

Abstract
The natural product Gambogic acid (GA) has been reported to have cytotoxic activity against tumor cells in culture, and was identified as an active compound in a cell-based high-throughput screening (HTS) assay for activators of caspases, proteases involved in apoptosis. Using the anti-apoptotic Bcl-2-family protein, Bfl-1, as a target for screening of a library of natural products, we identified GA as a competitive inhibitor that displaced BH3 peptides from Bfl-1 in a fluorescent polarization assay (FPA). Analysis of competition for BH3 peptide binding revealed that GA inhibits all 6 human Bcl-2-family proteins to various extents, with Mcl-1 and Bcl-B the most potently inhibited (concentrations required for 50% inhibition [IC50] <1 μM). Competition for BH3 peptide binding was also confirmed using a time-resolved fluorescence resonance energy transfer (TR-FRET) assay. GA functionally inhibited the anti-apoptotic Bcl-2-family proteins, as demonstrated by experiments using isolated mitochondria in which recombinant purified Bcl-2-family proteins suppress SMAC release in vitro, showing that GA neutralizes their suppressive effects on mitochondria in a concentration-dependent manner. GA killed tumor cell lines via an apoptotic mechanism, whereas analogs of GA with greatly reduced potency at BH3 peptide displacement showed little or no cytotoxic activity. However, GA retained cytotoxic activity against bax−/− bak−/− cells in which anti-apoptotic Bcl-2-family proteins lack a cytoprotective phenotype, implying that GA also has additional targets that contribute to its cytotoxic mechanism. Altogether, the findings suggest that suppression of anti-apoptotic Bcl-2-family proteins may be among the cytotoxic mechanisms by which GA kills tumor cells.
Keywords: Gambogic acid, natural product, Bcl-2, apoptosis
Introduction
Gambogic acid (GA) is a medicinal compound derived from the gamboges resin of the tree, Garcinia hanburyi GA has documented cytotoxic activity against tumor cell lines in culture, with concentrations required for killing 50% of cells (Lethal Dose 50% [LD50]) of ~ 1 μM (1, 2). This natural product also displays anti-tumor activity in preclinical mouse models involving human tumor xenografts (3–5). In contrast, GA is reportedly well tolerated in mice and rats (2, 4, 6), suggesting that a therapeutic window might be identified at which tumor but not normal cells are killed. It would therefore be interesting to know the cytotoxic mechanism of GA.
The mechanism by which GA kills tumor cells lines involves apoptosis, a cell death processing involving caspase-family proteases. In fact, GA was identified as an active compound in a cell-based high-throughput screening (HTS) assay that measured caspase activation (1). Among the regulators of apoptosis are Bcl-2-family proteins. Humans have 6 genes encoding distinct anti-apoptotic Bcl-2-family proteins: Bcl-2, Bcl-XL, Mcl-1, Bfl-1, Bcl-W, and Bcl-B (7, 8). These proteins typically localize to intracellular membranes, especially mitochondrial membranes, where they have been shown to block the release of apoptogenic proteins such as cytochrome c, SMAC Endonuclease G, and AIF (9–11). Several anti-apoptotic Bcl-2-family proteins are known to become pathologically over-expressed in human cancers, conferring apoptosis-resistant phenotypes (12–17).
The anti-apoptotic proteins are neutralized endogenously by proteins containing an α-helical interaction motif, known as BH3 (7, 18–20). Synthetic BH3 peptides bind anti-apoptotic Bcl-2-family proteins with nanomolar affinities, promoting apoptosis (21, 22). Non-peptidyl compounds have been identified that compete with BH3 peptides for binding to anti-apoptotic Bcl-2-family proteins, mimicking BH3 peptides and creating interest in development of these molecules as potential cancer therapeutics (23, 24).
We show here that GA has the ability to compete with BH3 peptides for binding to several anti-apoptotic Bcl-2-family proteins in vitro. GA also neutralizes the function of these proteins with respect to release of SMAC from isolated mitochondria. The cytotoxic activity of GA however appears to include Bcl-2-family-independent mechanisms, suggesting that suppression of Bcl-2 and related proteins represents only one of the cytotoxic mechanisms of this natural product.
Materials and Methods
Compounds
The MicroSource Spectrum Collection Library (Discovery Systems, Inc., Gaylordsville, CT) is a ~2000 compound collection of mostly pure natural products and their derivatives. Compounds were supplied as 10 mM stocks of Me2SO, stored at −20 °C and thawed immediately before analysis. Aliquots were dissolved to a final concentration of 10 μM for fluorescence polarization assays. Gambogic acid was purchased from Calbiochem. All the gambogic acid analogs were purchased from MicroSource.
Protein Purification
GST-fusion proteins containing Bcl-XL, Bcl-2, Bcl-W, Bcl-B, Bfl-1 and Mcl-1 lacking their C-terminal transmembrane domains (~ last 20 amino-acids) (“ΔTM”) were expressed from pGEX 4T-1 plasmid in XL-1 Blue cells (Stratagene, Inc.) as described previously (24). Briefly, cells were grown in 2 L of LB with 50 μg/mL ampicillin at 37 °C to an OD600 nm of 1.0., then IPTG (0.5 mM) was added, and the cultures were incubated at 25 °C for 6 h. Cells were then recovered in 20 mM phosphate buffer (pH 7.4), 150 mM NaCl, 1 mM DTT, 1 mM EDTA, 1 mM PMSF, followed by sonication. Cellular debris were sedimented by centrifugation at 27,500 g for 20 min, and the resulting supernatants were incubated with 10 mL of glutathionine-Sepharose (Pharmacia) at 4°C for 2 h. The resin was washed 3 times with 20 mM phosphate buffer (pH 7.4), 150 mM NaCl, and 1 mM DTT, and then 10 mM of reduced glutathione dissolved in 50 mM Tris-HCl (pH 8.0) was used to elute the GST-fusion proteins.
Other recombinant proteins used here, including His6-Bid and His6-caspase 8, were expressed and purified using methods similar to prior publications (25).
Fluorescence Polarization Assays (FPAs)
FPAs were performed as described previously using various Bcl-2-family proteins and fluorescein isothiocyanate (FITC)-conjugated Bid BH3 peptide (24, 26). Briefly, Bcl-2 proteins were incubated with 5 nM of FITC-Ahx-EDIIRNIARHLAQVGDSMDR in the dark. Fluorescence polarization was measured using an Analyst TM AD Assay Detection System (LJL Biosystem, Sunnyvale, CA) in phosphate-buffered saline (PBS) [pH 7.4]. IC50 determinations were performed using GraphPad Prism software (GraphPad, Inc., San Diego, CA).
Competitive Peptide Displacement Assays
Methods for competitive peptide displacement assays were similar to previous publications (24). Briefly, 100 nM of GST-Bcl-2 proteins were incubated with the compounds at various concentrations for 5 min at room temperature in PBS. Then 5 nM of FITC-Bid BH3 peptide was added and fluorescence polarization was measured after 10 min. IC50 determinations were generated by fitting the experimental data using a sigmoidal dose-response nonlinear regression model with GraphPad Prism software (GraphPad, Inc., San Diego, CA).
Time-Resolved-Fluorescence Resonance Energy Transfer (TR-FRET) Assays
For TR-FRET assays, GST-Bcl-XL and anti-GST-terbium (Invitrogen) were mixed together with the FITC-Bad BH3 peptide in PBS containing 0.005% tween 20 in 96 well plates in a total volume of 20 μl per well. After incubation at room temperature for 30 min, 2 μl of gambogic acid-containing solutions were added to the reaction mixtures containing 10 nM of Bcl-XL, 10 nM of FITC-Bad BH3 peptide and 2 nM of anti-GST-terbium for 30 min at room temperature. TR-FRET signals were measured with a SpectraMax M5 plate reader (Molecular Devices) using the following settings: excitation at 330nm, emission for FITC signal at 490 nm, and emission for terbium signal at 520 nm.
Mitochondria Purification and Protein Release Assays
HeLa cells were pelleted by centrifugation, and then washed once in HM buffer (10 mM HEPES, pH 7.4, 250 mM mannitol, 10 mM KCl, 5 mM MgCl2, 1 mM EGTA), containing 1 mM PMSF and a mixture of protease inhibitors (Roche Molecular Biochemicals). The cell pellet was then homogenized in HM buffer by 50 strokes of a dounce homogenizer, using a B-type pestle. The homogenate was centrifuged twice at 600g for 5 min to remove nuclei and debris. The resulting supernatant was centrifuged at 10,000g for 10 min, and the resulting mitochondria-containing pellet was washed twice with the HM buffer.
For mitochondrial protein release assays, 10 μl of mitochondria (50 μg) were added into a final volume of 50 μl HM buffer containing gambogic acid, tBid or tBid pre-incubated with gambogic acid or Bcl-2 family proteins at 30°C for 15 min. The reactions were further incubated at 30°C for 40–60 min, then mitochondria were pelleted by centrifugation and the supernatants were collected, boiled in Laemmli sample buffer, and analyzed by SDS-PAGE/immunoblotting using anti-SMAC antibody (25).
Peptide Synthesis
Peptides were synthesized using Fmoc solid-phase synthesis on an ACT 350 multiple peptide synthesizer. FITC conjugated-Bid were synthesized on Fmoc-Alanine Wang resin to give peptides with C-terminal carboxyl groups. FITC was linked to the Ahx. The crude peptides were purified with a Gilson HPLC instrument and analyzed by MALDI-TOF mass analysis with an Applied Biosystems Voyager System 6264.
Cell Culture, Transfection, and Apoptosis Assays
HeLa, HL60, Jurkat, PPC1 and MEF cells were maintained in Dulbecco’s modified Eagle’s medium (Irvine Scientific) supplemented with 10% fetal bovine serum (FBS), 1 mM L-glutamine, and antibiotics. Apoptosis was assessed using staining with Annexin V-FITC and propidium iodide (PI), followed by flow-cytometry analysis using FL-1 and FL-3 channels of a flow cytometer (Becton Dickinson; FACSort; San Jose, CA). Annexin V-positive/PI-negative cells were considered apoptotic.
For caspase assays, cell lysates were prepared, normalized for protein content, and 10 μg aliquots of cell lysates were incubated with 100 μM DEVD-AFC, measuring enzyme activity by the release of AFC-fluorescence. Data are reported as relative fluorescence units (RFU) of product produced per min per μg of total protein.
Results
Identification of GA as a Bfl-1-inhibitory compound by HTS
We devised a HTS in which binding of a FITC-conjugated BH3 peptide to recombinant purified Bfl-1 protein is measured by FPA. A library of ~2000 natural products was screened for suppression of BH3 peptide binding by ≥ 50% using this FPA, resulting in ~30 hits, among which was gambogic acid (GA).
GA competes with BH3 peptides for binding to the 6 anti-apoptotic Bcl-2-family proteins
The activity of GA against the 6 human anti-apoptotic Bcl-2-family proteins was contrasted, using FPAs we previously established (24). GA displaced to various extents FITC-BH3 peptide binding to all 6 proteins, with apparent IC50 1.47 μM for Bcl-XL, 1.21 μM for Bcl-2, 2.02 μM for Bcl-W, 0.66 μM for Bcl-B, 1.06 μM for Bfl-1, and 0.79 μM for Mcl-1. Thus, Mcl-1 and Bcl-B showing the greatest sensitivity (apparent IC50 < 1 μM) and Bcl-2 the least (Figure 1). Due to solubility, we were unable to increase the GA concentration to achieve complete BH3 peptide displacement for some Bcl-2 family proteins in these assays. Incubating GA with FITC-BH3 peptide in the absence of Bcl-2-family proteins only marginally affected baseline (background) fluorescence polarization (~Δ20 mP at concentrations ≥ 20 μM), excluding a direct effect of GA on the peptide probe as an explanation for the results (Figure 1). In contrast to Bcl-2-family proteins, GA did not inhibit in FPAs using rhodamine-SMAC peptide binding to BIR3 of XIAP (not shown) or FITC-ATP binding to Hsp70 (not shown), thus demonstrating the specificity for anti-apoptotic Bcl-2-family proteins.
Figure 1. Gambogic acid inhibits binding of FITC-BH3 peptide to anti-apoptotic Bcl-2 family proteins.

A, Structure of gambogic acid. B–D, For peptide competition experiments in FPA mode, 100 nM of Bcl-2 family proteins, including Bcl-XL, Bcl-2, Bcl-W, Bcl-B, Bfl-1 or Mcl-1, were incubated with various concentrations of gambogic acid (GA) for 2 min in PBS buffer, then 5 nM FITC-conjugated-Bid BH3 peptide was added. E, GA does not interfere with FITC-Bid BH3 peptide alone, tested as a control. Fluorescence polarization (milli-Polars [mP]) was measured after 10 min. Interaction of gambogic acid with FITC-conjugated Bid BH3 peptide alone was used as a control. Note that background FP for these assays from FITC-BH3 peptide in the absence of Bcl-2 family protein is ~50–70 mP (FP min), while the maximum FP ranges from ~130 to ~190 mP (FP max), depending on the Bcl-2-family member.
TR-FRET confirmation of GA competition for BH3-binding site
To confirm by an independent method the ability of GA to compete with BH3 peptides for binding to Bcl-2-family proteins, we devised a TR-FRET assay for Bcl-XL, in which FITC-Bad BH3 peptide bound to recombinant purified GST-Bcl-XL is excited by emissions from terbium conjugated anti-GST antibody. FITC-Bad BH3 peptide bound to GST-Bcl-XL (but not GST [not shown]) in a concentration and saturable manner, with apparent Kd’s of 2 nM (Figure 2A). GA inhibited FITC-Bad BH3 peptide binding to GST-Bcl-XL, as measured by TR-FRET, with apparent IC50’s of 1.5 μM (Figure 2B), thus yielding similar results as those obtained using FPAs.
Figure 2. TR-FRET assay confirms gambogic acid competes with BH3 peptide for binding to Bcl-XL.
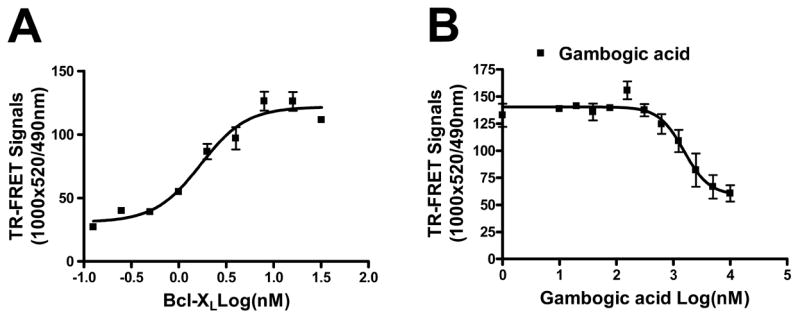
A, Binding of GST-Bcl-XL to FITC-BH3 peptide was assayed by TR-FRET. The final reaction mixtures contain 10 nM of FITC-Bad BH3 peptide, 2 nM of anti-GST-Turbium, and various concentrations of GST-Bcl-XL proteins in PBS buffer containing 0.005% Tween 20. B, Analysis of GA competition with BH3 peptide for Bcl-XL binding by TR-FRET. Reaction mixtures of final 20 μl volume contain 2 μl of various concentrations of GA with 10 nM FITC-Bad BH3 peptide, 2 nM anti-GST-Terbium, and 10 nM GST-Bcl-XL protein in PBS buffer containing 0.005% Tween 20. The mixtures are incubated for 30 min at room temperature. TR-FRET signals were measured using excitation at 330 nm, emission for FITC signal at 490 nm, and emission for terbium signal at 520 nm. Data are presented as 520 nm/490 nm ratio (mean ± std dev; n = 3).
GA neutralizes activity of Bcl-2-family proteins in vitro
An active, N-terminally truncated form of the pro-apoptotic BH3-containing protein Bid (tBid) induces release of apoptogenic proteins such as SMAC from isolated mitochondria in vitro (25, 27, 28). Using this assay, we showed that 5 of the 6 human anti-apoptotic Bcl-2 family proteins negate tBid-induced release of SMAC from isolated mitochondria (Figure 3). For these 5 proteins (Bcl-2, Bcl-XL, Bfl-1, Bcl-W, Mcl-1), adding GA restored tBid-induced SMAC release in a concentration-dependent manner (Figure 3). Complete restoration was typically achieved with 5 μM GA, representing an approximately 10:1 molar excess of GA relative to anti-apoptotic Bcl-2-family proteins. GA also enhanced tBid-induced release of SMAC from isolated mitochondria when recombinant Bcl-2-family proteins were not added (supplemental data), suggesting it may neutralize endogenous anti-apoptotic Bcl-2-family proteins associated with mitochondria.
Figure 3. Gambogic acid neutralizes ability of Bcl-2-family proteins to suppress tBid-induced mitochondrial leakage.

A–F, tBid (20 ng, cleaved by caspase 8) was pre-incubated with 2 μg of purified recombinant anti-apoptotic Bcl-2 family proteins, including Bcl-XL (A), Bcl-2 (B), Bcl-W (C), Bcl-B (D), Bfl-1 (E) or Mcl-1 (F) together with various concentrations of GA for 15 min in HM buffer, before adding 50 μg of isolated HeLa mitochondria for 1 hr at 30°C. Samples were centrifuged to generate supernatants that were analyzed by SDS-PAGE/immunoblotting using anti-SMAC antibody, comparing relative amounts of SMAC released from equivalent amounts of input mitochondria treated with tBid. The “Pellet” represents intact, untreated mitochondria, provided as a control.
GA overcomes cytoprotection of Bcl-2 in leukemia cells
GA has cytotoxic activity against various tumor cell lines in culture and induces apoptosis (1, 2). We confirmed the apoptosis-inducing activity of GA using Jurkat T-cell acute lymphoblastic leukemia and HL-60 acute promyelomonocytic leukemia cell lines, using Annexin V/propidium iodide (PI) staining, counting Annexin V+/PI- cells as apoptotic (Figure 4A). The concentration of GA required to induce apoptosis of ~50% of the cells within 20–24 hrs was ~0.2 μM and ~0.5 μM for Jurkat and HL-60, respectively. GA also induced clearly detectable proteolytic processing of pro-caspase-3, a marker of apoptosis, at concentrations ≥ 2 μM, as determined by immunoblotting using lysates from prostate cancer cell PPC1 treated with GA (Figure 4B). To assess whether GA affects expression of Bcl-2 family proteins, we preformed immunoblotting experiments in which HL-60 leukemia cells were treated with caspase inhibitor benozyl-valinyl-alainyl-asparatyl-fluoromethylketone (zVAD-fmk) to prevent apoptosis and thus avoid secondary declines in protein expression. Levels of Bcl-2 and Bcl-XL were not affected by treatment with GA (supplemental data, Figure S2).
Figure 4. Gambogic acid induces apoptosis of cancer cells.

A, Gambogic acid was used to treat HL-60 (black symbols) or Jurkat (white symbols) cells at concentrations of 0, 0.1, 0.2, 0.5, 2, 5 μM (from left to right). Cells were collected after 20 hrs, stained with FITC-Annexin V and PI, and the percentage of Annexin V-positive/PI-negative cells was determined. B, PPC1 cells were treated with gambogic acid at concentrations of 0, 0.5, 1, 2, 5, 10 μM. After 20 hrs, the cells were collected and the cell lysates were analyzed by SDS-PAGE/immunoblotting using anti-caspase 3 antibody. The positions of uncleaved proform and cleaved large subunit of the enzyme are indicated.
We tested the cytotoxic activity of GA against HL-60 cells that had been stably transfected with NEO-control or Bcl-2-expression plasmids (29). Over-expression of Bcl-2 reduced the sensitivity of HL-60 cells to GA, shifting the dose-response curve to the right, such that higher concentrations of GA were required to kill the cells (Figure 5A). Treating HL-60 NEO-control and Bcl-2-transfected cells with an upstream activator of the mitochondrial pathway for apoptosis, staurosporine (STS), confirmed that Bcl-2 over-expressing HL-60 cells have a block to apoptosis (Figure 5B), which GA overcomes. Experiments in which STS and GA were combined showed that GA overcomes Bcl-2-mediated resistance to STS (supplemental data, Figure S3).
Figure 5. Effects of Bcl-2 over-expression or Bax/Bak DKO on Gambogic acid-induced cytotoxicity.

A, B, Neo-control or Bcl-2 over-expressing HL-60 cells were treated with (A) gambogic acid or (B) Staurosporine at various concentrations, as indicated. C, D, MEF cells with wild-type (WT) (black symbols) or bax−/−/bak−/− (DKO) (white symbols) genotypes were treated with GA (C) or Staurosporine (D) at various concentrations. After 8 hrs, the cells were collected, stained with FITC-Annexin V and PI, and the percentage of live cells was determined (Annexin-negative/PI-negative). Data are representative of at least 3 independent experiments.
GA-induced cytotoxicity is only partly Bcl-2-dependent
Anti-apoptotic Bcl-2–family proteins suppress mitochondria-initiated cell death by inhibiting pro-apoptotic Bcl-2 members Bax and Bak (10, 20, 30). Consequently, when cells are genetically engineered to lack Bax and Bak, then Bcl-2 and related cytoprotective proteins no long display an anti-apoptotic phenotype (31). Cells doubly deficient in Bax and Bak therefore provide a context for assessing the mechanism of GA-induced toxicity. GA killed transformed mouse fibroblasts (MEFs) generated from bax+/+ bak+/+ embryos in a concentration-dependent manner (Figure 5C). However, GA also killed bax−/− bak−/− MEFs, requiring only approximately 2-fold higher concentrations of compound to reach the Lethal Dose 50% (LD50). Treating these MEFs with STS confirmed that Bak/Bak double knock-out cells have a profound block to cell death (Figure 5D), which GA overcomes. We conclude therefore that GA induces cytotoxicity through mechanisms that are only partly dependent on Bcl-2-family proteins.
GA analogs show differential activity against Bcl-2-family proteins, correlating with cytotoxicity
We examined the activity of analogs of GA with respect to displacement of Bid BH3 peptide from Bfl-1 in vitro (Figure 6A) and cytotoxic activity (Figure 6B). In the FPAs, dihydro-GA and acetyl-iso-GA displaced BH3 peptide from Bfl-1 with potencies only 3–6 fold less that GA (Figure 6A). In contrast, tetrahydro-GA and Garcinolic acid had greatly reduced activity in BH3 displacement assays. In agreement with the BH3 displacement data, dihydro-GA and acetyl-iso-GA exhibited cytotoxic activity against cultured leukemia cells, while tetrahydro-GA showed markedly reduced activity (~50-fold less potent) and Garcinolic acid was non-toxic (Figure 6B). Thus, the cytotoxic activity of GA analogs correlates roughly with their ability to compete with BH3 peptides for binding to an anti-apoptotic Bcl-2-family member.
Figure 6. Comparison of cytotoxic activity and BH3 peptide displacement activity of Gambogic acid and analogs.

A, Structures of GA and GA analogs are depicted, and IC50 values from FPAs are presented for competitive binding assays performed using Bfl-1 and FITC-Bid BH3 peptide. B, Jurkat cells were treated with GA and analogs at concentrations of 0, 1, 5, 10, 20, 50 μM. Cells were collected after 20 hrs, stained with FITC-Annexin V and PI, and the percentages of live cells (Annexin-negative/PI-negative) were determined. Data are representative of at least 3 experiments.
Discussion
We show here that the cytotoxic natural product GA competes for BH3 peptide binding sites on several anti-apoptotic members of the Bcl-2 family and neutralizes the ability of these proteins to suppress release of apoptogenic proteins from isolated mitochondria. Structure-function analysis (SAR) of GA using analogs suggested a general correlation between BH3 competition and cytoxicity activity, but experiments with bax/bak double knock-out cells suggest GA-induced cytotoxicity is only partially dependent on Bcl-2-family proteins.
GA has been reported to affect other molecular events relevant to cytotoxicity, including inducing increases in Bax protein levels, reductions in Bcl-2 protein levels, and suppressing transferrin receptor internalization, inhibiting the catalytic activity of human topoisomerase IIalpha, and modulating the nuclear factor-kappaB signaling pathway (2, 32–34). GA also reportedly induces G2/M-phase arrest of dividing cells (35), suggesting that this natural product hits targets involved in cell cycle. Thus, like many natural products, GA may have several targets in mammals, among which are anti-apoptotic members of the Bcl-2 family.
The vivo activity of GA and its therapeutic index has been previously explored in rodents (2, 6). Using a prostate cancer xenograft model, for example, it was recently reported that GA effectively inhibited tumor angiogenesis and suppressed tumor growth with few side effects, where GA was delivered by subcutaneous injection daily for 15 days (4). Also, using a mouse model of glioma, intravenous injection of GA daily for 14 days was reported to significantly reduce tumor volumes with little evidence of side effects (5). Anti-tumor activity was attributed to induction of apoptosis and to suppression of angiogenesis.
It is interesting to speculate why plants might produce compounds that neutralize Bcl-2 family proteins. Insects, nematodes, and other animal species that eat plants contain evolutionarily conserved anti-apoptotic Bcl-2 family proteins (reviewed in (36)). Consequently, plants might produce antagonists of these compounds as a mechanism to defend themselves against animal species. In this regard, several natural products from diverse plant species have been shown to competitively displace BH3 peptides and neutralize the activity of anti-apoptotic Bcl-2-family proteins, including epigallocatechin gallate (EGCG) from green tea, theaflavins from black tea, and gossypol from cotton seeds (37, 38). These compounds are structurally diverse, but all display < 1 μM activity against a variety of human anti-apoptotic Bcl-2 family proteins. It remains to be determined whether their affinity is greater for Bcl-2 family members from insects or other lower-organisms of the animal kingdom.
Supplementary Material
Acknowledgments
We thank Tessa Siegfried and Melanie Hanaii for manuscript preparation and the NIH for generous support (CA-113318; CA-55164).
References
- 1.Zhang HZ, Kasibhatla S, Wang Y, et al. Discovery, characterization and SAR of gambogic acid as a potent apoptosis inducer by a HTS assay. Bioorg Med Chem. 2004;12:309–17. doi: 10.1016/j.bmc.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 2.Zhao L, Guo QL, You QD, Wu ZQ, Gu HY. Gambogic acid induces apoptosis and regulates expressions of Bax and Bcl-2 protein in human gastric carcinoma MGC-803 cells. Biol Pharm Bull. 2004;27:998–1003. doi: 10.1248/bpb.27.998. [DOI] [PubMed] [Google Scholar]
- 3.Yang Y, Yang L, You QD, et al. Differential apoptotic induction of gambogic acid, a novel anticancer natural product, on hepatoma cells and normal hepatocytes. Cancer Lett. 2007;256:259–66. doi: 10.1016/j.canlet.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 4.Yi T, Yi Z, Cho SG, et al. Gambogic acid inhibits angiogenesis and prostate tumor growth by suppressing vascular endothelial growth factor receptor 2 signaling. Cancer Res. 2008;68:1843–50. doi: 10.1158/0008-5472.CAN-07-5944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qiang L, Yang Y, You QD, et al. Inhibition of glioblastoma growth and angiogenesis by gambogic acid: An in vitro and in vivo study. Biochem Pharmacol. 2008;75:1083–92. doi: 10.1016/j.bcp.2007.10.033. [DOI] [PubMed] [Google Scholar]
- 6.Guo Q, Qi Q, You Q, Gu H, Zhao L, Wu Z. Toxicological studies of gambogic acid and its potential targets in experimental animals. Basic Clin Pharmacol Toxicol. 2006;99:178–84. doi: 10.1111/j.1742-7843.2006.pto_485.x. [DOI] [PubMed] [Google Scholar]
- 7.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–56. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 8.Reed JC. Apoptosis-targeted therapies for cancer. Cancer Cell. 2003;3:17–22. doi: 10.1016/s1535-6108(02)00241-6. [DOI] [PubMed] [Google Scholar]
- 9.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 10.Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–9. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 11.Verhagen AM, Ekert PG, Pakusch M, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 12.Raffo AJ, Perlman H, Chen MW, Day ML, Streitman JS, Buttyan R. Overexpression of bcl-2 protects prostate cancer cells from apoptosis in vitro and confers resistance to androgen depletion in vivo. Cancer Res. 1995;55:4438–45. [PubMed] [Google Scholar]
- 13.Furuya Y, Krajewski S, Epstein JI, Reed JC, Isaacs JT. Expression of bcl-2 and the progression of human and rodent prostatic cancers. Clin Cancer Res. 1996;2:389–98. [PubMed] [Google Scholar]
- 14.Lomo J, Smeland EB, Krajewski S, Reed JC, Blomhoff HK. Expression of the Bcl-2 homologue Mcl-1 correlates with survival of peripheral blood B lymphocytes. Cancer Res. 1996;56:40–3. [PubMed] [Google Scholar]
- 15.Schimmer AD, Munk-Pedersen I, Minden MD, Reed JC. Bcl-2 and apoptosis in chronic lymphocytic leukemia. Curr Treat Options Oncol. 2003;4:211–8. doi: 10.1007/s11864-003-0022-y. [DOI] [PubMed] [Google Scholar]
- 16.Letai A, Sorcinelli MD, Beard C, Korsmeyer SJ. Antiapoptotic BCL-2 is required for maintenance of a model leukemia. Cancer Cell. 2004;6:241–9. doi: 10.1016/j.ccr.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 17.Andersen MH, Svane IM, Kvistborg P, et al. Immunogenicity of Bcl-2 in patients with cancer. Blood. 2005;105:728–34. doi: 10.1182/blood-2004-07-2548. [DOI] [PubMed] [Google Scholar]
- 18.Reed JC. Double identity for proteins of the Bcl-2 family. Nature. 1997;387:773–6. doi: 10.1038/42867. [DOI] [PubMed] [Google Scholar]
- 19.Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–6. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 20.Reed JC. Mechanisms of apoptosis. Am J Pathol. 2000;157:1415–30. doi: 10.1016/S0002-9440(10)64779-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuwana T, Bouchier-Hayes L, Chipuk JE, et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17:525–35. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 22.Chen L, Willis SN, Wei A, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 23.Reed JC, Pellecchia M. Apoptosis-based therapies for hematologic malignancies. Blood. 2005;106:408–18. doi: 10.1182/blood-2004-07-2761. [DOI] [PubMed] [Google Scholar]
- 24.Zhai D, Jin C, Satterthwait AC, Reed JC. Comparison of chemical inhibitors of antiapoptotic Bcl-2-family proteins. Cell Death Differ. 2006;13:1419–21. doi: 10.1038/sj.cdd.4401937. [DOI] [PubMed] [Google Scholar]
- 25.Zhai D, Luciano F, Zhu X, Guo B, Satterthwait AC, Reed JC. Humanin binds and nullifies Bid activity by blocking its activation of Bax and Bak. J Biol Chem. 2005;280:15815–24. doi: 10.1074/jbc.M411902200. [DOI] [PubMed] [Google Scholar]
- 26.Zhai D, Ke N, Zhang H, et al. Characterization of the anti-apoptotic mechanism of Bcl-B. Biochem J. 2003;376(Pt 1):229–36. doi: 10.1042/BJ20030374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Becattini B, Sareth S, Zhai D, Crowell KJ, Leone M, Reed JC, et al. Targeting apoptosis via chemical design: inhibition of bid-induced cell death by small organic molecules. Chem Biol. 2004;11:1107–17. doi: 10.1016/j.chembiol.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 28.Luciano F, Zhai D, Zhu X, et al. Cytoprotective peptide humanin binds and inhibits proapoptotic Bcl-2/Bax family protein BimEL. J Biol Chem. 2005;280:15825–35. doi: 10.1074/jbc.M413062200. [DOI] [PubMed] [Google Scholar]
- 29.Konopleva M, Tari AM, Estrov Z, et al. Liposomal Bcl-2 antisense oligonucleotides enhance proliferation, sensitize acute myeloid leukemia to cytosine-arabinoside, and induce apoptosis independent of other antiapoptotic proteins. Blood. 2000;95:3929–38. [PubMed] [Google Scholar]
- 30.Li P, Nijhawan D, Wang X. Mitochondrial activation of apoptosis. Cell. 2004;116(2 Suppl):S57–9. doi: 10.1016/s0092-8674(04)00031-5. 2 p following S9. [DOI] [PubMed] [Google Scholar]
- 31.Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–30. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kasibhatla S, Jessen KA, Maliartchouk S, et al. A role for transferrin receptor in triggering apoptosis when targeted with gambogic acid. Proc Natl Acad Sci U S A. 2005;102:12095–100. doi: 10.1073/pnas.0406731102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qin Y, Meng L, Hu C, et al. Gambogic acid inhibits the catalytic activity of human topoisomerase IIalpha by binding to its ATPase domain. Mol Cancer Ther. 2007;6:2429–40. doi: 10.1158/1535-7163.MCT-07-0147. [DOI] [PubMed] [Google Scholar]
- 34.Pandey MK, Sung B, Ahn KS, Kunnumakkara AB, Chaturvedi MM, Aggarwal BB. Gambogic acid, a novel ligand for transferrin receptor, potentiates TNF-induced apoptosis through modulation of the nuclear factor-kappaB signaling pathway. Blood. 2007;110:3517–25. doi: 10.1182/blood-2007-03-079616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu J, Guo QL, You QD, et al. Gambogic acid-induced G2/M phase cell-cycle arrest via disturbing CDK7-mediated phosphorylation of CDC2/p34 in human gastric carcinoma BGC-823 cells. Carcinogenesis. 2007;28:632–8. doi: 10.1093/carcin/bgl168. [DOI] [PubMed] [Google Scholar]
- 36.Reed JC, Doctor KS, Godzik A. The domains of apoptosis: a genomics perspective. Sci STKE 2004. 2004;(239):re9. doi: 10.1126/stke.2392004re9. [DOI] [PubMed] [Google Scholar]
- 37.Leone M, Zhai D, Sareth S, Kitada S, Reed JC, Pellecchia M. Cancer prevention by tea polyphenols is linked to their direct inhibition of antiapoptotic Bcl-2-family proteins. Cancer Res. 2003;63:8118–21. [PubMed] [Google Scholar]
- 38.Kitada S, Leone M, Sareth S, Zhai D, Reed JC, Pellecchia M. Discovery, characterization, and structure-activity relationships studies of proapoptotic polyphenols targeting B-cell lymphocyte/leukemia-2 proteins. J Med Chem. 2003;46:4259–64. doi: 10.1021/jm030190z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.