995;270:29209–29216. doi: 10.1074/jbc.270.49.29209. [DOI] [PubMed] [Google Scholar]
- Kaper JB, McDaniel TK, Jarvis KG, Gomez-Duarte O. Genetics of virulence of enteropathogenic E. coli. Adv Exp Med Biol. 1997;412:279–287. doi: 10.1007/978-1-4899-1828-4_47. [DOI] [PubMed] [Google Scholar]
- Kennedy MB. Origin of PDZ (DHR, GLGF) domains. Trends Biochem Sci. 1995;20:350. doi: 10.1016/s0968-0004(00)89074-x. [DOI] [PubMed] [Google Scholar]
- Kenny B, Jepson M. Targeting of an enteropathogenic Escherichia coli (EPEC) effector protein to host mitochondria. Cell Microbiol. 2000;2:579–590. doi: 10.1046/j.1462-5822.2000.00082.x. [DOI] [PubMed] [Google Scholar]
- Kenny B, Ellis S, Leard AD, Warawa J, Mellor H, Jepson MA. Coordinate regulation of distinct host cell signalling pathways by multifunctional enteropathogenic Escherichia coli effector molecules. Mol Microbiol. 2002;44:1095–1107. doi: 10.1046/j.1365-2958.2002.02952.x. [DOI] [PubMed] [Google Scholar]
- Lamprecht G, Seidler UE. The emerging role of PDZ adapter proteins for regulation of intestinal ion transport. Am J Physiol Gastrointest Liver Physiol. 2006 doi: 10.1152/ajpgi.00135.2006. [DOI] [PubMed] [Google Scholar]
- Lamprecht G, Heil A, Baisch S, Lin-Wu E, Yun CC, Kalbacher H, et al. The down regulated in adenoma (dra) gene product binds to the second PDZ domain of the NHE3 kinase A regulatory protein (E3KARP), potentially linking intestinal Cl-/HCO3- exchange to Na+/H+ exchange. Biochemistry. 2002;41:12336–12342. doi: 10.1021/bi0259103. [DOI] [PubMed] [Google Scholar]
- Lee-Kwon W, Kim JH, Choi JW, Kawano K, Cha B, Dartt DA, et al. Ca2+-dependent inhibition of NHE3 requires PKC alpha which binds to E3KARP to decrease surface NHE3 containing plasma membrane complexes. Am J Physiol Cell Physiol. 2003;285:C1527–1536. doi: 10.1152/ajpcell.00017.2003. [DOI] [PubMed] [Google Scholar]
- Levine SA, Montrose MH, Tse CM, Donowitz M. Kinetics and regulation of three cloned mammalian Na+/H+ exchangers stably expressed in a fibroblast cell line. J Biol Chem. 1993;268:25527–25535. [PubMed] [Google Scholar]
- Madara JL, Patapoff TW, Gillece-Castro B, Colgan SP, Parkos CA, Delp C, Mrsny RJ. 5′-adenosine monophosphate is the neutrophil-derived paracrine factor that elicits chloride secretion from T84 intestinal epithelial cell monolayers. J Clin Invest. 1993;91:2320–2325. doi: 10.1172/JCI116462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara BP, Donnenberg MS. A novel proline-rich protein, EspF, is secreted from enteropathogenic Escherichia coli via the type III export pathway. FEMS Microbiol Lett. 1998;166:71–78. doi: 10.1111/j.1574-6968.1998.tb13185.x. [DOI] [PubMed] [Google Scholar]
- McNamara BP, Koutsouris A, O’Connell CB, Nougayrede JP, Donnenberg MS, Hecht G. Translocated EspF protein from enteropathogenic Escherichia coli disrupts host intestinal barrier function. J Clin Invest. 2001;107:621–629. doi: 10.1172/JCI11138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muza-Moons MM, Schneeberger EE, Hecht GA. Enteropathogenic Escherichia coli infection leads to appearance of aberrant tight junctions strands in the lateral membrane of intestinal epithelial cells. Cell Microbiol. 2004;6:783–793. doi: 10.1111/j.1462-5822.2004.00404.x. [DOI] [PubMed] [Google Scholar]
- Nagai T, Abe A, Sasakawa C. Targeting of enteropathogenic Escherichia coli EspF to host mitochondria is essential for bacterial pathogenesis: critical role of the 16th leucine residue in EspF. J Biol Chem. 2005;280:2998–3011. doi: 10.1074/jbc.M411550200. [DOI] [PubMed] [Google Scholar]
- Nougayrede JP, Donnenberg MS. Enteropathogenic Escherichia coli EspF is targeted to mitochondria and is required to initiate the mitochondrial death pathway. Cell Microbiol. 2004;6:1097–1111. doi: 10.1111/j.1462-5822.2004.00421.x. [DOI] [PubMed] [Google Scholar]
- Reczek D, Berryman M, Bretscher A. Identification of EBP50: A PDZ-containing phosphoprotein that associates with members of the ezrin-radixin-moesin family. J Cell Biol. 1997;139:169–179. doi: 10.1083/jcb.139.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabolic I, Herak-Kramberger CM, Ljubojevic M, Biemesderfer D, Brown D. NHE3 and NHERF are targeted to the basolateral membrane in proximal tubules of colchicine-treated rats. Kidney Int. 2002;61:1351–1364. doi: 10.1046/j.1523-1755.2002.00266.x. [DOI] [PubMed] [Google Scholar]
- Shifflett DE, Clayburgh DR, Koutsouris A, Turner JR, Hecht GA. Enteropathogenic E. coli disrupts tight junction barrier function and structure in vivo. Lab Invest. 2005;85:1308–1324. doi: 10.1038/labinvest.3700330. [DOI] [PubMed] [Google Scholar]
- Simpson N, Shaw R, Crepin VF, Mundy R, FitzGerald AJ, Cummings N, et al. The enteropathogenic Escherichia coli type III secretion system effector Map binds EBP50/NHERF1: implication for cell signalling and diarrhoea. Mol Microbiol. 2006;60:349–363. doi: 10.1111/j.1365-2958.2006.05109.x. [DOI] [PubMed] [Google Scholar]
- Subramanya SB, Rajendran VM, Srinivasan P, Nanda Kumar NS, Ramakrishna BS, Binder HJ. Differential regulation of cholera toxin-inhibited Na-H exchange isoforms by butyrate in rat ileum. Am J Physiol Gastrointest Liver Physiol. 2007;293:G857–863. doi: 10.1152/ajpgi.00462.2006. [DOI] [PubMed] [Google Scholar]
- Taylor KA, O’Connell CB, Luther PW, Donnenberg MS. The EspB protein of enteropathogenic Escherichia coli is targeted to the cytoplasm of infected HeLa cells. Infect Immun. 1998;66:5501–5507. doi: 10.1128/iai.66.11.5501-5507.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomson FL, Viswanathan VK, Kanack KJ, Kanteti RP, Straub KV, Menet M, et al. Enteropathogenic Escherichia coli EspG disrupts microtubules and in conjunction with Orf3 enhances perturbation of the tight junction barrier. Mol Microbiol. 2005;56:447–464. doi: 10.1111/j.1365-2958.2005.04571.x. [DOI] [PubMed] [Google Scholar]
- Tse CM, Levine SA, Yun CH, Brant SR, Pouyssegur J, Montrose MH, Donowitz M. Functional characteristics of a cloned epithelial Na+/H+ exchanger (NHE3): resistance to amiloride and inhibition by protein kinase C. Proc Natl Acad Sci U S A. 1993;90:9110–9114. doi: 10.1073/pnas.90.19.9110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JR, Black ED, Ward J, Tse CM, Uchwat FA, Alli HA, et al. Transepithelial resistance can be regulated by the intestinal brush-border Na(+)/H(+) exchanger NHE3. Am J Physiol Cell Physiol. 2000;279:C1918–1924. doi: 10.1152/ajpcell.2000.279.6.C1918. [DOI] [PubMed] [Google Scholar]
- Viswanathan VK, Lukic S, Koutsouris A, Miao R, Muza MM, Hecht G. Cytokeratin 18 interacts with the enteropathogenic Escherichia coli secreted protein F (EspF) and is redistributed after infection. Cell Microbiol. 2004;6:987–997. doi: 10.1111/j.1462-5822.2004.00416.x. [DOI] [PubMed] [Google Scholar]
- Yun CH, Lamprecht G, Forster DV, Sidor A. NHE3 kinase A regulatory protein E3KARP binds the epithelial brush border Na+/H+ exchanger NHE3 and the cytoskeletal protein ezrin. J Biol Chem. 1998;273:25856–25863. doi: 10.1074/jbc.273.40.25856. [DOI] [PubMed] [Google Scholar]
- Yun CH, Oh S, Zizak M, Steplock D, Tsao S, Tse CM, et al. cAMP-mediated inhibition of the epithelial brush border Na+/H+ exchanger, NHE3, requires an associated regulatory protein. Proc Natl Acad Sci U S A. 1997;94:3010–3015. doi: 10.1073/pnas.94.7.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachos NC, Tse M, Donowitz M. Molecular physiology of intestinal Na+/H+ exchange. Annu Rev Physiol. 2005;67:411–443. doi: 10.1146/annurev.physiol.67.031103.153004. [DOI] [PubMed] [Google Scholar]
- Zizak M, Lamprecht G, Steplock D, Tariq N, Shenolikar S, Donowitz M, et al. cAMP-induced phosphorylation and inhibition of Na(+)/H(+) exchanger 3 (NHE3) are dependent on the presence but not the phosphorylation of NHE regulatory factor. J Biol Chem. 1999;274:24753–24758. doi: 10.1074/jbc.274.35.24753. [DOI] [PubMed] [Google Scholar]