

Abstract
Aims
Stretch is an important regulator of atrial function. The functional effects of stretch on human atrium, however, are poorly understood. Thus, we characterized the stretch-induced force response in human atrium and evaluated the underlying cellular mechanisms.
Methods and results
Isometric twitch force of human atrial trabeculae (n = 252) was recorded (37°C, 1 Hz stimulation) following stretch from 88 (L88) to 98% (L98) of optimal length. [Na+]i and pHi were measured using SBFI and BCECF epifluorescence, respectively. Stretch induced a biphasic force increase: an immediate increase [first-phase, Frank–Starling mechanism (FSM)] to ∼190% of force at L88 followed by an additional slower increase [5–10 min; slow force response (SFR)] to ∼120% of the FSM. FSM and SFR were unaffected by gender, age, ejection fraction, and pre-medication with major cardiovascular drugs. There was a positive correlation between the amplitude of the FSM and the SFR. [Na+]i rose by ∼1 mmol/L and pHi remained unchanged during the SFR. Inhibition of Na+/H+-exchange (3 µM HOE642), Na+/Ca2+-exchange (5 µM KB-R7943), or stretch-activated channels (0.5 µM GsMtx-4 and 80 µM streptomycin) did not reduce the SFR. Inhibition of angiotensin-II (AngII) receptors (5 µM saralasin and 0.5 µM PD123319) or pre-application of 0.5 µM AngII, however, reduced the SFR by ∼40–60%. Moreover, stretch increased phosphorylation of myosin light chain 2 (MLC2a) and inhibition of MLC kinase (10 µM ML-7 and 5 µM wortmannin) decreased the SFR by ∼40–85%.
Conclusion
Stretch elicits a SFR in human atrium. The atrial SFR is mediated by stretch-induced release and autocrine/paracrine actions of AngII and increased myofilament Ca2+ responsiveness via phosphorylation of MLC2a by MLC kinase.
Keywords: Stretch, Human, Atrium, Force, Angiotensin II
1. Introduction
Stretch is a key signal in the regulation of the heart. It modulates heart rate and rhythm, force of contraction, and gene expression. In supraventricular tissue, stretch directly modulates spontaneous beating rate in the sinoatrial node1 and force of contraction in the atrium.2 The latter effect is mediated via the Frank–Starling mechanism (FSM). This mechanism is important for adjusting cardiac output between the right and left cardiac chambers as well as for ventricular filling in settings of impaired cardiac function. Furthermore, acute or chronic stretch of the atria can induce atrial remodelling3 and atrial fibrillation.4 The cellular mechanisms causing these stretch-dependent alterations in atrial function, however, remain poorly understood.
Functional effects of stretch have been characterized in ventricular myocardium of animal and human hearts. Acute stretch of ventricular myocardium elicits a biphasic increase in developed force.5–15 The first phase occurs immediately after stretch and is attributed to increased myofilament Ca2+ sensitivity via the FSM. The second phase develops more slowly over a time course of 5–10 min and is termed slow force response (SFR). It is accompanied by an increased amplitude of the underlying [Ca2+]i-transient6–8,15 caused by elevation of SR-Ca2+ load via modulation of Na+/Ca2+-exchange (NCX).10,14 Modulation of NCX occurs as a result of an increase in [Na+]i8,15 which attenuates forward and favours reverse-mode NCX. The stretch-dependent increase in [Na+]i is mediated by stimulation of Na+/H+-exchange (NHE) and/or stretch-activated non-selective cation channels (SACs).8,11–15 How exactly NHE becomes activated through stretch, however, is still a matter of debate. While in rat, cat, and ferret myocardium stretch-dependent release of angiotensin-II (AngII) and endothelin-1 appears to stimulate NHE to contribute to the SFR,8–10,16 this mechanism is not involved in rabbit and human ventricle.12,14 Furthermore, even after blockade of NHE and NCX, a residual SFR remained implying that additional, as yet unidentified mechanisms are involved in the SFR.15
Despite extensive data obtained in ventricle, much less is known about stretch-induced force alterations in atrium. This is, however, of major pathophysiological relevance, because atrium is subject to even more pronounced mechanical stress than ventricle. A stretch-elicited biphasic force increase is observed in mammalian atrial myocardium.2,17,18 As in ventricle, the SFR in rat atrium is accompanied by a [Ca2+]i-transient increase.2,17 In addition, changes in action-potential morphology have been observed during the SFR. Based on modelling studies, it was suggested that increased Ca2+-affinity of troponin-C and activation of SACs contribute to action potential changes and SFR.2 Direct experimental evidence for this hypothesis, however, is scarce. Furthermore, it is unknown whether additional mechanisms might contribute to the SFR in atrium and the existence of this phenomenon in human atria has never been demonstrated. Thus, the cellular mechanisms underlying the SFR in atrium remain elusive. We, therefore, set out to elucidate the existence and mechanisms of the SFR in human atrium.
2. Methods
2.1. Myocardium
Right atrial appendages were obtained from hearts of patients (n = 125) undergoing cardiac surgery. The study was approved by the local ethics committee, and the patients gave informed written consent. The investigation conforms with the principles outlined in the Declaration of Helsinki.
Atrial trabeculae were prepared as previously described.19 Trabeculae (n = 252; diameter = 0.51 ± 0.01 mm) were mounted on hooks in a temperature-controlled (37°C) recording chamber and electrically stimulated at 1 Hz in Tyrode’s solution containing (mM): Na+ 152, K+ 3.6, Cl− 135, HCO3− 25, HEPES 5, Ca2+ 2.5, Mg2+ 0.6, H2PO4− 1.3, SO42− 0.6, pH 7.4. Isometric contractions were recorded using a force transducer and displayed on a chart recorder and a computer for further analysis.
2.2. Experimental protocol
The experimental protocol was essentially identical to the protocol previously employed for studying the stretch-dependent SFR in human ventricular myocardium.14 Briefly, trabeculae were gradually stretched to the length at which maximal force development was observed (Lmax). Afterwards, length was reduced to 88% of Lmax (L88). After 30 min, trabeculae were stretched acutely to 98% of Lmax (L98). This resulted in a biphasic increase in developed force: FSM and SFR, respectively. To assess the involvement of receptors, transporters, and ion channels in the stretch responses, various drugs were used (see below). All experiments involving drugs included paired stretch protocols, i.e. first a control stretch protocol was conducted in the absence of drug; then the muscle was released to L88 and drug was applied; after 25 min, a second stretch protocol was conducted in the same muscle strip in the presence of the respective drug.
2.3. Measurements of [Na+]i, pHi, and [Ca2+]i
[Na+]i and pHi were measured as described previously.15 Briefly, the fluorescent Na+ indicator SBFI was loaded into the trabeculae (n = 11) by incubation (180 min) in a solution containing 35 µM SBFI-AM. SBFI was excited alternately at 340 (F340) and 380 nm (F380) and fluorescence emission was collected by a photomultiplier at 505 nm. Excitation light was passed through a neutral density filter (1% transmission) and fluorescence recording restricted to ∼20 s/min to limit photobleaching. The ratio F340/F380 is a measure for [Na+]i. Following correction for background and autofluorescence, this ratio was converted to [Na+]i at the end of an experiment in each muscle-strip by an in vivo calibration approach using calibration solutions containing 0, 10, 20, or 30 mM NaCl, 10 mM HEPES, 1 mM EGTA, 0.2 µg/mL gramicidin D, 40 µM monensin, 100 µM strophanthidin, pH 7.4. KCl was added to yield a combined NaCl plus KCl concentration of 140 mM.
pHi was measured using BCECF.15 Trabeculae (n = 7) were incubated for 45 min in solution containing 15 µM BCECF-AM. Excitation wavelengths were 440 (F440) and 495 nm (F495) and fluorescence emission was collected at 535 nm. At the end of each experiment, the ratio F495/F440 was converted into pHi using calibration solutions with pH values of 7.6, 7.4, 7.2, 7.0, and 6.8. The calibration solution was composed of (mM): 140 KCl, 1.2 MgCl2, 5 HEPES, 30 2,3-butanedione-monoxime, and 0.05 nigericin.
[Ca2+]i was measured using Fura-2. Trabeculae (n = 6) were incubated for 120 min in solution containing 5 µM Fura-2-AM. Excitation wavelengths were 340 (F340) and 380 nm (F380) and fluorescence emission was collected at 505 nm. The ratio F340/F380 (arbitrary units) was taken as a measure for [Ca2+]i.
Experiments with SBFI, BCECF, and Fura-2 were performed at 30°C and 0.2 Hz stimulation.15 These conditions were found optimal for fluorescence recording. At higher temperature and stimulation frequency, dye loss from the trabeculae was accelerated and, therefore, recording of an entire experimental run in a muscle-strip was hampered. Control experiments (n = 3) revealed that the stretch-dependent force responses of atrial trabeculae obtained at 37°C/1 Hz stimulation or 30°C/0.2 Hz stimulation were comparable. Furthermore, the SFR measured in SBFI- or BCECF-loaded muscle-strips at 30°C/0.2 Hz stimulation was almost identical to the SFR obtained at 37°C/1 Hz stimulation.
2.4. Immunoblotting of atrial myosin light chain 2
Immunoblotting of myosin light chain 2 (MLC2a) was conducted using antibodies directed against phosphorylated and total MLC2a as described previously.20,21 Briefly, at pre-defined time points, stretched and non-stretched muscle strips were shock frozen in liquid nitrogen, homogenized, and centrifuged at 14 000 g for 10 min at 4°C. The pellet was dissolved in Laemmli buffer and subjected to SDS–PAGE and immunodetection using standard immunoblot techniques. The protein-specific antibody (1Ab040; MLC2a, 1:500) was kindly provided by the CBI Antibody Core at the Center for Biomedical Inventions, University of Texas Southwestern Medical School. The phospho-specific antibody (MLC2a-P, 1:3000) was a custom-made antibody from Eurogentec (Seraign, Belgium). Immunoreactive bands were quantified by densitometry using GeneTools (Syngene, Cambridge, UK).
2.5. Drugs and statistics
AngII, atrial natriuretic peptide (ANP), PD123319 (selective AT2R antagonist), and streptomycin (unspecific inhibitor of SACs) were from Sigma. ML-7 (specific inhibitor of MLCK) and wortmannin (inhibitor of MLCK) were from Calbiochem and KB-R7943 (inhibitor of NCX) and saralasin (unselective AT-R antagonist) from Tocris. HOE642 (cariporide, specific inhibitor of NHE1) and irbesartan (selective AT1R antagonist) were generous gifts from Sanofi-Aventis. Grammostola spatulata toxin, GsMtx-4 (specific inhibitor of SACs), was prepared as described previously.22,23
Data are presented as mean ± SEM. Experimental data were evaluated by paired t-tests. Linear regression analysis was performed between SFR (dependent variable) and FSM (independent variable). The impact of patient characteristics on FSM and SFR was analysed by a multifactorial univariate analysis of variance. Since there was a significant relationship between FSM and SFR (Figure 1B), the FSM was additionally considered as a covariate.
Figure 1.

Twitch kinetics and correlation between slow force response (SFR) and Frank–Starling mechanism (FSM). (A) Individual isometric twitches of an atrial trabecula obtained at L88, during FSM and SFR following stretch to L98. (B) Linear regression between SFR and FSM (SFR = 0.18 × FSM + 86; P < 0.01; R2 = 0.31). Mean values from 101 patients.
3. Results
3.1. Acute stretch elicits a reproducible, biphasic increase in developed force in human atrium
Figure 2A illustrates original recordings of isometric contractions of an atrial trabeculae immediately before and following acute stretch from L88 to L98. Two consecutive stretch protocols from the same muscle strip are shown. Each stretch protocol resulted in a biphasic force increase: an immediate increase (first phase, FSM) followed by a slower additional increase (SFR). The magnitudes of FSM and SFR were very similar in the two stretch protocols: 150 and 154% of twitch force developed at L88 for the FSM and 117 and 119% of twitch force developed during the FSM for the SFR. Figure 2B summarizes the results from 11 trabeculae. The amplitudes of FSM and SFR were highly reproducible amounting to 183.5 ± 9.5 and 193.8 ± 11.0% (both P < 0.01 vs. force at L88, F88) for the FSM and to 126.8 ± 4.3 and 129.9 ± 5.5% (both P < 0.01 vs. FSM) for the SFR.
Figure 2.

Stretch elicits a reproducible biphasic force response in human atrium. (A) Original recordings of isometric twitch force of an atrial trabecula during two consecutive stretch protocols. In this and the following original recordings, dashed lines mark systolic and diastolic tension of the Frank–Starling mechanism (FSM) and muscle length is indicated below the force trace. (B) Average values (n = 11 trabeculae) of FSM and slow force response obtained during a paired stretch protocol. In this figure and in Figures 4–6, SP1 and SP2 indicate first and second stretch protocol, respectively.
Almost identical stretch-induced biphasic force responses were observed in atrial trabeculae from male (n = 68) and female (n = 33) patients. Neither FSM nor SFR correlated with the age or left ventricular ejection fraction. FSM and SFR were unaffected by drug pre-treatment (Table 1). Atrial fibrillation increased the FSM, whereas there was no effect of cardiovascular diseases on the SFR (Table 1). Overall, the stretch-elicited biphasic force increase was a common phenomenon in human atrium.
Table 1.
Patient characteristics
| Drug/disease |
n |
FSM |
SFR |
|||||
|---|---|---|---|---|---|---|---|---|
| + | − | + | − | P | + | − | P | |
| ACE-inhibitors | 59 | 42 | 178.7 ± 4.8 | 185.7 ± 5.4 | 0.63 | 117.2 ± 1.6 | 120.3 ± 1.7 | 0.73 |
| AT1R-antagonists | 12 | 89 | 189.6 ± 10.1 | 180.5 ± 3.9 | 0.33 | 116.3 ± 2.7 | 118.8 ± 1.3 | 0.53 |
| β-blockers | 68 | 33 | 181.6 ± 4.6 | 181.5 ± 5.8 | 0.88 | 116.7 ± 1.3 | 122.1 ± 2.1 | 0.51 |
| Ca2+-antagonists | 24 | 77 | 174.6 ± 7.4 | 183.8 ± 4.1 | 0.40 | 118.6 ± 2.9 | 118.4 ± 1.2 | 0.92 |
| Nitrates | 38 | 63 | 180.0 ± 5.1 | 182.5 ± 4.9 | 0.19 | 118.7 ± 1.8 | 118.3 ± 1.5 | 0.72 |
| Diuretics | 56 | 45 | 180.8 ± 5.0 | 182.6 ± 5.3 | 0.27 | 117.8 ± 1.5 | 119.4 ± 1.8 | 0.23 |
| Insulin | 16 | 85 | 176.1 ± 7.7 | 182.6 ± 4.0 | 0.40 | 120.0 ± 3.6 | 118.2 ± 1.2 | 0.27 |
| Diabetes mellitus | 20 | 81 | 185.5 ± 9.3 | 180.6 ± 3.9 | 0.04 | 122.6 ± 3.5 | 117.5 ± 1.1 | 0.89 |
| Coronary artery disease | 68 | 33 | 182.8 ± 4.2 | 179.0 ± 6.8 | 0.12 | 118.0 ± 1.3 | 119.5 ± 2.3 | 0.32 |
| Mitral valve disease | 5 | 96 | 187.1 ± 11.8 | 181.3 ± 3.7 | 0.41 | 122.9 ± 5.3 | 118.2 ± 1.2 | 0.80 |
| Atrial fibrillation | 6 | 95 | 214.3 ± 13.2 | 179.5 ± 3.6 | <0.01 | 123.7 ± 4.5 | 118.1 ± 1.2 | 0.09 |
Effects of drug pre-treatment and diseases on Frank–Starling mechanism and slow force response. FSM and SFR of atrial trabeculae from patients (n = 101) with/without diabetes (taking insulin or oral anti-diabetics), coronary artery disease (undergoing bypass surgery), mitral valve disease (undergoing valve replacement), or atrial fibrillation pre-treated with various drugs. n indicates number of patients, +/− indicates with/without drug or disease. P indicates the level of significance.
3.2. Twitch kinetics following stretch and correlation between Frank–Starling mechanism and slow force response
In Figure 1A, individual twitches of an atrial trabecula obtained at L88, during the FSM and the SFR are superimposed and aligned to diastolic tension. Mean values of the twitch kinetics from 20 trabeculae are shown in Table 2. During the FSM, twitch force increased and the twitch was prolonged. Diastolic tension, systolic tension, developed force, maximal rates of force development and relaxation, time-to-peak tension (TPT), and time from peak tension to 50% relaxation (RT50) were all increased. During the SFR, there was a decrease in diastolic tension and additional increases in systolic tension, developed force, and maximal rates of force development and relaxation. Furthermore, TPT was diminished whereas RT50 was prolonged, indicating that the relaxation was slowed down during the SFR.
Table 2.
Kinetic parameters of isometric twitches before and following stretch
| Twitch parameter | L88 | FSM | Slow force response |
|---|---|---|---|
| Developed force (mN/mm2) | 7.07 ± 1.36 | 12.16 ± 2.08** | 15.23 ± 2.31## |
| Developed force (%) | 100.0 ± 0.0 | 181.5 ± 7.1** | 242.5 ± 15.1## |
| Developed force (%) | 56.6 ± 2.0 | 100.0 ± 0.0** | 132.1 ± 4.3## |
| Diastolic tension (mN/mm2) | 0.85 ± 0.19 | 4.86 ± 0.75** | 3.57 ± 0.57## |
| Systolic tension (mN/mm2) | 7.92 ± 1.47 | 17.02 ± 2.61** | 18.80 ± 2.74## |
| dF/dt(max) (mN/mm2/s) | 102.9 ± 19.3 | 170.2 ± 29.2** | 223.8 ± 34.7## |
| dF/dt(min) (mN/mm2/s) | −68.7 ± 12.8 | −92.2 ± 13.9** | −110.7 ± 15.4## |
| Time-to-peak tension (ms) | 140.9 ± 6.7 | 152.8 ± 6.4* | 142.0 ± 5.3## |
| RT50 (ms) | 69.4 ± 4.0 | 80.7 ± 4.0* | 85.3 ± 4.6## |
dF/dt(max): maximal rate of force development; dF/dt(min): maximal rate of relaxation; RT50: time from peak tension to 50% relaxation. Means ± SEM from 20 trabeculae.
*P < 0.05 vs. L88.
**P < 0.01 vs. L88.
#P < 0.05 vs. FSM.
##P < 0.01 vs. FSM.
Figure 1B illustrates the SFR as a function of the FSM. Mean values of SFR and FSM from a total of 101 patients are shown. It is evident from these data that the larger the FSM the larger the SFR. There was a positive correlation between SFR and FSM.
3.3. Effects of stretch on [Na+]i and pHi
The effects of stretch on [Na+]i and pHi were studied in trabeculae loaded with SBFI or BCECF, respectively. Figure 3 illustrates average results. Stretching the muscle-strips from L88 to L98 caused a SFR to 120.8 ± 2.3% and a [Na+]i increase of 1.5 ± 0.7 mM (Figure 3A, n = 6). Comparison with trabeculae stretched in the presence of the NHE inhibitor HOE642 (3 µM; n = 5) revealed no significant differences in either SFR (119.4 ± 5.1%) or [Na+]i increase (1.0 ± 0.3 mM; both P = N.S.). In BCECF-loaded muscle strips (Figure 3B, n = 7), the SFR amounted to 120.9 ± 4.1% whereas pHi remained almost unchanged (−0.04 ± 0.03 pH units).
Figure 3.

Effects of stretch on [Na+]i and pHi. (A) Mean values of slow force response (SFR) and changes in [Na+]i in trabeculae stretched in the absence (n = 6) or presence (n = 5) of 3 µM HOE642. (B) Mean values of SFR and stretch-induced changes in pHi from seven atrial trabeculae.
3.4. Inhibition of NHE, NCX, and SACs does not reduce the force response to stretch
Stretch-induced stimulation of NHE and/or SACs followed by reverse-mode NCX underlies the SFR in ventricle. Therefore, we studied the possible involvement of these transporters and channels in the stretch-induced force response in human atrium. Figure 4A illustrates an original recording of an atrial trabecula stretched from L88 to L98 first in the absence and then in the presence of 5 µM KB-R7943, an inhibitor of reverse-mode NCX that largely reduces the SFR in human ventricle.14 KB-R7943 decreased F88 by 14%. It did not, however, reduce the FSM or the SFR, which amounted to 169 and 118% before and to 214 and 125% after exposure to KB-R7943, respectively. On average (Figure 4B, n = 8), KB-R7943 reduced F88 to 79.6 ± 7.7% of control (P < 0.05) and left both FSM and SFR unaffected (both P = N.S.). NHE inhibition with 3 µM HOE642 neither affected F88 nor FSM or SFR (Figure 4B, n = 6). Specific inhibition of cationic SACs by 500 nM GsMtx-4 (n = 8) or non-specific inhibition by 80 µM streptomycin (n = 6) caused a reduction of F88 by ∼10–20% (Figure 4C; both P < 0.05). Neither substance, however, reduced FSM or SFR (Figure 4C). The same was true when the concentration of GsMtx-4 was raised to 1 µM (n = 3, not shown).
Figure 4.

Inhibition of NHE, NCX, and SACs does not reduce the stretch-induced increases in twitch force. (A) Original recording of stretch-induced alterations in isometric twitch force of an atrial trabecula before and following exposure to 5 µM KB-R7943. (B) Effects of NHE and NCX inhibition using 3 µM HOE642 (n = 6) and 5 µM KB-R7943 (n = 8), respectively, on F88, Frank–Starling mechanism (FSM), and slow force response (SFR). (C) Effects of SAC inhibition using 500 nM GsMtx-4 (n = 8) or 80 µM streptomycin (n = 6) on F88, FSM, and SFR. * and **P < 0.05 and 0.01 vs. control.
3.5. Saralasin and AngII reduce the SFR
In response to stretch cardiac myocytes release AngII.24 AT-R antagonists can suppress the SFR8 and autocrine/paracrine actions of AngII have been implicated in the SFR in ventricular myocardium of some species.8,9 Therefore, we tested whether the AT-R antagonist saralasin was able to reduce the SFR in human atrium. Figure 5A illustrates an original recording of an atrial trabecula stretched from L88 to L98 before, during, and after exposure to 5 µM saralasin. Saralasin did not affect F88 or FSM. However, it reversibly attenuated the SFR, which amounted to 128, 116, and 129%, respectively. On average (Figure 5B, n = 9), saralasin left F88 and the FSM unchanged and significantly reduced the SFR from 125.1 ± 5.7 to 110.9 ± 2.4% (P < 0.05) or by 57%. AngII (0.5 µM, Figure 5B, n = 8), which by itself augmented F88 to 246.7 ± 45.9% of control (P < 0.05), exerted similar effects on the stretch response, i.e. it left the FSM unaffected and significantly reduced the SFR from 120.1 ± 2.9 to 112.8 ± 3.5% (P < 0.05) or by 36%. Stretch may also directly release ANP. Therefore, we also studied the effects of ANP on the force response in human atrium. We reasoned that, if ANP was involved in the stretch response, the stretch-induced changes in force would be blunted in the presence of ANP, as observed for AngII. ANP (20 nM, Figure 5B, n = 6) reduced F88 to 86.8 ± 5.0% of control (P < 0.05) and slightly increased the FSM from 214.6 ± 13.0 to 238.4 ± 15.3% (P < 0.01). The SFR, however, was unchanged (Ctrl: 122.8 ± 3.0 vs. ANP: 133.7 ± 6.3%, P = N.S.).
Figure 5.
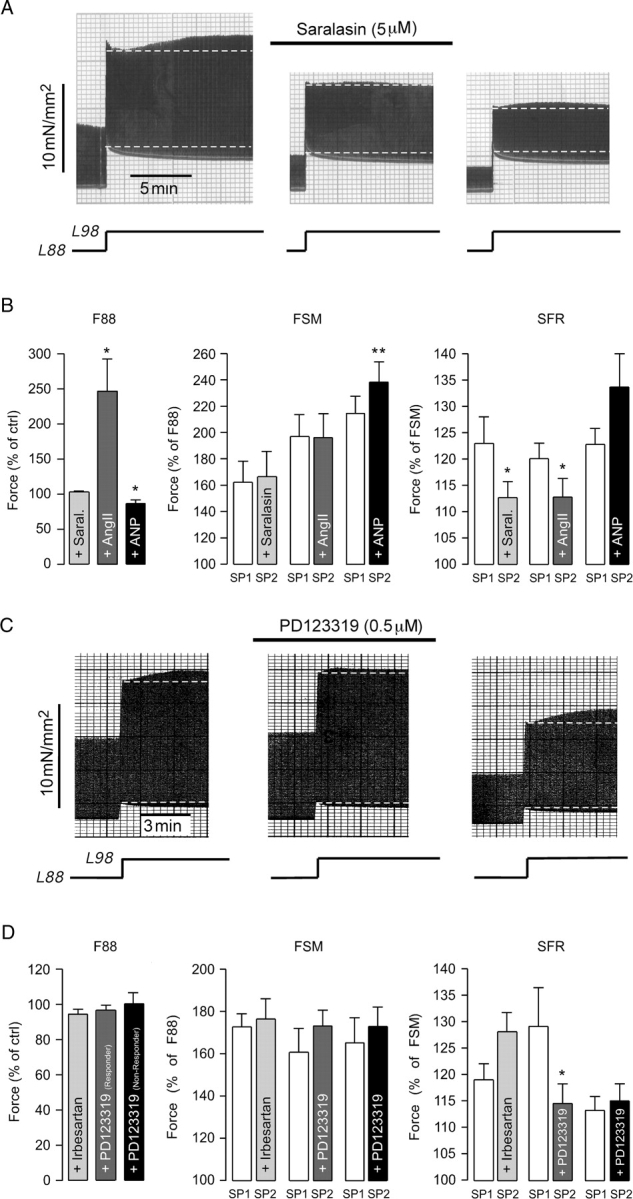
Effects of AT-R blockade, AngII, and ANP on the stretch-induced increases in twitch force. (A) Original recording of stretch-induced alterations in isometric twitch force of an atrial trabecula before, during, and following washout of 5 µM saralasin. (B) Effects of 5 µM saralasin (n = 9), 0.5 µM AngII (n = 8), and 20 nM ANP (n = 6) on F88, Frank–Starling mechanism (FSM), and slow force response (SFR). (C) Original recording of stretch-induced alterations in isometric twitch force of an atrial trabecula before, during, and following washout of 0.5 µM PD123319. Muscle length is indicated below the force trace. (D) Effects of 0.1 µM irbesartan (n = 8) and 0.5 µM PD123319 on F88, FSM, and SFR. PD123319-treated trabeculae were divided into responding and non-responding trabeculae (n = 6 each). * and **P < 0.05 and 0.01 vs. control.
To determine the AT-R subtype involved in the SFR, experiments were performed with 0.1 µM irbesartan, an AT1R antagonist, or 0.5 µM PD123319, an AT2R antagonist. Neither irbesartan nor PD123319 altered F88 or the FSM (Figure 5D). Irbesartan did not affect the SFR (Figure 5D). PD123319, however, reduced the SFR, as shown in the original recording in Figure 5C. The SFR in this trabecula was reduced from 112% under control conditions to 105% in the presence of 0.5 µM PD123319. This effect was completely reversible. Upon washout of PD123319, the SFR recovered to 120%. The inhibitory effect of PD123319, however, was only observed in 50% (6/12) of the trabeculae studied. In these ‘responding’ trabeculae, PD123319 reduced the SFR from 129.2 ± 7.3 to 114.6 ± 3.7% (P < 0.05) or by 50% (Figure 5D, medium grey). By contrast, in the other 50% of trabeculae, the ‘non-responding’ trabeculae, PD123319 did not affect the SFR (Figure 5D, black). Responding and non-responding trabeculae did not differ with respect to the (lack of) effect on F88 and FSM. The control SFR amounted to 129.2 ± 7.3% in responding and to 113.3 ± 2.6% in non-responding trabeculae (n = 6 each, P = 0.07).
3.6. Stretch stimulates MLCK to phosphorylate MLC2a
The results presented above suggested that autocrine/paracrine actions of AngII might underlie the SFR in human atrium. AngII activates Gq-coupled receptors. Recently, some Gq-coupled receptors have been shown to increase force via MLCK-mediated increases in myofilament Ca2+-sensitivity.20 We, therefore, tested whether the SFR in human atrium was caused by an increase in myofilament Ca2+-sensitivity via stimulation of MLCK. Figure 6 illustrates an original recording (A) as well as average results (C) obtained with the MLCK inhibitor ML-7. ML-7 (10 µM) abolished the SFR completely, an effect that was fully reversible. Before, during, and after ML-7, the SFR was 114, <100, and 113%, respectively. On average (Figure 6C), ML-7 reduced F88 to 82.0 ± 3.2% of control (n = 8, P < 0.01) and the SFR from 127.3 ± 5.4 to 117.2 ± 4.0% (n = 8, P < 0.05) or by 37% without affecting the FSM. Similarly, wortmannin (5 µM), a different MLCK inhibitor, also reduced F88 (to 54.8 ± 10.5% of control, n = 5, P < 0.05) and the SFR (from 120.3 ± 2.9 to 103.1 ± 1.0% or by 85%, n = 5, P< 0.01) without affecting the FSM (Figure 6C, n = 5).
Figure 6.

Involvement of MLCK in stretch-dependent slow force response (SFR). (A) Original recording of stretch-induced alterations in isometric twitch force of an atrial trabecula before, during, and following washout of 10 µM ML-7. (B) Effects of 10 µM ML-7 on twitch force and [Ca2+]i-transient in an atrial trabecula. (C) Effects of 10 µM ML-7 (n = 8) and 5 µM wortmannin (n = 5) on F88, Frank–Starling mechanism, and SFR. * and **P < 0.05/0.01 vs. control. (D) Immunoblots of phosphorylated (MLC2a-P) and total (MLC2a) myosin light chain 2 from 12 atrial trabeculae. Trabeculae were stretched for 10 min (+) or left at slack length (−). The ratio of MLC2a-P to MLC2a is given below the immunoblots. (E) Mean ratio of MLC2a-P to MLC2a in stretched and non-stretched muscle strips (n = 18 each; **P < 0.01).
Reduction of basal force by ML-7 was accompanied by a reduction of the [Ca2+]i-transient (Figure 6B). The decrease in [Ca2+]i-transient amplitude, however, was smaller than the decrease in force. In six trabeculae, ML-7 reduced basal force by 12.3 ± 2.5%, but [Ca2+]i-transient amplitude only by 6.1 ± 1.5% (P < 0.05 vs. force). Thus, ML-7 reduced force by a Ca2+-dependent and a Ca2+-independent mechanism consistent with inhibition of MLCK and reduction in myofilament Ca2+-sensitivity.
Taken together, these results suggest that MLCK activity contributes to basal force development and that stretch-dependent stimulation of MLCK is involved in the SFR. We tested this hypothesis further by determining phosphorylation of MLC2a, the major substrate of MLCK in human atrium, in stretched and non-stretched trabeculae. To this end, isometrically contracting atrial trabeculae were shock frozen either at slack length (no stretch) or following acute stretch for 10 min to muscle lengths corresponding to ∼L98 (stretch). Figure 6D shows immunoblots of 12 trabeculae, six shock frozen after 10 min of stretch (+) and six at slack length (−). Average results are presented in Figure 6E. Phosphorylation of MLC2a was elevated by stretch. The ratio of MLC2a-P to MLC2a increased from 0.56 ± 0.09 in non-stretched (n = 18) to 0.93 ± 0.10 in stretched muscle strips (n = 18; P < 0.01).
4. Discussion
The major findings of this study are: (i) the stretch-induced biphasic force response observed in human ventricle is present in human atrium. It is a common phenomenon unaffected by a number of cardiovascular diseases and drugs. (ii) In contrast to ventricle, the SFR in atrium is not mediated by SACs, NHE, and/or NCX. Rather, (iii) the atrial SFR is mediated, in part, by a pHi-independent mechanism involving autocrine/paracrine actions of AngII as well as increased myofilament Ca2+-sensitivity caused by stretch-induced stimulation of MLCK activity and subsequent phosphorylation of MLC2a. Thus, the present study identifies MLCK as an important novel target of the functional effects of stretch in human atrium.
4.1. Similarities and differences in stretch responses between atrium and ventricle
When stretched from L88 to L98, both atrium and ventricle exhibit a FSM of roughly 200% (this study14). Furthermore, both tissues are characterized by a SFR with a similar time course (∼5–10 min until maximum) and magnitude (120–125% of the FSM). pHi remained unchanged in physiological bicarbonate-buffered solution after stretch in both atrium and ventricle. The signalling pathways and cellular mechanisms underlying the SFR, however, appear to differ. There is ample evidence that in ventricle, the SFR is mediated in large part by stretch-dependent stimulation of non-selective cationic SACs and/or NHE, an increase in [Na+]I, and a subsequent increase in intracellular Ca2+ through reverse-mode NCX. Based on the results of the current study, however, it is concluded that these mechanisms do not play a prominent role in human atrium since selective inhibition of SACs (GsMtx-4), NHE (HOE642), or NCX (KB-R7943) did not affect the SFR. In line with these findings, stretch elicited large increases in [Na+]i of 3–6 mM in ventricle,8,15 but only significantly smaller increases in [Na+]i of ∼1.5 mM in human atrium. Thus, different mechanisms must account for the major part of the SFR in human atrium.
4.2. Autocrine/paracrine actions of AngII are involved in the slow force response in human atrium
Saralasin, PD123319, and AngII, but not ANP, reduced the SFR in human atrium. This argues against stretch-dependent ANP release as a mediator of the SFR in atrium and in favour of an involvement of autocrine/paracrine release and actions of AngII, as suggested for the SFR in rat, ferret, and cat ventricle.8–10 Although in human ventricle such a mechanism was excluded to underlie the SFR,14 it is nevertheless conceivable that it is operating in atrium. In this regard, it is noteworthy that there are distinct differences in the actions of AngII between human atrium and ventricle. In particular, AngII elicits a positive inotropic effect in human atrium but not ventricle.25 In fact, since both AngII and AT-R blockade by saralasin (AT1/2R) or PD123319 (AT2R) reduced the SFR, our results strongly suggest that stretch-dependent release of AngII followed by activation of AT-Rs contributes to the SFR in human atrium. Using the subtype-selective antagonists irbesartan (AT1R) and PD123319 (AT2R), the AT2R was identified as the main mediator of AngII action.
4.3. Stretch-dependent stimulation of MLCK contributes to the slow force response in human atrium
In human atrium, MLCK-dependent MLC2 phosphorylation increases force by increasing myofilament Ca2+-sensitivity.26 Furthermore, the positive inotropic effect of Gq-coupled receptors in human atrium is mediated in large part through stimulation of MLCK, subsequent phosphorylation of MLC2a, and increased myofilament Ca2+-sensitivity.20 According to our results, both functional, pharmacological, and immunoblot data indicated that stretch stimulated MLCK to phosphorylate MLC2a and, thereby, increased force development. Increased myofilament Ca2+-sensitivity should prolong relaxation. In line with this, RT50 was increased during the SFR when compared with the FSM. Thus, the present study has identified a novel mechanism underlying the SFR in human myocardium. The stretch-dependent increases in MLCK activity and MLC2a phosphorylation found in vitro are consistent with the observation that in vivo exercise-induced elevations in haemodynamic parameters are associated with increased MLC2 phosphorylation.27 Presumably, stretch-mediated stimulation of MLCK is not only of immediate functional relevance for force development under conditions of elevated mechanical load, it might also have important implications for the long-term effects of stretch on atrial structure. MLCK mediates sarcomere organization in response to hypertrophic stimuli such as AngII or endothelin.28 Reduction of the MLC2 phosphorylation level or mutations in MLC2 underlie some forms of hypertrophic cardiomyopathy.26 Furthermore, when MLC phosphorylation is prevented, the heart develops structural and functional abnormalities including ultrastructural changes of the myocytes and atrial hypertrophy and dilatation.29 Therefore, stretch-dependent stimulation of MLCK may serve a dual purpose in that it mediates the increase in force development necessary to maintain atrial function as short-term adaptive response and, in addition, that it contributes to structural changes in the atria during hypertrophy as long-term adaptive response to mechanical overload.
4.4. Limitations of the study
A possible limitation of the study is the development of core hypoxia in the atrial trabeculae. Careful experiments on rat trabeculae have shown that the critical diameter at which preparations develop core hypoxia is ∼0.2 mm.30 Trabeculae in this study had larger diameters (0.51 ± 0.01 mm, n = 252). Although we have no indication for functional impairment of the trabeculae (e.g. positive force–frequency relationship), we cannot exclude the possibility of core hypoxia. Nevertheless, this is unlikely to affect the general findings and conclusions of this study.
Conflict of interest: none declared.
Funding
The authors are grateful for financial support from the Deutsche Forschungsgemeinschaft (DFG PI414/1-3 and KFO155, TP6, to B.P. and J.K.), the Bundesministerium für Bildung und Forschung (BMBF, KNHI, TP8, to T.E. and B.P.), and the National Institutes of Health and the Oshei Foundation (F.S. and P.A.G.).
References
- 1.Cooper PJ, Lei M, Cheng LX, Kohl P. Selected contribution: axial stretch increases spontaneous pacemaker activity in rabbit isolated sinoatrial node cells. J Appl Physiol. 2000;89:2099–2104. doi: 10.1152/jappl.2000.89.5.2099. [DOI] [PubMed] [Google Scholar]
- 2.Tavi P, Han C, Weckstrom M. Mechanisms of stretch-induced changes in [Ca2+]i in rat atrial myocytes: role of increased troponin C affinity and stretch-activated ion channels. Circ Res. 1998;83:1165–1177. doi: 10.1161/01.res.83.11.1165. [DOI] [PubMed] [Google Scholar]
- 3.Schoonderwoerd BA, Van Gelder IC, Van Veldhuisen DJ, Van den Berg MP, Crijns HJ. Electrical and structural remodeling: role in the genesis and maintenance of atrial fibrillation. Prog Cardiovasc Dis. 2005;48:153–168. doi: 10.1016/j.pcad.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 4.Schotten U, Neuberger HR, Allessie MA. The role of atrial dilatation in the domestication of atrial fibrillation. Prog Biophys Mol Biol. 2003;82:151–162. doi: 10.1016/s0079-6107(03)00012-9. [DOI] [PubMed] [Google Scholar]
- 5.Parmley WW, Chuck L. Length-dependent changes in myocardial contractile state. Am J Physiol. 1973;224:1195–1199. doi: 10.1152/ajplegacy.1973.224.5.1195. [DOI] [PubMed] [Google Scholar]
- 6.Allen DG, Kurihara S. The effects of muscle length on intracellular calcium transients in mammalian cardiac muscle. J Physiol. 1982;327:79–94. doi: 10.1113/jphysiol.1982.sp014221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kentish JC, Wrzosek A. Changes in force and cytosolic Ca2+ concentration after length changes in isolated rat ventricular trabeculae. J Physiol. 1998;506:431–444. doi: 10.1111/j.1469-7793.1998.431bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alvarez BV, Perez NG, Ennis IL, Camilion de Hurtado MC, Cingolani HE. Mechanisms underlying the increase in force and Ca2+ transient that follow stretch of cardiac muscle: a possible explanation of the Anrep effect. Circ Res. 1999;85:716–722. doi: 10.1161/01.res.85.8.716. [DOI] [PubMed] [Google Scholar]
- 9.Calaghan SC, White E. Contribution of angiotensin-II, endothelin-1 and the endothelium to the slow inotropic response to stretch in ferret papillary muscle. Pflugers Arch. 2001;441:514–520. doi: 10.1007/s004240000458. [DOI] [PubMed] [Google Scholar]
- 10.Perez NG, de Hurtado MC, Cingolani HE. Reverse mode of the Na+–Ca2+ exchange after myocardial stretch: underlying mechanism of the slow force response. Circ Res. 2001;88:376–382. doi: 10.1161/01.res.88.4.376. [DOI] [PubMed] [Google Scholar]
- 11.Cingolani HE, Perez NG, Pieske B, von Lewinski D, Camilion de Hurtado MC. Stretch-elicited Na+/H+ exchanger activation: the autocrine/paracrine loop and its mechanical counterpart. Cardiovasc Res. 2003;57:953–960. doi: 10.1016/s0008-6363(02)00768-x. [DOI] [PubMed] [Google Scholar]
- 12.von Lewinski D, Stumme B, Maier LS, Luers C, Bers DM, Pieske B. Stretch-dependent slow force response in isolated rabbit myocardium is Na+ dependent. Cardiovasc Res. 2003;57:1052–1061. doi: 10.1016/s0008-6363(02)00830-1. [DOI] [PubMed] [Google Scholar]
- 13.Calaghan S, White E. Activation of Na+–H+ exchange and stretch-activated channels underlies the slow inotropic response to stretch in myocytes and muscle from the rat heart. J Physiol. 2004;559:205–214. doi: 10.1113/jphysiol.2004.069021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.von Lewinski D, Stumme B, Fialka F, Luers C, Pieske B. Functional relevance of the stretch-dependent slow force response in failing human myocardium. Circ Res. 2004;94:1392–1398. doi: 10.1161/01.RES.0000129181.48395.ff. [DOI] [PubMed] [Google Scholar]
- 15.Luers C, Fialka F, Elgner A, Zhu D, Kockskamper J, von Lewinski D, et al. Stretch-dependent modulation of [Na+]i, [Ca2+]i, and pHi in rabbit myocardium—a mechanism for the slow force response. Cardiovasc Res. 2005;68:454–463. doi: 10.1016/j.cardiores.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 16.Cingolani HE, Alvarez BV, Ennis IL, Camilion de Hurtado MC. Stretch-induced alkalinization of feline papillary muscle: an autocrine–paracrine system. Circ Res. 1998;83:775–780. doi: 10.1161/01.res.83.8.775. [DOI] [PubMed] [Google Scholar]
- 17.Tavi P, Han C, Weckstrom M. Intracellular acidosis modulates the stretch-induced changes in E–C coupling of the rat atrium. Acta Physiol Scand. 1999;167:203–213. doi: 10.1046/j.1365-201x.1999.00615.x. [DOI] [PubMed] [Google Scholar]
- 18.Tavi P, Weckstrom M, Ruskoaho H. cAMP- and cGMP-independent stretch-induced changes in the contraction of rat atrium. Pflugers Arch. 2000;441:65–68. doi: 10.1007/s004240000403. [DOI] [PubMed] [Google Scholar]
- 19.Meyer M, Lehnart S, Pieske B, Schlotthauer K, Munk S, Holubarsch C, et al. Influence of endothelin 1 on human atrial myocardium–myocardial function and subcellular pathways. Basic Res Cardiol. 1996;91:86–93. doi: 10.1007/BF00788869. [DOI] [PubMed] [Google Scholar]
- 20.Grimm M, Haas P, Willipinski-Stapelfeldt B, Zimmermann WH, Rau T, Pantel K, et al. Key role of myosin light chain (MLC) kinase-mediated MLC2a phosphorylation in the alpha 1-adrenergic positive inotropic effect in human atrium. Cardiovasc Res. 2005;65:211–220. doi: 10.1016/j.cardiores.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 21.Grimm M, Mahnecke N, Soja F, El-Armouche A, Haas P, Treede H, et al. The MLCK-mediated alpha1-adrenergic inotropic effect in atrial myocardium is negatively modulated by PKCepsilon signaling. Br J Pharmacol. 2006;148:991–1000. doi: 10.1038/sj.bjp.0706803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oswald RE, Suchyna TM, McFeeters R, Gottlieb P, Sachs F. Solution structure of peptide toxins that block mechanosensitive ion channels. J Biol Chem. 2002;277:34443–34450. doi: 10.1074/jbc.M202715200. [DOI] [PubMed] [Google Scholar]
- 23.Ostrow KL, Mammoser A, Suchyna T, Sachs F, Oswald R, Kubo S, et al. cDNA sequence and in vitro folding of GsMTx4, a specific peptide inhibitor of mechanosensitive channels. Toxicon. 2003;42:263–274. doi: 10.1016/s0041-0101(03)00141-7. [DOI] [PubMed] [Google Scholar]
- 24.Sadoshima J, Xu Y, Slayter HS, Izumo S. Autocrine release of angiotensin-II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell. 1993;75:977–984. doi: 10.1016/0092-8674(93)90541-w. [DOI] [PubMed] [Google Scholar]
- 25.Holubarsch C, Hasenfuss G, Schmidt-Schweda S, Knorr A, Pieske B, Ruf T, et al. Angiotensin I and II exert inotropic effects in atrial but not in ventricular human myocardium. An in vitro study under physiological experimental conditions. Circulation. 1993;88:1228–1237. doi: 10.1161/01.cir.88.3.1228. [DOI] [PubMed] [Google Scholar]
- 26.Morano I. Tuning the human heart molecular motors by myosin light chains. J Mol Med. 1999;77:544–555. doi: 10.1007/s001099900031. [DOI] [PubMed] [Google Scholar]
- 27.Fitzsimons DP, Bodell PW, Baldwin KM. Myocardial functional correlates of cardiac myosin light chain 2 phosphorylation. J Appl Physiol. 1990;68:2426–2433. doi: 10.1152/jappl.1990.68.6.2426. [DOI] [PubMed] [Google Scholar]
- 28.Aoki H, Sadoshima J, Izumo S. Myosin light chain kinase mediates sarcomere organization during cardiac hypertrophy in vitro. Nat Med. 2000;6:183–188. doi: 10.1038/72287. [DOI] [PubMed] [Google Scholar]
- 29.Sanbe A, Fewell JG, Gulick J, Osinska H, Lorenz J, Hall DG, et al. Abnormal cardiac structure and function in mice expressing nonphosphorylatable cardiac regulatory myosin light chain 2. J Biol Chem. 1999;274:21085–21094. doi: 10.1074/jbc.274.30.21085. [DOI] [PubMed] [Google Scholar]
- 30.Schouten VJA, ter Keurs HEDJ. The force-frequency relationship in rat myocardium. Pflugers Arch. 1986;407:14–17. doi: 10.1007/BF00580714. [DOI] [PubMed] [Google Scholar]