

Abstract
Rho-kinase is a serine threonine kinase that increases vasomotor tone via its effects on both endothelium and smooth muscle. Rho-kinase inhibition reduces cerebral infarct size in wild type, but not endothelial nitric oxide synthase deficient (eNOS−/−) mice. The mechanism may be related to Rho-kinase activation under hypoxic/ischemic conditions and impaired vasodilation because of downregulation of eNOS activity. To further implicate Rho-kinase in impaired vascular relaxation during hypoxia/ischemia, we exposed isolated vessels from rat and mouse to 60 mins of hypoxia, and showed that hypoxia reversibly abolished acetylcholine-induced eNOS-dependent relaxation, and that Rho-kinase inhibitor hydroxyfasudil partially preserved this relaxation during hypoxia. We, therefore, hypothesized that if hypoxia-induced Rho-kinase activation acutely impairs vasodilation in ischemic cortex, in vivo, then Rho-kinase inhibitors would acutely augment cerebral blood flow (CBF) as a mechanism by which they reduce infarct size. To test this, we studied the acute cerebral hemodynamic effects of Rho-kinase inhibitors in ischemic core and penumbra during distal middle cerebral artery occlusion (dMCAO) in wild-type and eNOS−/− mice using laser speckle flowmetry. When administered 60 mins before or immediately after dMCAO, Rho-kinase inhibitors hydroxyfasudil and Y-27632 reduced the area of severely ischemic cortex. However, hydroxyfasudil did not reduce the area of CBF deficit in eNOS−/− mice, suggesting that its effect on CBF within the ischemic cortex is primarily endothelium-dependent, and not mediated by its direct vasodilator effect on vascular smooth muscle. Our results suggest that Rho-kinase negatively regulates eNOS activity in acutely ischemic brain, thereby worsening the CBF deficit. Therefore, rapid nontranscriptional upregulation of eNOS activity by small molecule inhibitors of Rho-kinase may be a viable therapeutic approach in acute stroke.
Keywords: focal cerebral ischemia, hydroxyfasudil, hypoxia, isolated vessels, laser speckle flowmetry, Rho-kinase
Introduction
Despite decades of intense research, therapeutic options for acute stroke remain limited. Therapies that reinstate cerebral blood flow (CBF) to the ischemic territory (i.e., clot dissolution by thrombolysis or angioplasty) are efficacious in acute stroke, suggesting that CBF is a critical determinant of final stroke outcome. Cerebrovascular regulation during ischemia is poorly understood. Autoregulation is impaired in ischemic brain, and vessels may be maximally dilated because of reduced perfusion pressure and metabolic acidosis. However, vasodilator drugs such as nitric oxide donors or Ca2+ channel blocker nimodipine reportedly increase CBF during acute stroke suggesting that there is residual cerebrovascular reserve that can be recruited by vasodilators (Zhang et al, 1994). Endothelial nitric oxide synthase (eNOS) is an important enzyme regulating vascular tone, platelet aggregation, and inflammatory cell migration in cerebral ischemia. Under most conditions, stimulation of eNOS activity is protective (Limbourg et al, 2002), whereas its inhibition or genetic deletion is detrimental in animal models of stroke (Huang et al, 1996). Takemoto et al (2002) found that hypoxia downregulates eNOS expression in human saphenous and pulmonary artery endothelial cells, by decreasing eNOS mRNA half-life as early as a few hours after exposure to hypoxia. Rho-kinase, a serine threonine protein kinase and upstream negative regulator of eNOS activity, is implicated in this downregulation. Besides post-transcriptional downregulation of eNOS expression, Rho-kinase also decreases eNOS activity via phosphatidylinositol-3-kinase/Akt pathway, a faster post-translational mechanism of eNOS regulation occurring within minutes (Wolfrum et al, 2004).
Rho-kinase is activated in cerebral ischemia (Rikitake et al, 2005), and increases the sensitivity of the contractile apparatus to intracellular Ca2+ ([Ca2+]i) in smooth muscle by enhancing myosin light chain (MLC) phosphorylation (Amano et al, 2000), or suppressing eNOS activity (Ming et al, 2002). In addition to the regulation of vasomotor tone, Rho-kinase inhibitors reduce inflammation (Satoh et al, 2001), inhibit NADPH oxidase and superoxide production in neutrophils (Arai et al, 1993), and activate KATP channels in cardiac myocytes (Terzic and Kurachi 1996). We recently reported that Rho-kinase inhibitors reduce infarct size in focal cerebral ischemia, and this protection was critically dependent on the expression of eNOS (Rikitake et al, 2005). We now present evidence showing that Rho-kinase is rapidly activated in ischemic cortex, and its inhibitors can acutely enhance CBF in both core and penumbra in an eNOS-dependent manner in a mouse model of focal ischemia. The mechanism of this CBF augmentation by Rho-kinase inhibitors appears related to preservation of eNOS function under hypoxic conditions.
Materials and methods
Isolated Vessel Experiments
Mice or rats were killed by decapitation. Brains with attached arteries or aortas were removed and immersed in physiologic solution (composition, mmol/L: NaCl, 118; KCl, 4.6; NaHCO3, 25; MgSO4, 1.2; KH2PO4, 1.2; CaCl2, 1.25; glucose, 10; EDTA, 0.025; pH 7.4 at 37°C). Arteries were dissected, cut into segments (1.5 to 2 mm long), threaded onto wires (25 μm diameter for rat basilar artery, 40 μm diameter for mouse aorta), and mounted in an isometric myograph (610 M, Danish Myo Technology, Aarhus, Denmark). After mounting, each preparation was stabilized for 30 mins in physiologic solution, and aerated with 95% O2/5% CO2 at 37°C. The normalized passive resting force and corresponding diameter were then determined for each preparation from its own length–pressure curve. Contractile responses were recorded into a computer, by using a data acquisition and recording software (Myodaq and Myodata, Danish Myo Technology, Aarhus, Denmark).
After normalization and 30-mins equilibration in physiologic solution, the arteries were preconstricted with either phenylephrine (1 μmol/L in mouse aorta) or serotonin (1 μmol/L in rat basilar artery). Once preconstriction reached a steady state, endothelium-dependent relaxations were studied by adding increasing concentrations of acetylcholine (ACh, 1 nmol/L to 1 μmol/L) in mouse aorta. In rat basilar arteries, a single concentration of ACh (3 μmol/L) was tested. Endothelium-independent relaxations were studied by sodium nitroprusside (0.1 μmol/L). After washout, the aerating gas mixture was switched to 95% N2/5% CO2 to simulate severe hypoxia. The pO2 in the organ bath was 20 ± 2 mm Hg 15 mins after the onset of aeration with 95% N2/5% CO2, measured in 0.1 ml samples using an arterial blood gas analyzer (n = 8; Corning 178 blood gas/pH analyzer, Ciba Corning Diagnostics, Medford, MA, USA); measurements made at 60 mins did not differ significantly. The vasodilator agents were tested after 30 and 60 mins of hypoxia. After the second washout, the aerating gas mixture was again switched to 95% O2/5% CO2, and the vasodilator agents were tested in the same manner after reoxygenation. In a separate group, arteries were incubated with hydroxyfasudil (3 μmol/L) for 30 min, after the first ACh concentration–response. Then hypoxia was instituted for 60 mins in the presence of hydroxyfasudil as described above, and ACh concentration–response was repeated. Hypoxic vessels were then incubated with NG-nitro-l-arginine methyl ester (l-NAME, 0.3 mmol/L) in the presence of hydroxyfasudil, and ACh concentration–response was repeated. In a separate group, hypoxic vessels were incubated with hydralazine (0.3 mmol/L) instead of hydroxyfasudil to test whether a nonspecific reduction in phenylephrine preconstriction tone augments ACh concentration–response.
General Surgical Preparation
Mice (C57BL/6J, and eNOS deficient, 23 to 28 g) were housed under diurnal lighting conditions and allowed food and tap water ad libitum. Endothelial NOS deficient mice were backcrossed on a C57BL/6J wild-type background for more than 10 generations. Mice were anesthetized with 2% isoflurane (in 70% N2O and 30% O2) and intubated transorally. Femoral artery was catheterized for the measurement of blood pressure (BP) and heart rate (HR) (ETH 400 transducer amplifier, CB Sciences, Milford, MA, USA). Anesthesia was maintained on 1% isoflurane, the depth of anesthesia was checked by the absence of cardiovascular changes in response to tail pinch. Rectal temperature was kept at 36.8°C to 37.1°C using a thermostatically controlled heating mat (FHC, Brunswick, ME, USA). Mice were paralyzed (pancuronium bromide, 0.4 mg/kg/h, i.p.), mechanically ventilated (CWE, SAR-830, Ardmore, PA, USA), and placed in a stereotaxic frame (David Kopf, Tujunga, CA, USA). Arterial blood gases and pH were measured once every hour in 30 μL blood samples (Corning 178 blood gas/pH analyzer, Ciba Corning Diagnostics, Medford, MA, USA). The data were continuously recorded using a data acquisition and analysis system (PowerLab, AD Instruments, Medford, MA, USA) and stored in a computer. The arterial blood gas and pH were within previously reported normal limits (Table 1) (Huang et al, 1995).
Table 1.
Physiological parameters in mice with distal middle cerebral artery occlusion
| Strain | Treatment (time from dMCAO) | n | BP | pH | pO2 | pCO2 |
|---|---|---|---|---|---|---|
| Wild-type | Vehicle (−60 or +5 mins) | 21 | 76±9 | 7.36±0.05 | 164±42 | 38±6 |
| Hydroxyfasudil | ||||||
| −60 mins | 5 | 67±8 | 7.38±0.06 | 176±10 | 36±3 | |
| +5 mins | 9 | 71±10 | 7.34±0.07 | 166±41 | 36±6 | |
| Y-27632 (−60 mins) | 8 | 63±3 | 7.36±0.06 | 155±34 | 39±4 | |
| l-NIO (−60 mins) | 6 | 93±4 | 7.35±0.03 | 175±26 | 39±4 | |
| Hydroxyfasudil+l-NIO (−60 mins) | 4 | 82±5 | 7.36±0.03 | 140±2 | 38±4 | |
| Hydralazine (−60 mins) | 3 | 66±2 | 7.36±0.03 | 121±24 | 37±3 | |
| eNOS (−/−) | Vehicle | 5 | 101±5 | 7.37±0.06 | 167±17 | 32±2 |
| Hydroxyfasudil (−60 mins) | 5 | 84±7 | 7.37±0.09 | 164±10 | 33±3 |
BP (mean arterial blood pressure at ischemia onset), pO2, and pCO2 are expressed in mm Hg. Data from wild-type mice receiving preischemic or postischemic vehicle were combined.
Laser Speckle Flowmetry
Laser speckle flowmetry (LSF) was used to study the spatiotemporal characteristics of CBF changes during focal cerebral ischemia (Ayata et al, 2004). Briefly, a CCD camera (Cohu, San Diego, CA, USA) was positioned above the head, and a laser diode (780 nm) was used to illuminate the intact skull surface in a diffuse manner. The penetration depth of the laser is approximately 500 μm. Raw speckle images were used to compute speckle contrast, which is a measure of speckle visibility related to the velocity of the scattering particles, and therefore, CBF. The speckle contrast is defined as the ratio of the standard deviation of pixel intensities to the mean pixel intensity in a small region of the image. Ten consecutive raw speckle images were acquired at 15 Hz (an image set), processed by computing the speckle contrast using a sliding grid of 7 × 7 pixels, and averaged to improve signal to noise ratio. Speckle contrast images were converted to images of correlation time values, which represent the decay time of the light intensity autocorrelation function. The correlation time is inversely and linearly proportional to the mean blood velocity. Relative CBF images (percentage of baseline) were calculated by computing the ratio of a baseline image of correlation time values to subsequent images. Laser speckle perfusion images were obtained every 7.5 secs.
Laser speckle flowmetry imaging was started 1 min before distal middle cerebral artery occlusion (dMCAO) and continued throughout the experiment. Ischemic CBF deficit was analyzed two-dimensionally over time by quantifying the area of cortex (mm2) with either severe (0% to 20% residual CBF, representing core) or moderate CBF reduction (21% to 30% residual CBF, representing penumbra) using a thresholding paradigm. The effect of Rho-kinase inhibitors on resting CBF in nonischemic brain was determined over time by placing five cortical regions of interest (250 × 250 μm) away from large surface vessels, and averaging CBF changes within these regions of interest.
Focal Cerebral Ischemia
Focal cerebral ischemia was induced by dMCAO. After general surgical preparation, mice were placed in a stereotaxic frame, and skull surface was prepared for LSF as described above. The temporalis muscle was separated from the temporal bone and removed. A burr hole (2 mm diameter) was drilled under saline cooling in the temporal bone overlying the MCA just above the zygomatic arch. The dura was kept intact and MCA was occluded using a microvascular clip.
In Vivo Experimental Protocols
The following drugs were tested in wild-type or eNOS−/− mice: saline (5 ml/kg, i.p., n = 15 wild-type and 5 eNOS−/−), hydroxyfasudil (10 mg/kg, i.p., n = 5 wild-type and 5 eNOS−/−), Y-27632 (10 mg/kg, i.p., n = 8), N5-(1-Iminoethyl)-l-ornithine (l-NIO, 20 mg/kg, i.p., n = 6), hydroxyfasudil (10 mg/kg, i.p.) plus l-NIO (20 mg/kg, i.p., n = 4), and hydralazine (0.7 mg/kg, i.p., n = 3). All drugs were administered 60 mins before dMCAO; in addition, saline (i.v., n = 6) and hydroxyfasudil (10 mg/kg, i.v., n = 9) were also tested when administered 5 mins after dMCAO. The doses of hydroxyfasudil and Y-27632 were chosen based on previously reported lowest systemic doses that reduce infarct size in cerebral and coronary ischemia models (Bao et al, 2004; Rikitake et al, 2005). The dose of hydralazine was determined in pilot experiments to obtain a BP reduction comparable to Rho-kinase inhibitors. Data from preischemic vehicle-treated wild-type mice were pooled into a single control group (n = 15) for statistical comparisons to preischemic drug-treated groups. Post-ischemic vehicle-treated mice (n = 6) served as control for post-ischemic hydroxyfasudil-treated group (n = 9).
The systemic and cerebrovascular effects of Rho-kinase inhibitors under resting conditions in nonischemic brain were studied in a separate group of mice by LSF, BP and HR monitoring for 1 h, after injection of hydroxyfasudil (10 mg/kg, i.p., in wild-type and eNOS−/− mice, n = 4 each) or Y-27632 (10 mg/kg, i.p., in wild-type mice, n = 4). These data were expressed as % change in BP, HR and CBF.
Rho-kinase Activity Assay
Rho-kinase phosphorylates the myosin-binding subunit (MBS) of MLC phosphatase at Thr853 (Kawano et al, 1999). A rabbit polyclonal antibody was raised against MBS phosphorylated at Thr853 with the use of the following synthetic peptide: Cys845-Pro-Arg-Glu-Lys-Arg-Arg-Ser-phospho-Thr853-Gly-Val-Ser-Phe857 (BioSource International, Carlsbad, CA, USA). The antiserum was affinity purified with a column containing immobilized phospho-peptide.
For immunoblotting, brains were fresh frozen 60 mins after dMCAO, and severely ischemic core (determined using LSF images) and corresponding contralateral nonischemic cortex were dissected. Tissues were homogenized in 1500 μl 10% trichloroacetic acid and 10 mmol/L of dithiothreitol on ice three times for 10 secs each. After centrifugation, pellets were dissolved in 25 μL of 1 mol/L Tris base and then mixed with 250 μl of extraction buffer (8 mol/L urea, 2% SDS, 5% sucrose, and 5% 2-mercaptoethanol). Equal amounts of cell extracts were subjected to 7.5% SDS-PAGE and transferred onto PVDF membrane (Immobilon-P, Millipore, Billerica, MA, USA). Membranes were incubated with rabbit anti-phospho-specific Thr853-MBS polyclonal antibody (1:1000) or rabbit anti-MBS polyclonal antibody (1:5000, Covance, Princeton, NJ, USA). Bands were visualized with the use of the ECL detection kit (Amersham Pharmacia Biotech, Piscataway, NJ, USA). Rho-kinase activity was expressed as the ratio of phospho-Thr853 MBS/total MBS.
Infarct Size Determination
Infarct size was determined in a separate group of mice without physiologic monitoring or mechanical ventilation. Mice were treated with either vehicle (n = 5) or hydroxyfasudil (10 mg/kg, i.p. 1 h before dMCAO, n = 5), placed in stereotaxic frame, and dMCAO was performed during LSF as described above. One hour after dMCAO, the microvascular clip was carefully removed and reperfusion was confirmed using LSF for an additional 10 mins. The surgical wound was sutured and mice were allowed to recover from anesthesia. Mice were killed 48 h after dMCAO and brains rapidly removed. Whole brain was incubated in 2,3,5-triphenyltetrazolium chloride for 40 mins, and then stored in 4% paraformaldehyde. Images of the dorsal surface of topically stained whole brain were acquired using a CCD camera, and then the brain was cut into 1 mm thick coronal slices for infarct volume measurement as described before (Shin et al, 2006). Sufficient penetration of topically applied 2,3,5-triphenyltetrazolium chloride stain into deeper cortical layers was visually confirmed for each brain.
Data Analysis
The data were expressed as mean±standard deviation. Statistical comparisons were performed using paired or unpaired Student's t-test, and one-way analysis of variance (ANOVA) or two-way ANOVA for repeated measures followed by Fisher's protected least significant difference test. P < 0.05 was considered statistically significant.
Results
Isolated Vessels
Acetylcholine relaxed isolated mouse aorta in a concentration-dependent manner under normoxic conditions; this relaxation was completely abolished by l-NAME (0.3 mmol/L, not shown) indicating that it is eNOS mediated. Hypoxia did not significantly alter resting tension (e.g., 97%±4% and 100%±1% of baseline, during hypoxia and reoxygenation, respectively), or the magnitude of phenylephrine preconstriction (1.8±0.7 and 1.5±0.6 mN/mm, during normoxia and hypoxia, respectively; P > 0.05, paired t-test), but strongly inhibited ACh-induced relaxation in mouse aorta (n = 12; Figures 1A and 1B). Inhibition was partial at 30 mins and complete after 60 mins of hypoxia. Similar results were obtained in rat femoral arteries (not shown) and in rat basilar arteries (3 μmol/L ACh, n = 12) (Figures 1C and 1D). In contrast, endothelium-independent relaxation to sodium nitroprusside (0.1 μmol/L) was augmented during hypoxia (65%±11% versus 95%±6% relaxation in mouse aorta, during normoxia and hypoxia, respectively, P < 0.05, two-way ANOVA for repeated measures, n = 4). Hypoxic endothelial impairment was reversible on reoxygenation in all arteries studied (Figure 1).
Figure 1.

Acetylcholine-induced endothelium-dependent relaxations are reversibly abolished during hypoxia. (A) Representative tracings show that the addition of increasing concentrations of ACh (1 nmol/L to 1 mmol/L, arrows) relaxed mouse aorta concentration-dependently in vessel segments preconstricted with phenylephrine (1 μmol/L) under normoxic conditions (95%O2/5%CO2). When the aerating gas is switched to 95%N2/5%CO2 for 60 mins, ACh-induced relaxations were completely abolished. On reoxygenation with 95%O2/5%CO2 ACh-induced relaxations were largely restored suggesting that endothelium was not injured during hypoxia. (B) Averaged data from 12 preparations show that hypoxia reversibly impaired eNOS-dependent ACh-induced relaxations in mouse aorta. **P < 0.01 versus normoxia, two-way ANOVA for repeated measures. Error bars indicate standard deviations, and are shown unidirectional for clarity. (C) Representative tracings showing that ACh (3 μmol/L, arrow) completely relaxed rat basilar arteries preconstricted with serotonin (1 μmol/L) under normoxic conditions. Hypoxia, induced by switching the aerating gas to 95%N2/5%CO2 for 60 mins, abolished ACh-induced relaxations. On reoxygenation with 95%O2/5%CO2 ACh-induced relaxations were largely restored. (D) Averaged data from 12 preparations show that hypoxia reversibly abolished eNOS-dependent relaxations in rat basilar arteries. **P < 0.01 versus normoxia, one-way ANOVA for repeated measures. Vertical bars indicate standard deviations.
Incubation with a high concentration of hydroxyfasudil (100 μmol/L) completely abolished phenylephrine or serotonin constriction in both mouse aorta and rat cerebral arteries (not shown); this precluded testing of endothelium dependent relaxations. Therefore, we preincubated mouse aorta with a lower concentration of hydroxyfasudil (3 μmol/L for 90 mins including 60 mins of hypoxia, n = 21), which did not significantly alter the resting tension, and reduced the magnitude of phenylephrine-induced constriction by 50% (1.5±0.6 versus 0.8±0.3 mN/mm, in control and hydroxyfasudil groups, respectively, during hypoxia; P < 0.05, t-test). At this concentration, hydroxyfasudil partially restored endothelium-dependent ACh-induced relaxations in hypoxic mouse aorta (P < 0.05 versus hypoxia alone, two-way ANOVA for repeated measures; Figure 2). The restored ACh-induced relaxations were completely abolished by subsequent incubation with l-NAME (0.3 mmol/L, n = 8), confirming that they are eNOS-mediated (Figure 2). To test whether the recovery of ACh-induced relaxations by hydroxyfasudil was due to a nonspecific reduction in phenylephrine preconstriction, we incubated a separate group of hypoxic vessels with hydralazine (0.3 mmol/L, n = 8). Hydralazine reduced phenylephrine preconstriction tone by approximately 40%, but did not restore ACh-induced relaxations in hypoxic vessels (Figure 2). These data suggest that hypoxic eNOS inhibition is, at least in part, mediated by Rho-kinase.
Figure 2.
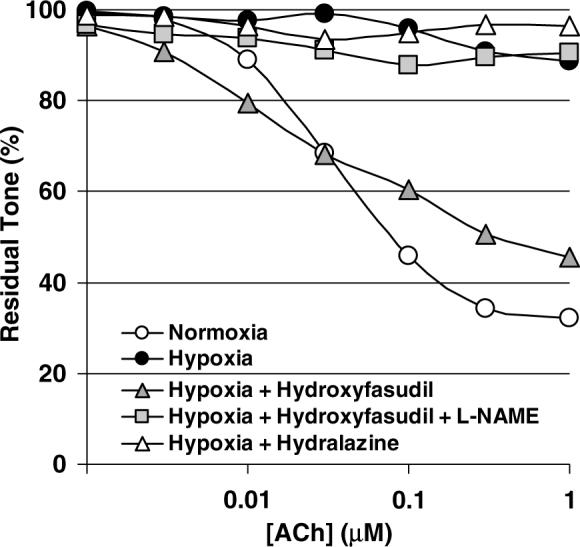
Hydroxyfasudil preserved endothelium-dependent relaxations during hypoxia. Hypoxia abolished ACh-induced endothelium-dependent relaxations in phenylephrine-preconstricted (1 μmol/L) mouse aorta (white and black circles, normoxia and hypoxia, respectively; n = 15). Preincubation with hydroxyfasudil (3 μmol/L, gray triangles, n = 29) preserved concentration-dependent relaxation to ACh during hypoxia (P < 0.05 versus hypoxia alone, two-way ANOVA for repeated measures). Subsequent incubation with L-NAME (0.3 mmol/L) abolished ACh-induced relaxations in the presence of hydroxyfasudil during hypoxia (square, n = 8). In contrast to hydroxyfasudil, incubation of hypoxic vessels with hydralazine (white triangles, n = 8) did not restore ACh-induced relaxations. Standard deviation bars were omitted for clarity.
Systemic Physiology
Hydroxyfasudil and Y-27632 caused mild hypotension in mice with dMCAO (Table 1), as well as in a separate group of nonischemic mice (Figures 3B and 3E; n = 4 each). This was not due to bradycardia, since HR slightly increased in both groups (Figures 3C and 3F). Both drugs also reduced resting CBF (Figures 3A and 3D). The reduction in resting CBF by hydroxyfasudil completely recovered after 30 mins (5% reduction), whereas the reduction in BP resolved only partially (15% reduction, P > 0.05). In contrast, the effects of Y-27632 on both CBF and BP were longer lasting (20% and 30% reduction in CBF and BP, respectively, at 60 min, P < 0.05 versus baseline; Figures 3D and 3E). Interestingly, eNOS knockout mice also developed hypotension after hydroxyfasudil (Figure 3B; n = 4). The magnitude of hypotension in eNOS knockouts was similar to wild type, although its onset was more gradual.
Figure 3.

Rho-kinase inhibitors caused hypotension and reduced resting CBF in nonischemic brain. (A) Hydroxyfasudil (10 mg/kg, i.p.) caused a small and reversible decrease in resting CBF in nonischemic wild-type (eNOS+/+ , n = 4) and eNOS knockout mice (eNOS−/−, n = 4). (B) This effect corresponded to a BP reduction of approximately 20 to 25% in both wild-type and eNOS knockout mice (P < 0.05 versus baseline) that was partially reversible over 60 mins. The onset of hypotension appeared to be slower in eNOS knockouts (P > 0.05). (C) There was a small increase in HR accompanying the hypotension in both strains (eNOS+/+, P < 0.05; eNOS−/−, P > 0.05 versus baseline). (D–F) Y-27632 (10 mg/kg, i.p., n = 4) caused a larger decrease in resting CBF and BP, and increase in HR in wild-type mice (eNOS+/+ ), which were not reversible over 60 mins (P < 0.05 versus baseline, as well as hydroxyfasudil-treated mice, two-way ANOVA for repeated measures). Vertical bars indicate standard deviations.
Focal Cerebral Ischemia
Hydroxyfasudil, administered 60 mins before dMCAO, attenuated the CBF deficit in both core and penumbra. The area of cortex with ≤20% residual CBF (i.e., core) was 50% smaller in hydroxyfasudil group compared with vehicle controls (n = 5 and 15, respectively, P < 0.05; Figure 4). This effect was apparent 10 mins after dMCAO and persisted throughout 60 mins of imaging (Figure 4D). The area of moderately ischemic cortex with 21% to 30% residual CBF (i.e., penumbra) was also smaller by 25% in the hydroxyfasudil group (Figure 5A). When administered 5 mins after dMCAO, hydroxyfasudil still reduced the area of core and penumbra by 33% and 25%, respectively (P < 0.05, n = 9 and 6 for hydroxyfasudil and vehicle groups; Figure 5B). Y-27632, a Rho-kinase inhibitor that is structurally and pharmacologically distinct from hydroxyfasudil, also attenuated the CBF deficit when administered 60 mins before dMCAO (40% reduction in the area of ischemic core, n = 8; Figure 5A). In contrast, hydralazine worsened the CBF deficit when administered 1 h before dMCAO. The area of severely ischemic cortex with ≤20% residual CBF was 5.7±0.5 mm2 in hydralazine-treated mice (n = 3), compared with 4.1±1.7 mm2 in the control group (P < 0.05), suggesting that the improvement in ischemic CBF by Rho-kinase inhibition is not due to a nonspecific vasodilator effect.
Figure 4.
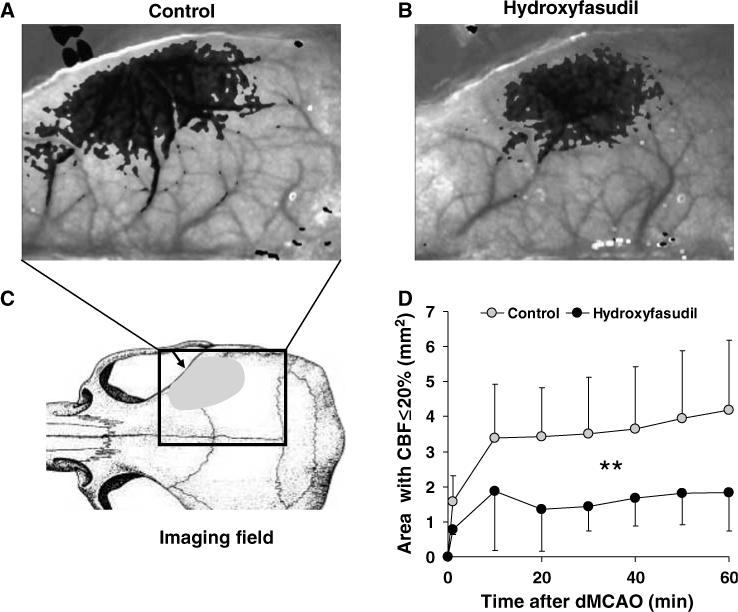
Rho-kinase inhibitor hydroxyfasudil preserved CBF after dMCAO. Representative speckle contrast images taken 60 mins after dMCAO are shown from control (A) and hydroxyfasudil-pretreated mice (B). Superimposed in gray are pixels with residual CBF ≤30%; the more severe the CBF deficit, the darker the superimposed gray. Hydroxyfasudil (10 mg/kg, i.p.) was administered 60 mins before dMCAO. (C) Imaging field (rectangle) included the entire right hemisphere and CBF was measured through intact skull. Arrow indicates the location of dMCAO using a microvascular clip, and shaded area represents the distribution of CBF deficit in this mouse dMCAO model. (D) The time course of changes in cortical area with severe CBF deficit (residual CBF ≤20%) in control (gray circles, n = 15) and hydroxyfasudil-pretreated mice (black circles, n = 5). On dMCAO (time 0), the area of severely ischemic cortex expanded rapidly during the first 10 mins, and continued to enlarge over 60 mins of imaging in controls. In contrast, this area was smaller in hydroxyfasudil group at all time points compared with controls (P < 0.05, two-way ANOVA for repeated measures), and did not expand after 10 mins. The area of CBF deficit was quantified using a thresholding paradigm (see Materials and methods). Vertical bars indicate standard deviations of the mean, and are shown unidirectional for clarity.
Figure 5.

Rho-kinase inhibitors hydroxyfasudil and Y-27632 reduced the area of hypoperfused cortex. (A) Hydroxyfasudil (10 mg/kg, i.p., 60 mins before dMCAO, n = 5) reduced the area of severe CBF deficit (i.e., core with residual CBF ≤20%) by more than 50%, when measured 60 mins after dMCAO; the area of moderate CBF deficit (i.e., 21 to 30% residual CBF) was reduced by 25%. Y-27632 (10 mg/kg, i.p., 60 mins before dMCAO, n = 8) also reduced the area of severe CBF deficit. (B) When administered 5 mins after dMCAO, hydroxyfasudil (10 mg/kg, slow i.v. bolus over 10 mins, n = 9) still reduced the area of CBF deficit; however, the effect was weaker in core compared with preischemic treatment. *P < 0.05 versus control group, two-way ANOVA. Vertical bars indicate standard deviations.
Rho-kinase activity (i.e., phospho-Thr853 MBS/total MBS ratio) was increased almost three-fold in ischemic core compared with corresponding contralateral cortex, when measured 60 mins after dMCAO (273%±145% of nonischemic cortex, n =3, P < 0.05). Both hydroxyfasudil and Y-27632 (10 mg/kg, administered 60 mins before dMCAO) abolished this increase to a similar degree (119%±50% and 125%±62% of nonischemic cortex, n = 3 each).
Rho-kinase inhibitors can dilate cerebral vessels and improve CBF directly by reducing MLC phosphorylation, or indirectly via disinhibition of eNOS. To distinguish these two mechanisms, we tested hydroxyfasudil in eNOS−/− mice. Endothelial NOS knockout mice were hypertensive (n = 5; Table 1), and developed worse CBF deficit and larger area of ischemic cortex after dMCAO compared with wild-type controls (48% and 31% larger core and penumbra, respectively, P < 0.01; Figure 6). Hydroxyfasudil did not improve CBF in eNOS−/− mice (n = 5), suggesting that its cerebral hemodynamic effects are dependent on eNOS (Figure 6). Consistent with these data, a relatively specific eNOS inhibitor L-NIO expanded the area of core by 60% in wild-type mice, when administered 60 mins before dMCAO (n = 6; P < 0.01 versus vehicle; Figure 6); by contrast, administering the relatively nNOS selective inhibitor 7-nitroindazole (50 mg/kg, i.p.; peanut oil) did not significantly increase this area (P > 0.05, n = 6, not shown). Because genetic knockout of eNOS may cause compensatory changes in other potential molecular targets for hydroxyfasudil, we tested in a separate group of mice whether acute pharmacologic eNOS inhibition also prevents CBF improvement by hydroxyfasudil. Consistent with the results in eNOS−/− mice, hydroxyfasudil failed to improve CBF when administered together with l-NIO (area of cortex with ≤20% and 21% to 30% residual CBF, 5.2±0.2 and 5.8±0.7 mm2, respectively; P > 0.05 versus l-NIO alone, n = 4). When administered in eNOS−/− mice, l-NIO did not alter BP or expand the area of ischemic cortex, suggesting that its systemic and cerebrovascular effects are eNOS-specific in this model (n = 3, data not shown).
Figure 6.
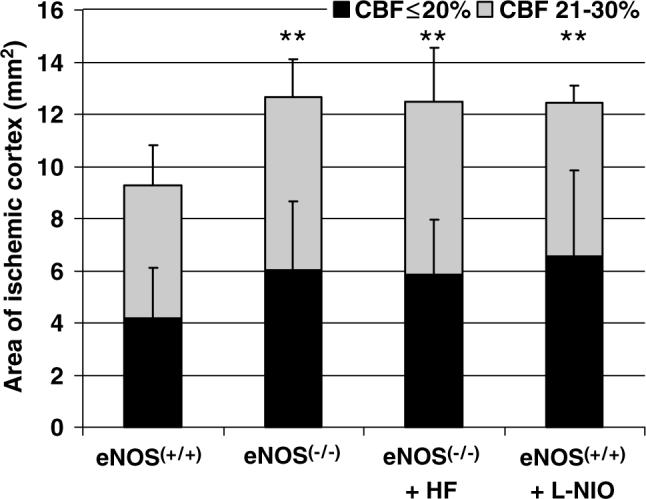
Hydroxyfasudil did not improve CBF deficit in eNOS knockout mice. Endothelial NOS knockout mice (eNOS−/−, n = 5) developed larger area of CBF deficits after dMCAO compared with wild-type controls (eNOS+/+ , n = 15). Hydroxyfasudil did not reduce the area of either severely (i.e., core with residual CBF ≤20%) or moderately (i.e., 21 to 30% residual CBF) hypoperfused cortex in eNOS knockouts (eNOS−/− + HF, n = 5). N5-(1-Iminoethyl)-l-ornithine (l-NIO, 20 mg/kg, i.p., n = 6), a relatively eNOS selective inhibitor, significantly enlarged the area of hypoperfused cortex (eNOS+/+ + l-NIO, n = 6), confirming the critical role of eNOS in maintaining CBF in ischemic cortex. **P < 0.01 versus control group, two-way ANOVA. Vertical bars indicate standard deviations.
To determine whether increased CBF by hydroxyfasudil during acute cerebral ischemia improved tissue outcome, we performed 1 h transient dMCAO and measured the infarct size at 48 h using 2,3, 5-triphenyltetrazolium chloride staining. Consistent with its CBF effect, hydroxyfasudil (10 mg/kg, i.p., 1 h before dMCAO) reduced infarct volume by 45% in this model (P < 0.05, n = 5 each in control and hydroxyfasudil groups; Figure 7).
Figure 7.
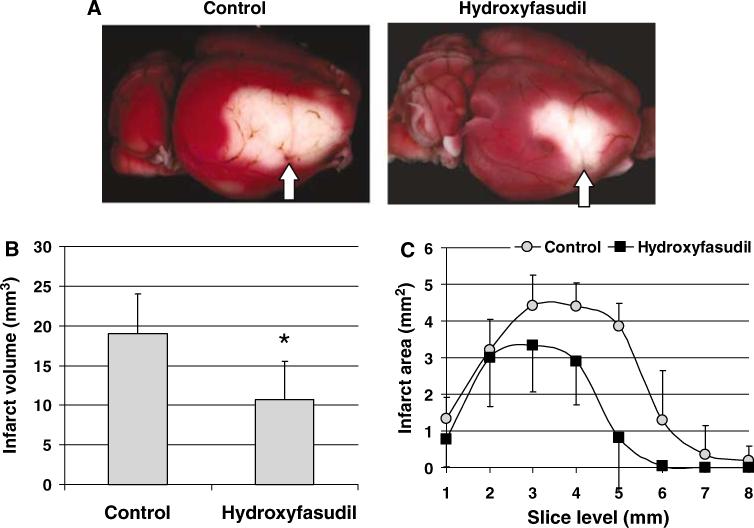
Hydroxyfasudil improved tissue outcome in wild-type mice. (A) Topical 2,3,5-triphenyltetrazolium chloride stained brains from representative control and hydroxyfasudil-treated (10 mg/kg, i.p., 1 h before dMCAO) mice demonstrating the infarct in the dorsolateral cortex. Arrow indicates the site of dMCAO using a microsurgical clip (see Methods). (B) Hydroxyfasudil reduced infarct volume by 45% when measured 48 h after 1 h transient dMCAO (*P < 0.05 versus control, t-test, n = 5 in each group). Infarct volume was calculated by integrating the infarct area in 1 mm thick coronal slices; because of the relatively small volume of infarct, only direct infarct volume was determined. (C) Graph showing the infarct area at each coronal slice level. Hydroxyfasudil reduced the infarct size mainly in the posterior cortical regions. Vertical bars indicate standard deviations of the mean.
Peri-infarct depolarizations negatively impact CBF in core and penumbra, and their suppression improves perfusion in focal ischemia (Shin et al, 2006). Therefore, we determined the frequency of peri-infarct depolarizations during dMCAO, as detected by the attendant CBF changes on LSF, and showed that neither their frequency nor associated CBF changes differed between groups (data not shown).
Discussion
Rho-kinase is a key regulator of endothelial and smooth muscle cell function and morphology, and is increasingly implicated in the pathophysiology of cardiovascular diseases such as hypertension, atherosclerosis, and vasospasm (Shimokawa 2002). Rho-kinase inhibitors are neuroprotective in experimental cerebral ischemia (Rikitake et al, 2005; Satoh et al, 2001; Toshima et al, 2000); however, it is not known whether this is due to a vasodilator effect on cerebral vessels leading to an acute augmentation of CBF in ischemic brain, or related to other mechanisms, such as a direct neuroprotective action, reduced blood viscosity (Hitomi et al, 2000), or inhibition of neutrophil accumulation (Satoh et al, 2001). In this study, we showed that two structurally unrelated Rho-kinase inhibitors, hydroxyfasudil (isoquinolinesulfonic acid derivative) and Y-27632 (pyridine derivative) (Uehata et al, 1997), acutely improve CBF in focal cerebral ischemic core and penumbra, and do so in an eNOS-dependent fashion. In vitro experiments on systemic and cerebral arteries showed that eNOS function is reversibly impaired under hypoxic conditions, and that Rho-kinase inhibition partially prevents this impairment, providing a mechanism by which Rho-kinase inhibitors may selectively increase CBF in acutely ischemic brain.
Several recent studies have suggested that hypoxia or ischemia may downregulate eNOS activity via Rho-kinase activation (Fagan et al, 2004; Takemoto et al, 2002), through both transcriptional and post-transcriptional mechanisms (Liao et al, 1995). Consistent with this, we found that hypoxia abolished eNOS-dependent ACh-induced relaxations in isolated systemic and cerebral arteries. This was not due to endothelial injury, since relaxations were completely restored on reoxygenation. The inhibition started within 30 mins and was complete after 60 mins of hypoxia, implicating post-translational mechanisms such as altered eNOS phosphorylation. Impaired eNOS-dependent relaxation by severe hypoxia has previously been reported for coronary arteries in response to bradykinin (Vedernikov et al, 1991). Consistent with the hypothesis that acute hypoxic downregulation of eNOS activity may occur because of Rho-kinase activation, hydroxyfasudil administration restored eNOS-dependent relaxations during severe hypoxia. The partial rather than complete restoration of eNOS-dependent relaxations may be because of incomplete Rho-kinase inhibition by a relatively low concentration of hydroxyfasudil (3 μmol/L) chosen to minimize the direct vasodilator effects of the drug. Overall, our data support the notion that Rho-kinase is acutely activated in hypoxic/ischemic vasculature, both in isolated vessels, in vitro, and in cerebral ischemia, in situ, and inhibits eNOS activity thereby worsening CBF.
In support of the hypothesis that hypoxia acutely upregulates Rho-kinase, cortical homogenates obtained from ischemic core 60 mins after dMCAO showed an almost three-fold increase in Rho-kinase activity, as determined by Thr853-MBS phosphorylation. These data are consistent with previous reports showing Rho-kinase upregulation 24 h after filament occlusion of MCA (Rikitake et al, 2005; Yagita et al, 2005), and suggest that this upregulation in ischemic cortex takes place acutely within 60 mins. Rho-kinase is widely expressed in most cell types in brain (Noma et al, 2006); therefore, measurement of Rho-kinase activity in cortical homogenates is not specific for vascular cells. However, Rho-kinase activity, as measured by adducin phosphorylation, was increased in penumbral microvessels after permanent MCAO (Yagita et al, 2005). Taken together, these results suggest that Rho-kinase is acutely upregulated in ischemic endothelium and worsens the CBF deficit.
Neuroprotection in cerebral ischemia was previously reported for fasudil (Hitomi et al, 2000; Satoh et al, 1999), hydroxyfasudil (Satoh et al, 2001), and Y-27632 (Rikitake et al, 2005; Satoh et al, 1996). For example, hydroxyfasudil (10 mg/kg) reduced infarct size and neurological deficits in rat microembolic ischemia when administered 5 mins after ischemia onset; however, intraischemic CBF reduction was not studied, and neuroprotection was in part attributed to the anti-inflammatory effects of hydroxyfasudil leading to a significant reduction in neutrophil infiltration (Satoh et al, 2001). In a model of endothelial injury and microvascular thrombosis induced by intracarotid injection of sodium laurate, fasudil (10 mg/kg) reduced infarct size and neurological deficits when administered 5 mins after ischemia onset and then repeated daily for 2 days; a lower dose of 1 mg/kg was ineffective, and CBF changes, again, were not reported (Toshima et al, 2000). Our laboratory has recently shown that Rho-kinase inhibitors fasudil and Y-27632 reduced infarct size in a transient MCA occlusion model in mice, when administered 10 mg/kg/day for 2 days before ischemia (Rikitake et al, 2005); this protection was associated with increased resting as well as intraischemic CBF in the treatment group. Our data are consistent with previous studies, and extend them by showing that Rho-kinase inhibitors augment CBF in both ischemic core and penumbra, and do that acutely within 60 mins, as demonstrated by LSF with high spatiotemporal resolution.
Rho-kinase inhibitors may augment CBF via their effects on endothelium and/or smooth muscle cells. In vascular smooth muscle, Rho-kinase modulates the sensitivity of contractile apparatus to intracellular Ca2+ by increasing MLC phosphorylation either directly or via phosphorylation and inhibition of myosin binding subunit of MLC phosphatase (MLCP) (Amano et al, 2000; Kimura et al, 1996; Shimokawa 2002); the degree of MLC phosphorylation determines the degree of vasoconstriction by augmenting actin–myosin interaction. In endothelial cells, Rho-kinase is a negative upstream regulator of eNOS via both transcriptional and post-transcriptional mechanisms, including changes in eNOS mRNA stability (Laufs and Liao 1998), subcellular translocation of eNOS because of reorganization of actin cytoskeleton, and eNOS phosphorylation at serine 1179 via phosphatidylinositol 3-kinase/Akt pathway (Ming et al, 2002). Therefore, Rho-kinase inhibition may cause vasodilation and acutely augment CBF in ischemic brain by at least two distinct mechanisms (i.e., direct inhibition of smooth muscle contraction and increased eNOS activity). We explored the relative contribution of these two mechanisms to the observed CBF improvement in ischemic core and penumbra by testing hydroxyfasudil in eNOS−/− mice, and showed that hydroxyfasudil did not augment CBF in eNOS−/− mice. These data indicate that eNOS plays an obligatory role in this response, and that the direct vascular smooth muscle relaxing effect of Rho-kinase inhibitors does not augment CBF in this model of focal ischemia. Furthermore, cerebral hemodynamic benefit of Rho-kinase inhibitors developed rapidly suggesting that nontranscriptional mechanisms are involved in acute upregulation of eNOS activity, such as increased eNOS S1179 phosphorylation (Fulton et al, 1999), which occurs within less than an hour in response to high-dose corticosteroids or statins (Amin-Hanjani et al, 2001; Endres et al, 1998).
The endothelial and smooth muscle mechanisms of vasodilation by Rho-kinase inhibitors appear to be differentially active in systemic and cerebral circulation. This is because hydroxyfasudil did not improve ischemic CBF in eNOS−/− mice, but still caused hypotension similar to wild type in magnitude, albeit more gradual in onset. Hence, direct smooth muscle relaxant effect of hydroxyfasudil decreased systemic, but not cerebrovascular resistance in focal ischemia. In light of the potential detrimental effect of systemic vasodilation and hypotension on cerebral perfusion in acute stroke, new Rho-kinase inhibitors with higher selectivity towards cerebral over systemic vasculature, perhaps by targeting endothelium (i.e., eNOS) rather than smooth muscle (i.e., MLC/MLCP), may be more efficacious in stroke therapy.
Hydroxyfasudil, the active metabolite of fasudil, has longer plasma elimination half-life than fasudil (Satoh et al, 2001), and is 50, 100, and 1000 times more selective towards Rho-kinase than protein kinases A and C, and MLCK, respectively (Rikitake et al, 2005). It is freely cell-permeable, and peak plasma concentrations are attained rapidly after both intravenous and intraperitoneal administration of the drug (Satoh et al, 2001), suggesting that pharmacokinetic properties of the drug are favorable for use in acute stroke therapy. Y-27632 is structurally unrelated to hydroxyfasudil, and also shows higher selectivity towards Rho-kinase than protein kinases A and C, as well as MLCK compared with fasudil (Uehata et al, 1997). Therefore, acute cerebral hemodynamic improvement by hydroxyfasudil and Y-27632 in focal ischemia is likely due to inhibition of Rho-kinase rather than other protein kinases.
Despite a significant CBF augmentation in ischemic cortex, Rho-kinase inhibitors did not increase resting CBF in nonischemic brain, when measured acutely for 1 h after a single dose. Indeed, resting CBF in nonischemic brain decreased by 15% and 25% by hydroxyfasudil and Y-27632, respectively. This appeared to correspond to their hypotensive effect (Figure 3), and probably accounted for the lower efficacy of Y-27632 in improving ischemic CBF deficit, which caused more severe and longer lasting hypotension compared with hydroxyfasudil. In contrast to our data, both fasudil and hydroxyfasudil have reportedly increased resting CBF within minutes by as much as 20% in canine cortex, although changes in BP were not provided in that study (Satoh et al, 2001). Rho-kinase inhibitors are potent hypotensive drugs (Shimokawa 2002; Takahara et al, 2003; Uehata et al, 1997), which may limit their use in high doses for the treatment of acute stroke. It is noteworthy, however, that the improvement in ischemic CBF by Rho-kinase inhibitors occurred despite a reduction in cerebral perfusion pressure because of hypotension, and without an effect on resting CBF, suggesting that Rho-kinase activity is selectively upregulated in ischemic vasculature, and worsens the CBF deficit.
In summary, our data suggest that the rapid nontranscriptional upregulation of eNOS activity by Rho-kinase inhibitors, shared by a number of other neuroprotective strategies including statins (Endres 2005) and high-dose corticosteroids (Limbourg et al, 2002), can acutely augment blood flow in cerebral ischemic core and penumbra. We speculate that Rho-kinase inhibitors may provide greater CBF benefit in acute stroke in the presence of pathologic activation of vascular Rho/Rho-kinase, such as in hyperlipidemia, diabetes, hypertension, and hyperhomocysteinemia (Rikitake and Liao 2005; Seko et al, 2003; Shah and Singh 2006; Zhu et al, 2003). Therefore, Rho-kinase inhibition may present a novel therapeutic opportunity during acute stroke.
Acknowledgments
This work was supported by the American Heart Association (0335519N, Ayata) and National Institutes of Health (P50 NS10828 and PO1 NS35611, Moskowitz; R01EB00790-01A2, Boas; RO1 HL052233, Liao).
References
- Amano M, Fukata Y, Kaibuchi K. Regulation and functions of Rho-associated kinase. Exp Cell Res. 2000;261:44–51. doi: 10.1006/excr.2000.5046. [DOI] [PubMed] [Google Scholar]
- Amin-Hanjani S, Stagliano NE, Yamada M, Huang PL, Liao JK, Moskowitz MA. Mevastatin, an HMG-CoA reductase inhibitor, reduces stroke damage and upregulates endothelial nitric oxide synthase in mice. Stroke. 2001;32:980–6. doi: 10.1161/01.str.32.4.980. [DOI] [PubMed] [Google Scholar]
- Arai M, Sasaki Y, Nozawa R. Inhibition by the protein kinase inhibitor HA1077 of the activation of NADPH oxidase in human neutrophils. Biochem Pharmacol. 1993;46:1487–90. doi: 10.1016/0006-2952(93)90116-e. [DOI] [PubMed] [Google Scholar]
- Ayata C, Dunn AK, Gursoy OY, Huang Z, Boas DA, Moskowitz MA. Laser speckle flowmetry for the study of cerebrovascular physiology in normal and ischemic mouse cortex. J Cereb Blood Flow Metab. 2004;24:744–55. doi: 10.1097/01.WCB.0000122745.72175.D5. [DOI] [PubMed] [Google Scholar]
- Bao W, Hu E, Tao L, Boyce R, Mirabile R, Thudium DT, Ma XL, Willette RN, Yue TL. Inhibition of Rho-kinase protects the heart against ischemia/reperfusion injury. Cardiovasc Res. 2004;61:548–58. doi: 10.1016/j.cardiores.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Endres M. Statins and stroke. J Cereb Blood Flow Metab. 2005;25:1093–110. doi: 10.1038/sj.jcbfm.9600116. [DOI] [PubMed] [Google Scholar]
- Endres M, Laufs U, Huang Z, Nakamura T, Huang P, Moskowitz MA, Liao JK. Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 1998;95:8880–5. doi: 10.1073/pnas.95.15.8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan KA, Oka M, Bauer NR, Gebb SA, Ivy DD, Morris KG, McMurtry IF. Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rho-kinase. Am J Physiol Lung Cell Mol Physiol. 2004;287:L656–64. doi: 10.1152/ajplung.00090.2003. [DOI] [PubMed] [Google Scholar]
- Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitomi A, Satoh S, Ikegaki I, Suzuki Y, Shibuya M, Asano T. Hemorheological abnormalities in experimental cerebral ischemia and effects of protein kinase inhibitor on blood fluidity. Life Sci. 2000;67:1929–39. doi: 10.1016/s0024-3205(00)00781-5. [DOI] [PubMed] [Google Scholar]
- Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–42. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, Moskowitz MA. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J Cereb Blood Flow Metab. 1996;16:981–7. doi: 10.1097/00004647-199609000-00023. [DOI] [PubMed] [Google Scholar]
- Kawano Y, Fukata Y, Oshiro N, Amano M, Nakamura T, Ito M, Matsumura F, Inagaki M, Kaibuchi K. Phosphorylation of myosin-binding subunit (MBS) of myosin phosphatase by Rho-kinase in vivo. J Cell Biol. 1999;147:1023–38. doi: 10.1083/jcb.147.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science. 1996;273:245–8. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem. 1998;273:24266–71. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- Liao JK, Zulueta JJ, Yu FS, Peng HB, Cote CG, Hassoun PM. Regulation of bovine endothelial constitutive nitric oxide synthase by oxygen. J Clin Invest. 1995;96:2661–6. doi: 10.1172/JCI118332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limbourg FP, Huang Z, Plumier JC, Simoncini T, Fujioka M, Tuckermann J, Schutz G, Moskowitz MA, Liao JK. Rapid nontranscriptional activation of endothelial nitric oxide synthase mediates increased cerebral blood flow and stroke protection by corticosteroids. J Clin Invest. 2002;110:1729–38. doi: 10.1172/JCI15481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming XF, Viswambharan H, Barandier C, Ruffieux J, Kaibuchi K, Rusconi S, Yang Z. Rho GTPase/Rho kinase negatively regulates endothelial nitric oxide synthase phosphorylation through the inhibition of protein kinase B/Akt in human endothelial cells. Mol Cell Biol. 2002;22:8467–77. doi: 10.1128/MCB.22.24.8467-8477.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma K, Oyama N, Liao JK. Physiological role of ROCKs in the cardiovascular system. Am J Physiol Cell Physiol. 2006;290:C661–8. doi: 10.1152/ajpcell.00459.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rikitake Y, Kim HH, Huang Z, Seto M, Yano K, Asano T, Moskowitz MA, Liao JK. Inhibition of Rho kinase (ROCK) leads to increased cerebral blood flow and stroke protection. Stroke. 2005;36:2251–7. doi: 10.1161/01.STR.0000181077.84981.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rikitake Y, Liao JK. Rho-kinase mediates hyperglycemia-induced plasminogen activator inhibitor-1 expression in vascular endothelial cells. Circulation. 2005;111:3261–8. doi: 10.1161/CIRCULATIONAHA.105.534024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh S, Ikegaki I, Suzuki Y, Asano T, Shibuya M, Hidaka H. Neuroprotective properties of a protein kinase inhibitor against ischaemia-induced neuronal damage in rats and gerbils. Br J Pharmacol. 1996;118:1592–6. doi: 10.1111/j.1476-5381.1996.tb15579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh S, Kobayashi T, Hitomi A, Ikegaki I, Suzuki Y, Shibuya M, Yoshida J, Asano T. Inhibition of neutrophil migration by a protein kinase inhibitor for the treatment of ischemic brain infarction. Jpn J Pharmacol. 1999;80:41–8. doi: 10.1254/jjp.80.41. [DOI] [PubMed] [Google Scholar]
- Satoh S, Utsunomiya T, Tsurui K, Kobayashi T, Ikegaki I, Sasaki Y, Asano T. Pharmacological profile of hydroxy fasudil as a selective rho kinase inhibitor on ischemic brain damage. Life Sci. 2001;69:1441–53. doi: 10.1016/s0024-3205(01)01229-2. [DOI] [PubMed] [Google Scholar]
- Seko T, Ito M, Kureishi Y, Okamoto R, Moriki N, Onishi K, Isaka N, Hartshorne DJ, Nakano T. Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. Circ Res. 2003;92:411–8. doi: 10.1161/01.RES.0000059987.90200.44. [DOI] [PubMed] [Google Scholar]
- Shah DI, Singh M. Involvement of Rho-kinase in experimental vascular endothelial dysfunction. Mol Cell Biochem. 2006;283:191–9. doi: 10.1007/s11010-006-2679-6. [DOI] [PubMed] [Google Scholar]
- Shimokawa H. Rho-kinase as a novel therapeutic target in treatment of cardiovascular diseases. J Cardiovasc Pharmacol. 2002;39:319–27. doi: 10.1097/00005344-200203000-00001. [DOI] [PubMed] [Google Scholar]
- Shin HK, Dunn AK, Jones PB, Boas DA, Moskowitz MA, Ayata C. Vasoconstrictive neurovascular coupling during focal ischemic depolarizations. J Cereb Blood Flow Metab. 2006;26:1018–30. doi: 10.1038/sj.jcbfm.9600252. [DOI] [PubMed] [Google Scholar]
- Takahara A, Sugiyama A, Satoh Y, Yoneyama M, Hashimoto K. Cardiovascular effects of Y-27632, a selective Rho-associated kinase inhibitor, assessed in the halothane-anesthetized canine model. Eur J Pharmacol. 2003;460:51–7. doi: 10.1016/s0014-2999(02)02929-1. [DOI] [PubMed] [Google Scholar]
- Takemoto M, Sun J, Hiroki J, Shimokawa H, Liao JK. Rho-kinase mediates hypoxia-induced downregulation of endothelial nitric oxide synthase. Circulation. 2002;106:57–62. doi: 10.1161/01.cir.0000020682.73694.ab. [DOI] [PubMed] [Google Scholar]
- Terzic A, Kurachi Y. Actin microfilament disrupters enhance K(ATP) channel opening in patches from guinea-pig cardiomyocytes. JPhysiol. 1996;492(Part 2):395–404. doi: 10.1113/jphysiol.1996.sp021316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toshima Y, Satoh S, Ikegaki I, Asano T. A new model of cerebral microthrombosis in rats and the neuroprotective effect of a Rho-kinase inhibitor. Stroke. 2000;31:2245–50. doi: 10.1161/01.str.31.9.2245. [DOI] [PubMed] [Google Scholar]
- Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, Narumiya S. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–4. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- Vedernikov YP, Graser T, Leisner H, Tiedt N. Effect of hypoxia on endothelium-dependent relaxation of porcine coronary arteries and veins. Biomed Biochim Acta. 1991;50:257–63. [PubMed] [Google Scholar]
- Wolfrum S, Dendorfer A, Rikitake Y, Stalker TJ, Gong Y, Scalia R, Dominiak P, Liao JK. Inhibition of Rho-kinase leads to rapid activation of phosphatidylinositol 3-kinase/protein kinase Akt and cardiovascular protection. Arterioscler Thromb Vasc Biol. 2004;24:1842–7. doi: 10.1161/01.ATV.0000142813.33538.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagita Y, Kitagawa K, Sasaki T, Sugiura S, Todo K, Omura-Matsuoka E, Matsushita K, Hori M. Activation of Rho/Rho kinase system after focal cerebral ischemia. J Cereb Blood Flow Metab. 2005;25:S264. [Google Scholar]
- Zhang F, White JG, Iadecola C. Nitric oxide donors increase blood flow and reduce brain damage in focal ischemia: evidence that nitric oxide is beneficial in the early stages of cerebral ischemia. J Cereb Blood Flow Metab. 1994;14:217–26. doi: 10.1038/jcbfm.1994.28. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Liao HL, Niu XL, Yuan Y, Lin T, Verna L, Stemerman MB. Low density lipoprotein induces eNOS translocation to membrane caveolae: the role of RhoA activation and stress fiber formation. Biochim Biophys Acta. 2003;1635:117–26. doi: 10.1016/j.bbalip.2003.10.011. [DOI] [PubMed] [Google Scholar]