

Abstract
A significant number of patients infected with human immunodeficiency virus-1 (HIV-1) suffer cognitive impairment ranging from mild to severe HIV-associated dementia (HAD), a result of neuronal degeneration in the basal ganglia, cerebral cortex and hippocampus. Mononuclear phagocyte dysfunction is thought to play an important role in the pathogenesis of HAD. Glutamate neurotoxicity is triggered primarily by massive Ca2+ influx arising from over-stimulation of the NMDA subtype of glutamate receptors. The underlying mechanisms, however, remain elusive. We have tested the hypothesis that mitochondrial glutaminase in HIV-infected macrophages is involved in converting glutamine to glutamate. Our results demonstrate that the concentration of glutamate in HIV-1 infected conditioned media was dependent on glutamine dose, and HIV-1 infected conditioned medium mediated glutamine-dependent neurotoxicity. These results indicate HIV-infection mediates neurotoxicity through glutamate production. In addition, glutamate-mediated neurotoxicity correlated with caspase activation and neuronal cell cycle re-activation. Inhibition of mitochondrial glutaminase diminished the HIV-induced glutamate production, and attenuated NMDA over-stimulation and subsequent neuronal apoptosis. These data implicate mitochondrial glutaminase in the induction of glutamate-mediated neuronal apoptosis during HIV-associated dementia, and provides a possible therapeutic strategy for HAD treatment.
Keywords: apoptosis, glutamate, glutaminase, HIV-1-associated dementia, macrophages
Human immunodeficiency virus, the agent responsible for acquired immunodeficiency syndrome (AIDS) also induces a neurological disease culminating in frank dementia (Kaul et al. 2005). HIV-associated dementia (HAD) affects 7–15% of AIDS patients and is a consequence of neuronal damage and dysfunction. How HIV infection mediates neuronal degeneration remains a controversial topic. The neuropathological hallmark of HIV-1 brain infection is HIV-encephalitis (HIVE), and is associated with neuronal dendritic damage, apoptosis, and necrosis, all of which are thought to result from release of neurotoxins from HIV-infected macrophages and other activated glial cells (Dawson et al. 1993; Gelbard et al. 1995; Petito and Roberts 1995; Adamson et al. 1996; Kaul et al. 2001). Neurotoxins present in the HIV-encephalitic brain include viral proteins, such as gp120, gp41 and Tat, and host inflammatory products including platelet-activating factor, arachidonic acid and its metabolites, proinflammatory cytokines and glutamate.
Glutamate is the predominant excitatory neurotransmitter in the mammalian CNS, mediating numerous physiological functions through activation of multiple receptors. HIV-1-infected macrophages have been shown to be an important cellular source of extracellular glutamate and glutamate concentrations in the CSF of HIV-1 infected patients are higher as compared to uninfected controls (Droge et al. 1987; Ollenschlager et al. 1988; Espey et al. 1999; Ferrarese et al. 2001; Jiang et al. 2001b; Zhao et al. 2004; Erdmann et al. 2007). Production of excess glutamate by HIV-infected macrophages in HAD may contribute to neuronal cell death, but how HIV-1 infection influences glutamate production and neuronal cell death remains unclear.
Proper regulation of glutamate is critical to normal synaptic function and CNS homeostasis. Phosphate-activated glutaminase (PAG) is a mitochondrial enzyme that catalyzes the deamination of glutamine to glutamate and ammonia (Ward et al. 1983; Nicklas et al. 1987; Wurdig and Kugler 1991; Curthoys and Watford 1995; Zhao et al. 2004). Glutamine provides an abundant substrate in vivo and PAG is the predominant glutamine-utilizing enzyme of the brain. Our previous work demonstrated overproduction of extracellular glutamate in HIV-1-infected macrophages (Zhao et al. 2004), and inhibition of mitochondrial glutaminase using novel small molecule inhibitors or siRNA decreased glutamate production in vitro (Erdmann et al. 2007). In this report, we propose mitochondrial glutaminase contributes to the neuronal toxicity typical of HIV infection of the CNS. To investigate the effects of mitochondrial glutaminase activity on glutamate generation and glutamate-mediated neurotoxicity during HIV-1 infection, we examined the glutamate concentration in monocyte-derived macrophages (MDM) conditioned media (MCM) with or without HIV-1 infection, and determined the effect of glutaminase inhibition on glutamate-mediated neuronal apoptosis. We found HIV-1 infection increases glutamate generation by human macrophages and potentiates neuronal apoptosis. In addition, glutamate-mediated neurotoxicity correlated with caspase activation and neuronal cell cycle re-activation and novel inhibitors of mitochondrial glutaminase efficiently blocked the increase in glutamate, attenuating neuronal apoptosis. These results indicate mitochondrial glutaminase plays a role in HIV-infected MDM-mediated neuronal apoptosis, and identifies a possible therapeutic strategy for HAD treatment.
Materials and methods
Isolation and culture of primary monocytes
Human monocytes were recovered from peripheral blood mononuclear cells of HIV-1, -2 and hepatitis B seronegative donors after leukopheresis, and then purified by counter-current centrifugal elutriation. Monocytes were cultured as adherent mono-layers (1.1 × 106 cells/well in 24-well plates) and differentiated for 7 days in Dulbecco’s modified Eagle’s medium (GIBCO, Invitrogen Co., Carlsbad, CA, USA) supplemented with 10% heat-inactivated pooled human serum, 50 μg/mL gentamicin (Sigma-Aldrich, St. Louis, MO, USA), 10 μg/mL ciprofloxacin (Sigma), and macrophage colony stimulating factor (1000 U/mL highly purified recombinant human MCSF; a generous gift from Wyeth Pharmaceutical Inc., Cambridge, MA, USA). All tissue reagents were screened and found negative for endotoxin (< 10 pg/mL; Associates of Cape Cod Inc., Woods Hole, MA, USA) and mycoplasma contamination (Gen-probe II; Gen-probe Inc., San Diego, CA, USA).
Infection of monocyte-derived macrophages and collection of MDM conditioned media
Seven days after plating, MDM were infected with HIV-1ADA at a multiplicity of infection of 0.1 virus/target cell. Viral stocks were screened for mycoplasma and endotoxin using hybridization and limulus amebocyte lysate assays, respectively. In some experiments, cells were treated at 7 days after HIV-1 infection in Neurobasal™ medium (GIBCO, Invitrogen Co.) supplemented with 0.01% bovine serum albumin, 25 μg/mL penicillin–streptomycin with or without the addition of 5 mmol/L glutamine for an additional 24 h at 37°C prior to collection of supernatants. Cells were washed with Dulbecco’s modified Eagle’s medium before Neurobasal™ medium was added, for 24 h incubation. MCM is then harvested, centrifuged at 1000 g to remove dead cells and debris, and was then filtered through 3 KD CENTRIPLUS® Centrifugal Filter Devices (YM-3, Millipore Corporation, Bedford, MA, USA) by centrifugation at 4°C, 3000 g. The flow-through was then collected for analysis and treatment.
Isolation and culture of rat cortical neurons
Primary cortical neurons were prepared from rat cortices as previously described (Zheng et al. 2001). Briefly, rat cortex was dissected from embryonic day 17 Sprague–Dawley rat fetuses. Individual cells were mechanically dissociated by trituration in Neurobasal™ medium without additives. Tissue was digested with 0.1% trypsin at 37°C for 30 min, then reaction was neutralized with 10% fetal bovine serum on ice for 15 min and the cell suspension was washed three times in Hank’s Buffered Salt Solution. Cells were resuspended in Neurobasal™ medium containing B27 supplement, 0.5 mmol/L glutamine, 25 μg/mL penicillin–streptomycin, and filtered through 70 μm sterile nylon. Neurons were plated onto poly-d-lysine-coated six-well plates at a density of 1.5 × 106/well. Cultured cells were deemed mature 7–12 days after plating.
MTT reduction assay
Cell viability was assessed by methylthiazolyldiphenyl-tetrazolium bromide (MTT) reduction as described previously (Jiang et al. 2001a; Zhao et al. 2004). Cells were incubated with 200 μL of 10% MTT solution in Neurobasal media for 1 h at 37°C. The extent of MTT conversion to formazan by mitochondrial dehydrogenase, indicating cell viability, was determined by measuring absorbance at 490 nm using a microplate reader.
Cell cycle assay
Cell cycle was examined by propidium iodide (PI, Sigma–Aldrich Inc.) staining (Liao et al. 2003). In brief, 5 × 106 cells were harvested and washed twice with Ca2+/Mg2+ free phosphatebuffered saline (PBS), fixed overnight in 70% cold ethanol, digested with RNase A (Sigma–Aldrich Inc.) and stained with PI (100 μg/mL). Data were obtained and analyzed by flow cytometry using the CellQuest software on a FACScan (BD, Franklin Lakes, NJ, USA) from a cell population from which debris was gated out.
Detection of caspase activation
Detection of caspase activation was performed using the “CaspACE™ FITC-VAD-FMK In Situ Marker” (Promega, San Luis Obispo, CA, USA, Cat. G7461) as described previously (Guaragnella et al. 2006) with minor modification. Briefly, 1 × 106 cells were washed in PBS, re-suspended in 100 μL staining solution containing 10 μmol/L of the fluorescein isothiocyanate conjugate of z-VAD-fmk (FITC-VAD-fmk) and incubated for 20 min at RT in the dark. Cells were then washed once and re-suspended in PBS. Flow cytometric analysis was performed using an Epics® XLMCL™ (Beckman Coulter, Fullerton, CA, USA) flow cytometer. Twenty thousand events were acquired for each analysis. Data were analyzed using WinMDI 2.8 software.
Intracellular calcium measurements
Cells cultured on glass coverslips were loaded with 7.1 μmol/L fura-2AM for 30 min at 37°C in Ringer’s solution of the following composition:145 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgCl2, 10 mmol/L HEPES, 2 mmol/L CaCl2, and 10 mmol/L d-glucose, pH 7.4. Cells were then washed twice and incubated again for 20 min in Ringer’s solution to allow for intracellular dye cleavage. The coverslips were inserted into the chamber and fura-2 was excited at wavelengths of 340 and 380 nm using a PTI Deltascan System as previously described (Munsch and Deitmer 1995; Zheng et al. 1999).
SDS–PAGE and immunoblotting
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis and immunoblotting were performed as described (Chen et al. 2003). Briefly, the cells were resuspended in NP-40 lysis buffer (10 mmol/L HEPES (pH 7.4), 2 mmol/L EGTA, 0.5% NP-40, 1 mmol/L NaF, 1 mmol/L NaVO4, 1 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L dithiothreitol, 50 mg/mL trypsin inhibitor, 10 mg/mL aprotinin and leupeptin) and placed on ice for 30 min. The lysates were centrifuged at 12 000 g for 10 min at 4°C, and then protein concentration was determined with BCA™ Protein Assay Kit (Pierce, Rockford, IL, USA Prod# 23225). Equivalent samples (30 μg protein) were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis on 12% gel. The proteins were then transferred onto nitrocellulose membranes, and probed with caspase-3 (Cell Signaling, Danvers, MA, USA pAb, #9661), caspase-9 (Cell Signaling, mAb, #9508) and β-actin (Sigma, mAb, A5441) antibodies, separately, followed by the appropriate secondary antibodies conjugated to horseradish peroxidase (KPL, Gaithersburg, MD, USA). Immunoreactive bands were visualized using enhanced chemiluminescence (Pierce). The molecular sizes of the developed proteins were determined by comparison with pre-stained protein markers (Invitrogen).
Analyses of glutamate in conditioned media by RP-HPLC
RP-HPLC analysis was performed using an HP series II 1090 liquid chromatograph and HP1046A fluorescence detector (Hewlett Packard, Palo Alto, CA, USA) as described previously (Zhao et al. 2004). In brief, 250 μL of sample was mixed with equal volumes of 3% perchloric acid (Sigma-Aldrich, Milwaukee, WI, USA), and then immediately neutralized with 11.5 μL saturated potassium carbonate (Sigma-Aldrich). Samples were centrifuged at 12 000 g for 15 min at 4°C, and then injected into an RP-HPLC system. Glutamate detection was monitored using a fluorescence detector with wavelengths of excitation at 340 nm and emission at 450 nm.
Statistical analysis
Data were analyzed as mean ± standard deviation (SD). The data were evaluated statistically by the analysis of variance (anova), followed by the student’s t-test for paired observations. Significance was determined as p < 0.05 and p < 0.01.
Results
HIV-1 infected macrophages increase glutamate production in a glutamine-dependant manner
We first examined the dose-effect of glutamine on glutamate production in HIV-1 infected MDM. Seven days postinfection, culture medium was removed and fresh neurobasal medium containing 0 to 25 mmol/L glutamine was added to MDM culture for overnight incubation. Supernatants were collected from control and HIV-infected cultures and the total concentration of glutamate was measured by RP-HPLC (Fig. 1a). Our results demonstrated that following the addition of glutamine, uninfected MDM showed a nominal dose-dependent increase in glutamate production. However, HIV-1-infected MDM showed a dramatic increase in glutamate production, an effect dependent on the concentration of glutamine (0–5 mmol/L). When statistically analyzed, a negative correlation was found between glutamine and glutamate concentrations in supernatants taken from uninfected and HIV-1-infected MDM (r2 = 0.02353, p = 0.0011) (Fig. 1b), suggesting HIV-1-mediated glutamate production is dependent on the presence of glutamine.
Fig. 1.
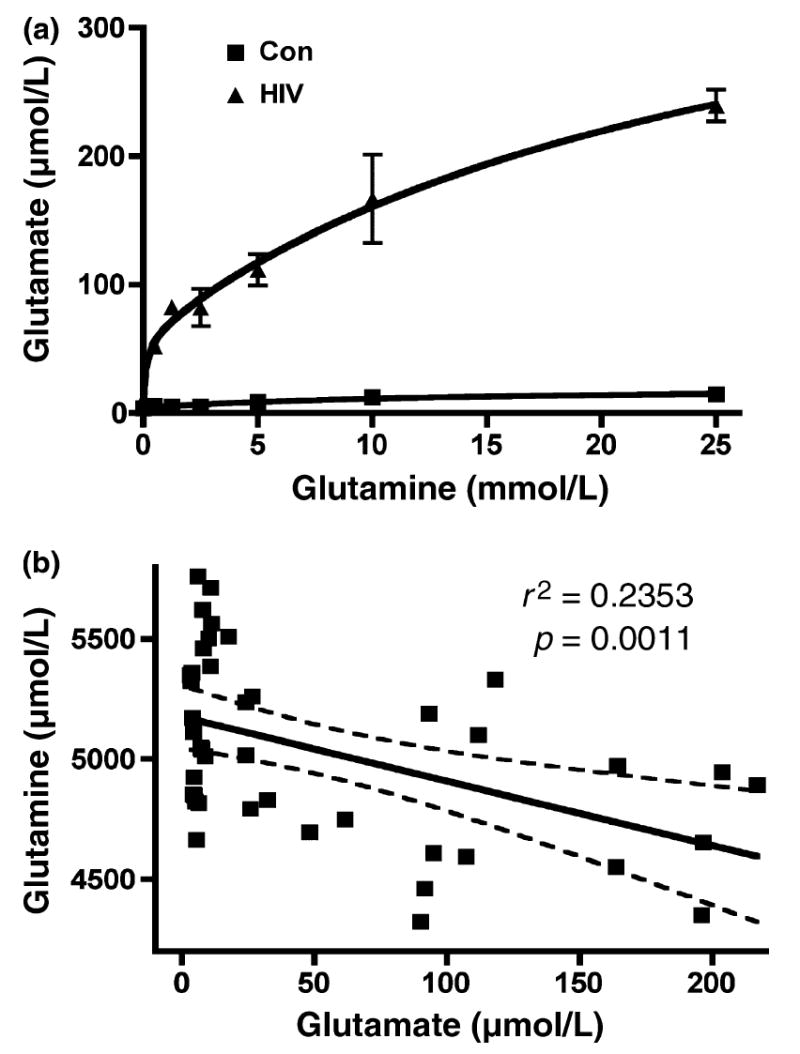
HIV-infected macrophage produce higher levels of glutamate, and glutamate production is dependent on glutamine addition. (a) HIV-1-mediated glutamate production is dependent on the addition of glutamine. Human MDM were infected with HIV-1ADA for 7 days. Cells were washed three times and incubated for 48 h in serum-free neurobasal media with 0–25 mmol/L glutamine. Samples were stabilized with 3% perchloric acid and potassium carbonate treatment. The concentration of glutamate in cell-free supernatants was determined by RP-HPLC. All data are expressed as absolute concentration of glutamate (μmol/L). Results are expressed as average ± SD of triplicate samples and are representative of three independent experiments. (b) Glutamate up-regulation is correlated with glutamine down-regulation in HIV infected MCM. Human MDM from seven different donors were infected with HIV-1ADA for 7 days. Cells were washed three times and incubated for 24 h in serum-free neurobasal media with 5 mmol/L glutamine. Samples were stabilized with 3% perchloric acid and potassium carbonate treatment. The concentrations of glutamate and glutamine in both control and HIV samples were determined by RP-HPLC. The correlation between glutamine and glutamate was analyzed.
HIV-1-mediated neurotoxicity is dependent on glutamine and NMDA over-stimulation
We investigated the effects of MCM from HIV-infected MDM on rat cortical neurons (RCN). Control and HIVinfected macrophages were treated with neurobasal medium containing glutamine free or 5 mmol/L glutamine and then conditioned media were collected. Neuron cultures were treated with MCM and cell viability was then measured by MTT assay (Fig. 2). No obvious neurotoxic effect was observed in RCN treated with glutamine-free MCM collected from both uninfected (No detection, ND) and HIV-infected (10.26 μmol/L glutamate) MDM. RCN treated with conditioned media from uninfected MDM treated with 5 mmol/L glutamine (1.62 μmol/L glutamate) likewise showed little toxicity. However, MCM from HIV-1 infected MDM treated with 5 mmol/L glutamine (37.16 μmol/L glutamate) induced a significant neurotoxic effect on RCN. These data suggest HIV-1 infected MDM treated with glutamine have increased glutamate and enhanced neurotoxicity towards rat cortical neurons.
Fig. 2.
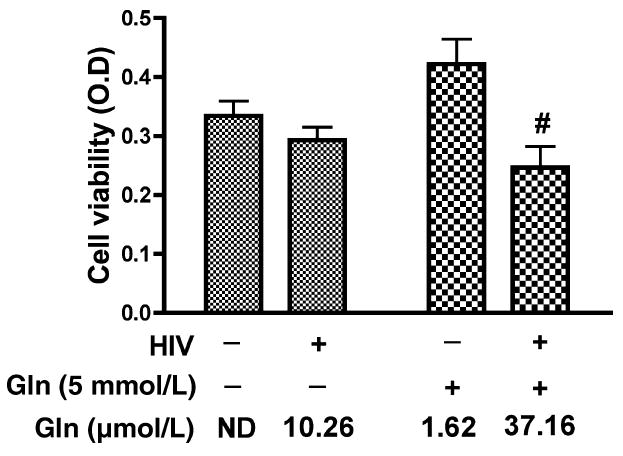
Neurotoxic effect of HIV-infected MDM conditioned media on RCN is dependent on the addition of glutamine and the production of glutamate. Human MDM were infected with HIV-1ADA for 7 days. Cells were washed three times and incubated for 24 h in serum-free neurobasal media with and without 5 mmol/L glutamine. Samples were stabilized with 3% perchloric acid and potassium carbonate treatment. The concentration of glutamate in cell-free supernatants was determined by RP-HPLC. All data are expressed as absolute concentration of glutamate (μmol/L) in MCM. Results shown are the mean of triplicate samples. Rat cortical neurons were treated with unfiltered MCM (Con Gln free, HIV Gln free, Con 5 mmol/L Gln and HIV 5 mmol/L Gln) and neuron viability was measured by MTT assay. HIV-infected MCM with 5 mmol/L glutamine was significantly neurotoxic as compared to control MCM with 5 mmol/L glutamine and HIV-MCM without 5 mmol/ L glutamine (#denotes p < 0.05). Glutamine free MCM with low concentration of glutamate showed no toxicity even in the HIV infected MCM.
Glutamate neurotoxicity is triggered primarily by massive Ca2+ influx arising from over-stimulation of the NMDA subtype of glutamate receptors, and NMDA receptor antagonists block most excitotoxic effects of glutamate (Choi et al. 1988; Li et al. 1995). N-methyl-d-aspartate receptors (NMDAR) are tetra-heteromeric structures permeable to sodium, potassium, zinc, and calcium (Hollmann and Heinemann 1994). The findings in Fig. 2, in addition to previous works (Jiang et al. 2001) using HIV-infected MDM established excitotoxicity as the primary mediator of decreased viability in MCM-treated rat cortical neurons. To demonstrate that MCM-mediated neurotoxicity is through over-stimulation of NMDA receptors, we examined calcium influx in RCN. To conduct this and the following experiments, MCM was filtered to remove infectious particles. RCN were loaded with Fura-2 and monitored for calcium influx by micro-fluorescent imaging. Our results found that MCM from both control and HIV-infected MDM treated with 0 mmol/L glutamine induced minimal calcium responses. However, MCM from HIV-infected MDM treated with 5 mmol/L glutamine showed significantly higher levels of calcium influx relative to MCM from control MDM (Fig. 3a and b, p < 0.001). To further confirm that the neurotoxicity seen in RCN treated with MCM was caused by over-stimulation of NMDAR, we pre-incubated RCN with a selective NMDA receptor antagonist, MK801 (10 μmol/L), before MCM treatment of RCN and then detected cell viability. The results showed the addition of MK801 blocked the neurotoxicity (Fig. 3c), suggesting NMDA receptor over-stimulation is involved in HIV-infected MCM mediated neurotoxicity.
Fig. 3.
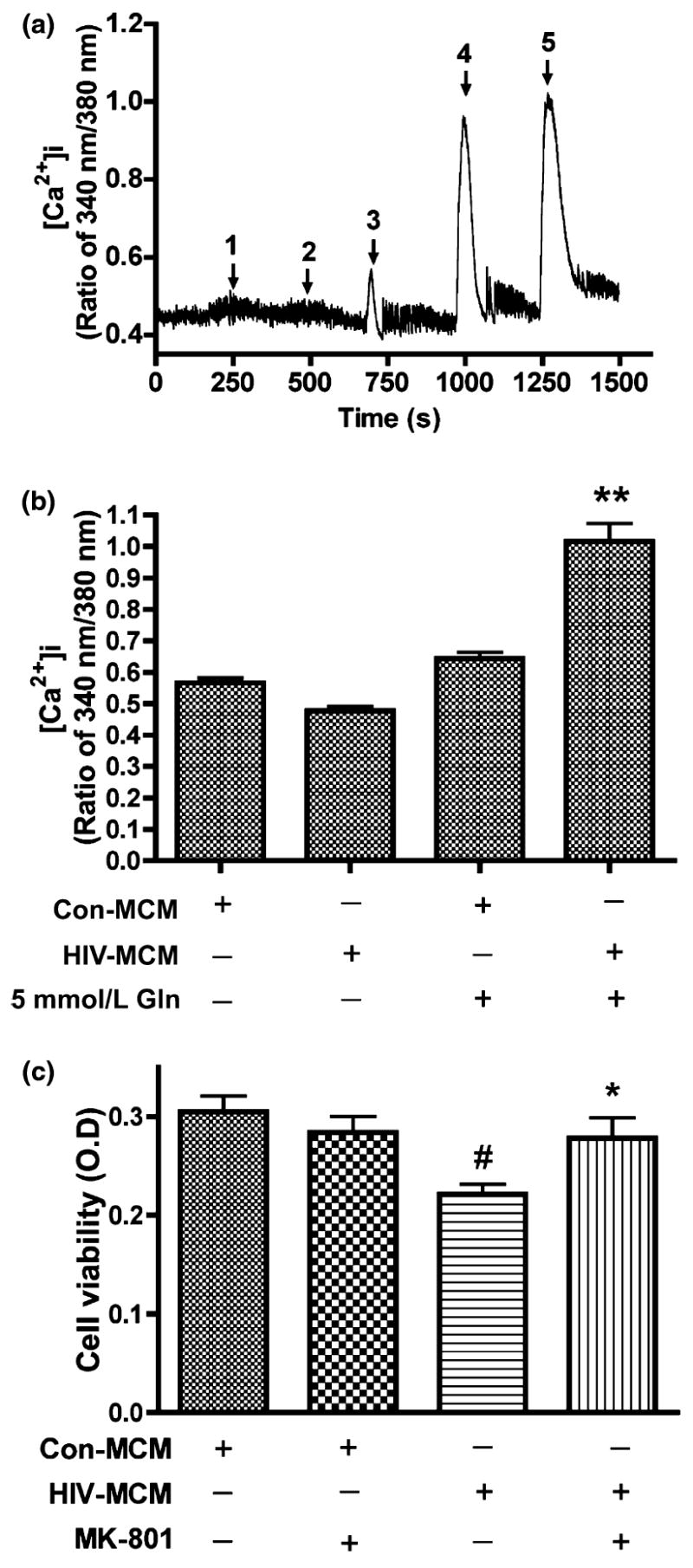
HIV-MCM mediates neurotoxicity through over-stimulation of NMDA Receptors and NMDAR antagonist (MK801) blocks HIV-MCM-mediated neurotoxicity. Rat cortical neurons were loaded with Fura-2 and monitored for calcium influx by micro-fluorescent imaging. Filtered control MCM and HIV-1 MCM (including Glutamine free or 5 mmol/L glutamine) were tested to see the responses of NMDA receptor (a), 1: Con Gln free MCM; 2: HIV Gln free MCM; 3: Con 5 mmol/L Gln MCM; 4: HIV 5 mmol/L Gln MCM; 5: 100 μmol/L Glu. Panel B expressed as mean ± SD. Data represent five independent experiments. **denotes p < 0.001 in comparison to control MCM with 5 mmol/L glutamine and HIV-MCM without 5 mmol/L glutamine. (c) NMDA receptor antagonist MK801 blocks the neurotoxic effect of HIV-1 infected MCM. Rat cortical neurons were pre-treated with 10 μmol/L MK801 for 15 min before MCM treatment. HIV-MCM induced significant neuronal death and this kind of neurotoxic effect was blocked by MK801. The results are expressed as mean ± SD. #denotes p < 0.05 compared with related control MCM; *denotes p < 0.01 in comparison with HIV-MCM.
Inhibition of phosphate-activated glutaminase blocks glutamate production and prevents over-stimulation of NMDA receptors
We previously suggested that inhibition of glutaminase activity decreases glutamate production by HIV-infected MDM using glutaminase small-molecule inhibitors and glutaminase-specific siRNA (Erdmann et al. 2007). To further describe the role of mitochondrial glutaminase in HIV-1 infection-mediated glutamate production and subsequent glutamate-mediated calcium influx, we tested whether a glutaminase inhibitor (Property of MGI Pharma) blocks neurotoxicity by HIV-1 infected human MDM. 19560 is a small-molecule glutaminase inhibitor designed to specifically inhibit glutaminase activity (Erdmann et al. 2007). Inhibitor 19560 was added at a concentration of 10 μmol/L to MDM culture for overnight incubation before conditioned media was collected and glutamate concentration was determined by RP-HPLC. Our results indicate glutamate in MCM from HIV-infected MDM without 19560 increased about fourfold as compared to uninfected MDM conditioned media. However, in the presence of inhibitor 19560, glutamate production was significantly reduced in infected macrophages and comparable with control glutamate levels (Fig. 4a). Subsequently, we analyzed the Ca2+ response of RCN to MCM, and found the inhibition of glutaminase activity significantly attenuated Ca2+ influx (Fig. 4b and c). These data suggest mitochondrial glutaminase is essential to glutamate-mediated over-stimulation of NMDA receptors and neurotoxicity.
Fig. 4.
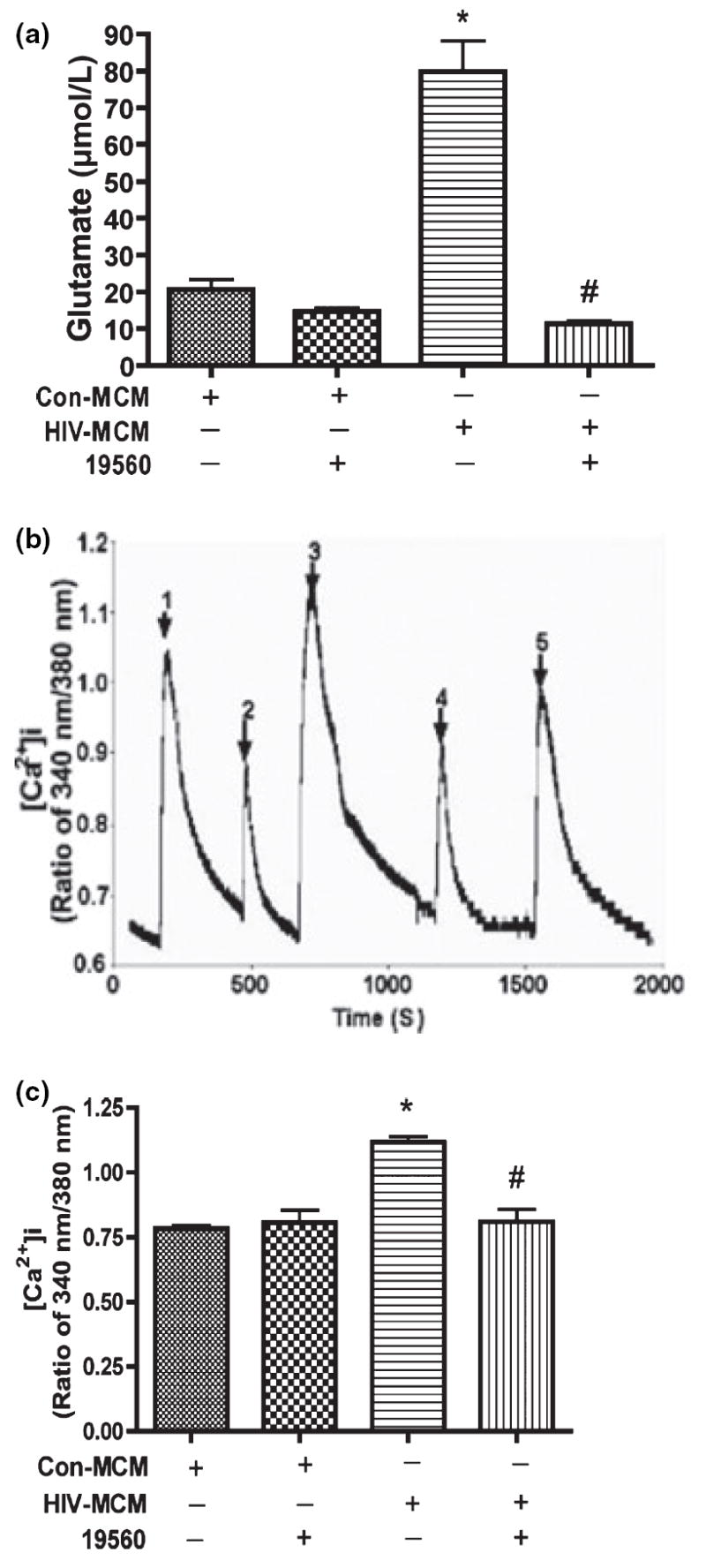
The inhibition of glutaminase activity decreases glutamate production and prevents over-stimulation of NMDA receptors by glutamate. Human MDM were infected with HIV-1ADA for 7 days. Cells were washed three times and incubated for 24 h in serum-free neurobasal media which contains 5 mmol/L glutamine and 10 μmol/L glutaminase inhibitor (19560) as indicated in Fig. 4a. Samples were stabilized with 3% perchloric acid and potassium carbonate treatment. The concentrations of glutamate were determined by RP-HPLC. All data are expressed as absolute concentration of glutamate (μmol/L) in MCM, and results showed are the mean of triplicate samples, *denotes p < 0.001 in comparison to control; #denotes p < 0.001 in comparison with HIV-MCM. Rat cortical neurons were loaded with Fura-2 and monitored for calcium influx by micro-fluorescent imaging. (b) Filtered control and HIV-1 MCM which contains glutaminase inhibitors (19560 and 20638) were tested to see the responses of NMDA receptor, 1: Con-MCM; 2: Con-MCM+19560; 3: HIV-MCM; 4: HIV-MCM+19560; 5: HIV-MCM+20638. (c) The graph showed the statistical analysis results, and these results are expressed as average ±SD and are representative of three independent experiments, *denotes p < 0.01 in comparison to control; #denotes p < 0.01 in comparison with HIV-MCM.
Cell cycle re-activation is involved in HIV-1-mediated caspase activation and neuronal apoptosis
We treated RCN with 100 μmol/L glutamate at different time points, and then analyzed the cell cycle by flow cytometry (Fig. 5a–c). The cell cycle data demonstrate that RCN treated with 100 μmol/L glutamate for 48 h have a typical subG1 peak (Fig. 5a-vi), indicative of an apoptotic response. NMDA receptor antagonist MK801 blocked the glutamateinduced cell cycle changes (Fig. 5a-vii). RCN treated with 100 μmol/L glutamate for 6 or 24 h lacked an obvious subG1 peak (Fig. 5a-i–v), indicating neurons initiate apoptosis after 24 h treatment with glutamate. The statistical results are shown in Fig. 5c.
Fig. 5.
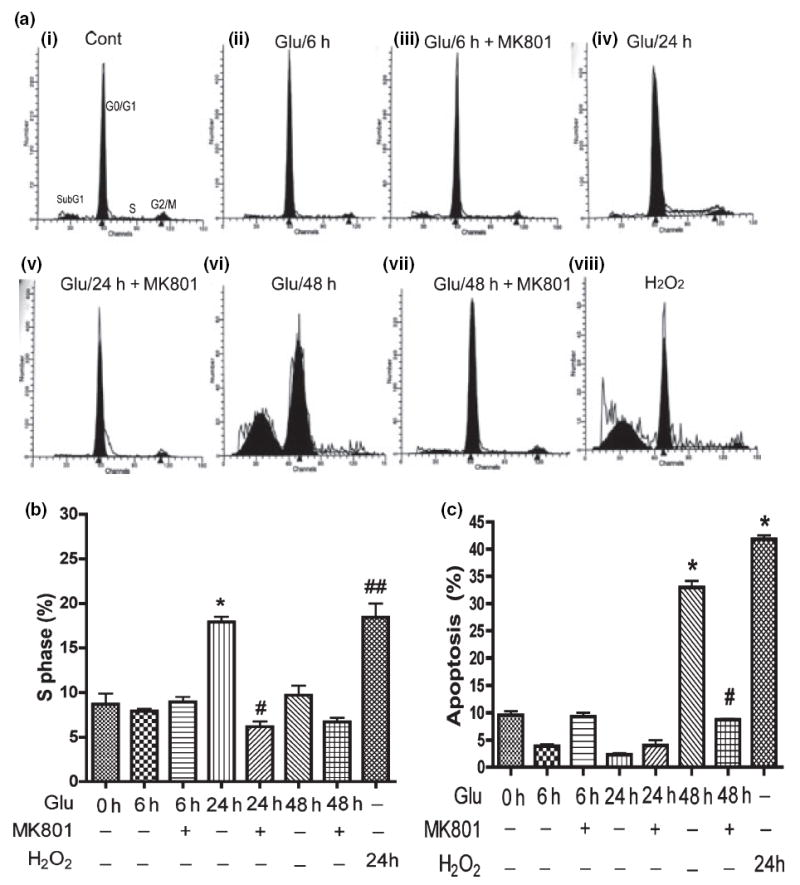
Glutamate induces neuronal cell cycle re-activation and neuronal apoptosis and is blocked by NMDA receptor antagonist (MK801). Rat neurons were pre-treated with NMDA receptor antagonist (10 μmol/L MK801) for 15 min, then treated with 100 μmol/L glutamate for different times before cell cycle assays were performed. (a) Representative cell cycle assay showing sub G1 population in rat neurons after 48 h treatment. MK801 is NMDA receptor-specific antagonist; hydrogen peroxide (H2O2) as a positive control. (b) The population of S phase in cell cycle was analyzed by Flow Cytometry. The results are expressed as average ±SD of triplicate samples and are representative of three independent experiments. H2O2 as a positive control, *denotes p < 0.001 in comparison with control; #denotes p < 0.001 in comparison to Glu 24 h; ##denotes p < 0.001 compared with control. (c) The results are expressed as average ±SD of triplicate samples and are representative of three independent experiments, H2O2 treatment (24 h) as positive control, *denotes p < 0.001 in comparison to control; #denotes p < 0.001 in comparison with Glu 48 h.
Recently, cell cycle re-activation in apoptotic neuronal cell death has been reported during development and disease states (Becker and Bonni 2004). To explore whether HIV-mediated neuronal apoptosis is associated with cell cycle re-entry, RCN were treated with glutamate with or without MK801 preincubation (Fig. 5b). Cells in S phase have initiated DNA synthesis indicating cell cycle re-entry. Our results demonstrated that the population of cells in S phase after glutamate treatment for 24 h significantly increased, and MK801blocked the transition from G0/G1 to S phase. However, 48 h treatment with glutamate had no obvious increase in the S phase population (Fig. 5b). These data indicate glutamate-treated neurons initiated apoptosis after the transition from G0/G1 to S phase in response to glutamate stimulation.
The ability of caspase inhibitors to block neuronal cell death induced by trophic factor deprivation and other cytotoxic conditions has provided solid evidence for the role of caspases in neuronal cell death (Cryns and Yuan 1998; Yuan and Yankner 2000). We assayed caspase activity in treated RCN by flow cytometry and western blotting. Our results demonstrated caspase activity significantly increased as compared to control cells after 24 h glutamate treatment (Fig. 6a), specifically caspase-9 and caspase-3 (Fig. 6b), and MK801 blocked caspase activation (Fig. 6a and b). These results suggest over-stimulation of NMDA receptors by glutamate promotes cell cycle reactivation, activates the caspase cascade resulting in neuronal apoptosis.
Fig. 6.
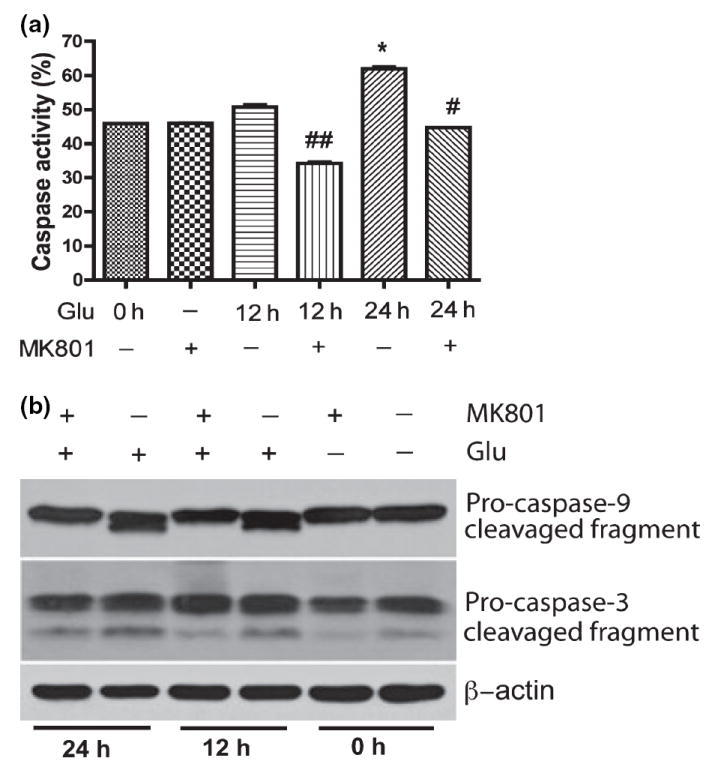
Caspase activation was involved in glutamate-mediated neuronal apoptosis. Rat neurons were treated with 100 μmol/L glutamate and/or 10 lmol/L MK801 as indicated in (a), and then incubated with FITC-VAD-fmk, a FITC-conjugated inhibitory substrate of caspases, and analyzed by Flow Cytometry. The results are expressed as average ±SD of triplicate samples, *denotes p < 0.001 in comparison to control; #denotes p < 0.05 in comparison with glutamate treatment (24 h); ##denotes p < 0.05 in comparison to Glu 12 h. (b) Rat neurons were collected at the indicated times after being treated with 100 μmol/L Glutamate and/or pretreated with 10 μmol/L for 30 min, and were analyzed by western blotting for caspase-9 and caspase-3 cleavage. β-actin was used as a protein loading control.
Inhibition of mitochondrial glutaminase prevents HIV-induced cell cycle re-activation and neuronal apoptosis
Inhibition of glutaminase activity prevents excess glutamate production by HIV-infected MDM and blocked MCM-mediated neurotoxicity. To investigate the effects of inhibiting phosphate-activated glutaminase activity on neurotoxicity, we assessed the toxicity of 19560 treatment on RCN by MTT assay. Our results indicated 19560 had no toxic effects on neuron viability (Fig. 7a), but attenuated MCM-mediated neurotoxicity through inhibition of glutaminase activity (data not shown), similar to the effect of MK801 (Fig. 3c). We next asked whether inhibition of glutaminase also prevents MCM-mediated cell cycle activation and neuronal apoptosis. Utilizing the glutaminase-specific inhibitor 19560, we treated RCN with MCM as indicated in Fig. 7b and c and analyzed cell cycle changes by flow cytometry. Our results demonstrated glutaminase inhibition significantly decreased the S phase population in cell cultures treated with MCM from HIV-infected MDM, similar to the effect of MK801 treatment (Fig. 7b), and consistent with the observed cell death (Fig. 7c). These data suggest cell cycle re-activation plays an important role in HIV-infected MCM-mediated neuronal apoptosis.
Fig. 7.
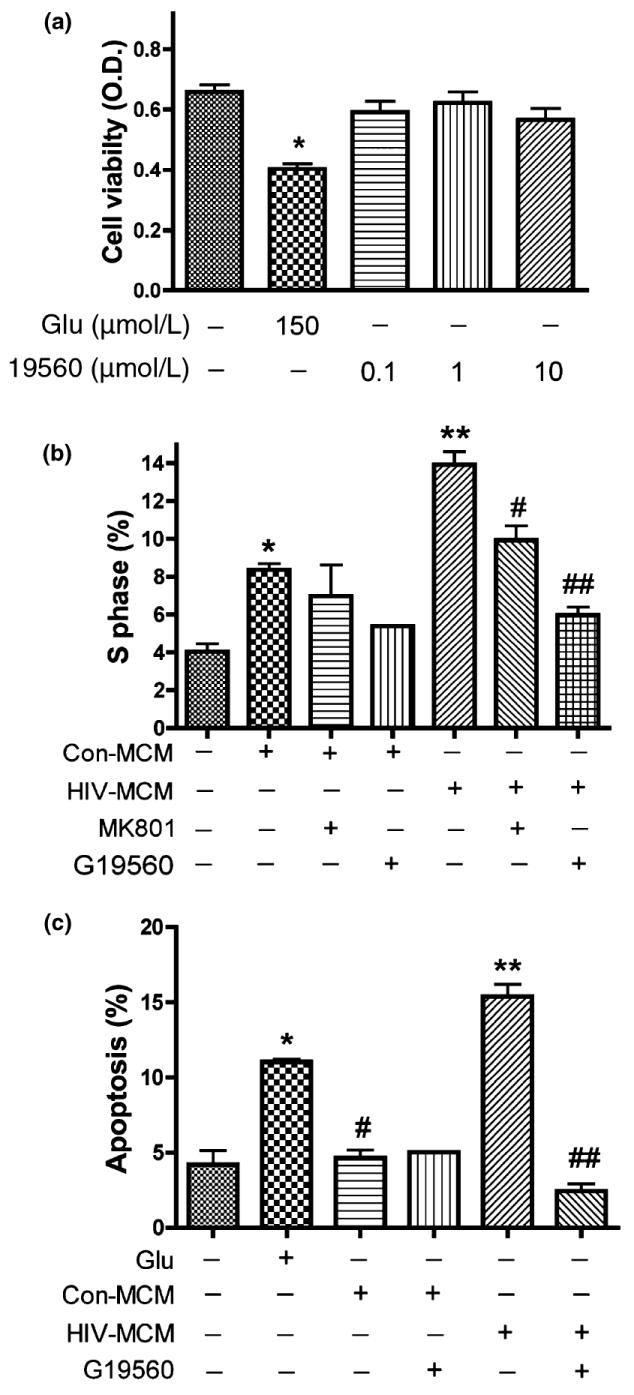
Glutaminase inhibitor attenuates glutamate-mediated neuronal apoptosis by preventing cell cycle re-activation. Rat neuronal cultures were treated 24 h in neurobasal medium with no stimulation, 150 μmol/L glutamate, or 0.1 μmol/L, 1 μmol/L, or 10 μmol/L of glutaminase inhibitor 19560. Cell viability was assessed with MTT assay in triplicate. Data presented is from a representative donor, *indicates p < 0.05 (a). After 10 days in culture, rat neurons were treated with conditioned media and glutamate as indicated (b) for 24 h. The population of S phase in cell cycle was analyzed by Flow Cytometry, *denotes p < 0.05 in comparison with control; **denotes p < 0.01 in comparison to Con-MCM; #denotes p < 0.05 in comparison to HIV-MCM; ##denotes p < 0.001 in comparison with HIV-MCM +19560. (c) The results are expressed as average ±SD of triplicate samples, glutamate treatment as positive control. **denotes p < 0.001 in comparison to Con-MCM; ##denotes p < 0.001 in comparison with HIV-MCM; *denotes p < 0.05 in comparison with control.
Discussion
In this report, we have demonstrated excessive glutamate produced by HIV-1 infected macrophages stimulates NMDA receptors leading to increased calcium influx and activation of caspases-3 and -9, causing neuronal apoptosis. The observed glutamate production by HIV-1-infected macrophages is dependent on the presence of glutamine. Inhibition of mitochondrial glutaminase blocks glutamate production by HIV-1 infected MDM, and conditioned media from treated cultures showed decreased neuronal apoptosis. Further, we have evidence suggesting cell cycle re-activation is involved in the observed excitotoxic neuronal apoptosis. These findings provide insights into the excitotoxic mechanisms of HIV-1 and the role of infected macrophages during HIV-associated dementia.
HIV infection within the brain triggers a cascade of events leading to neuronal damage and death, and many in vitro and in vivo studies support mononuclear phagocytes as primary mediators of inflammation and excitotoxicity (Brenneman et al. 1988; Dreyer et al. 1990; Tardieu et al. 1992; Brew et al. 1995; Power et al. 1998; Epstein and Gelbard 1999; Kaul et al. 2001, 2005; Valle et al. 2004; O’Donnell et al. 2006). HIV-infected macrophages release various neurotoxins that directly or indirectly alter neuronal function, including glutamate, quinolinic acid, platelet activating factor, reactive oxygen species (ROS), NTox, Tat, and gp120. Our studies confirm HIV-1 infection of human macrophages results in a pathogenic increase of glutamate production, an effect dependent on glutamine (see Fig. 1a), and that the increase in glutamate correlates to a decrease in glutamine (see Fig. 1b).
Neuronal injury caused by over-stimulation of glutamate receptors is well-documented, and has been implicated in a variety of neurodegenerative conditions (Lipton 1994; Leist and Nicotera 1998). NMDA receptors play a significant role in HIV-induced neurotoxicity in vitro (Lipton 1993; Chen et al. 2002; O’Donnell et al. 2006). In our model, excessive exposure of RCN to glutamate or MCM from HIV-infected MDM causes neurotoxicity (Fig. 2), an effect dependent on glutamine and triggered by massive Ca2+ influx arising from over-stimulation of the NMDA subtype of glutamate receptors (Fig. 3a and b). The ability of MK801 treatment to prevent neuronal cell death (Fig. 3c) further supports the notion that glutamate in HIV-1-infected MCM is responsible for neurotoxicity via over-stimulation of NMDA receptors.
Excessive NMDA receptor stimulation initiates detrimental intracellular signals that contribute to neuronal injury and death by apoptosis or necrosis (Bonfoco et al. 1995). Brain tissue analysis of HIV positive patients, as well as with in vitro experiments demonstrate that neurons undergo a form of programmed cell death called apoptosis. Caspase activation, mitochondrial alterations, and DNA fragmentation consistent with apoptosis have been documented in affected brain regions of HIV dementia patients (Petito and Roberts 1995). Caspases have previously been implicated in HIV-related neuronal damage (Adle-Biassette et al. 1995; Yuan and Yankner 2000). Because glutamate plays an important role in HIV-1-mediated neurotoxicity, we used glutamate to treat RCN and then assessed the extent of caspase activation and apoptosis. Our results demonstrate that glutamate induces typical neuronal apoptosis, and is blocked by a NMDA receptor antagonist (Fig. 5a and c). Moreover, neuronal apoptosis is accompanied by caspase-3 and -9 activation (Fig. 6). We further confirm HIV-1-infection causes neuronal apoptosis by over-stimulation of NMDA receptors through massive Ca2+ influx (Fig. 3a) and subsequent caspase activation.
Although apoptosis is recognized as an important component in many progressive and acute neurodegenerative diseases, the extracellular signals and intracellular mechanisms triggering and regulating apoptosis in neuronal cells are still a focus of investigation. Recently, aberrant cell cycle re-entry has been implicated in a number of mouse and human neurodegenerative disease models (Herrup and Busser 1995; Nagy et al. 1997; Husseman et al. 2000; Evans et al. 2007; McShea et al. 2007). Cell cycle-related mechanisms contribute to neuronal cell death in a wide variety of circumstances including neurotrophic factor deprivation (Park et al. 1997), DNA damage (Park et al. 1997, 1998), exposure to Aβ (Giovanni et al. 1999) and excitotoxicity (Ino and Chiba 2001; Giardina and Beart 2002; Verdaguer et al. 2002). Our findings demonstrate that the transition from G0/G1 to S phase is involved in glutamate-induced apoptosis (see Fig. 5b). These findings identify cell cycle re-activation as a potential mechanism in the pathogenesis of HAD. However, which cell cycle-related molecules are involved in HIV-1-infected MDM-mediated neuronal apoptosis requires further investigation.
We previously demonstrated that increased glutamate production from HIV-1-infected macrophages was dependent upon mitochondrial glutaminase (Zhao et al. 2004; Erdmann et al. 2007). Glutaminase, which catalyzes the enzymatic conversion of glutamine to glutamate, is the predominant glutamine-utilizing enzyme of the brain. Glutamine is the most abundant amino acid present in the extracellular fluid of the brain, providing substrate for glutaminase in vivo (Newcomb et al. 1997; Holcomb et al. 2000). Here we assessed the role of glutaminase in HIV-infected macrophage-mediated neurotoxicity. First, the removal of glutamine from HIV-infected macrophage conditioned media led to decreased glutamate and a significant reduction in subsequently observed neuronal calcium influx (Fig. 3a) and neurotoxicity (Fig. 2). Second, we used a small molecule glutaminase inhibitor to treat HIV-1-infected macrophage and found glutamate production was efficiently blocked and MCM-mediated calcium response in neurons was greatly reduced (Fig. 4). Third, glutaminase inhibitor treatment of macrophage prevented MCM-mediated cell cycle activation and subsequent neuronal apoptosis (Fig. 7). Pharmacologic inhibition of glutaminase efficiently blocked glutamate elevation in MCM, and decreased glutamate directly correlated to neuronal survival, however, at this stage of discovery, secondary targets of glutaminase inhibitors cannot be completely ruled out. Collectively, these findings identify glutaminase as an important component of HIV-infected macrophage-mediated neurotoxicity, mainly through over-production of glutamate.
Various processes occur during HIV infection to sensitize neuronal populations to excitotoxic insults, including inflammatory cytokine over-stimulation, decreased astrocyte function and neuronal support, impaired mitochondrial function and others (Erdmann et al. 2006). We previously observed HIV-1-MCM-mediated Ca2+ influx in rat neuronal culture and NMDA-dependent neurotoxicity (Jiang et al. 2001; Zheng et al. 2001). This phenomena was tightly associated with glutamine-dependent glutamate production in HIV-1 infected macrophage (Zhao et al. 2004). Based on these data, we concluded glutamate production is a primary factor for induction of neurotoxicity mediated by HIV-infected macrophages. In this study, we first demonstrated glutamine-dependent neurotoxicity in the MCM of HIV-infected MDM. Next, we used filtered MCM from HIV-infected macrophages to determine the mechanism behind HIV-mediated neurotoxicity. We demonstrated that blocking excess glutamate generation through glutaminase inhibition prevented cell cycle re-activation in rat neurons. The reduction in neurotoxicity following glutaminase inhibition was in fact greater than the effect observed following MK801 treatment, an NMDAR-specific inhibitor (Fig. 5b). These results suggest glutamate is a fundamental component of HIV-1 infected macrophage-mediated neurotoxicity through NMDAR, and also indicates non-NMDA receptors may be involved in HIV-MCM-mediated neurotoxicity.
Glutamate-mediated neuronal apoptosis is associated with NMDA receptor over-stimulation, resulting in Ca2+ overload (see Fig. 3 and Fig. 4). Excessive intracellular Ca2+ can over-stimulate nNOS and protein kinase cascades resulting in generation of free radicals, including reactive oxygen species and NO (Nicotera et al. 1997; Urushitani et al. 2001; Raines et al. 2006), resulting in mitochondrial stress. Multiple intracellular pathways initiated by excitotoxic stimuli converge on the mitochondria. Calcium overload resulting from glutamate receptor stimulation can compromise mitochondrial function as well as the high energetic demands of ion pumps to remove excess calcium and sodium place a great burden on mitochondrial output. Mitochondrial stress leads to radical production and energy shortages in the cell (Wang et al. 1994; Castilho et al. 1999; Nicholls et al. 1999; Krieger and Duchen 2002). The mitochondria initiate powerful signaling cascades that can rapidly lead to disassembly of the whole cell. Glutaminase is a mitochondrial enzyme and how its function is manipulated by HIV infection and excitotoxic stress may be an important question to understanding neurodegenerative mechanisms. In summary, mitochondrial glutaminase plays an important role in mediating glutamate over-generation in HIV-infected macrophages and subsequent caspase-mediated neuronal apoptosis, and the inhibition of glutamate production by blocking mitochondrial glutaminase activity may prevent neurotoxicity during HIV-1 infection.
Acknowledgments
This work was supported in part by research grants by the National Institutes of Health: R01 NS 41858, P20 RR15635 and P01 NS043985 (JZ). We kindly thank Dr Norman Curthoys and Lynn Taylor (Colorado State University) and Drs. Takashi Tsukamoto and Dana Ferraris (MGI Pharma Inc.) provided support for this work. Drs Howard E. Gendelman and Yunlong Huang provided valuable comments and suggestions about the manuscript. Dr Charles Kuszynski and Linda Wilkie performed the Fluorescence Activated Cell Sorter (FACS) analyses. The water-soluble, small-molecule inhibitor designed to specifically block glutaminase named 19560 was kindly provided by MGI Pharma Inc., Baltimore, MD, USA, Julie Ditter, Emilie Scoggins, Johnna Belling, Robin Taylor, Myhanh Che and Na Ly and provided outstanding administrative and secretarial support.
Abbreviations used
- HAD
HIV-associated dementia
- MCM
MDM conditioned media
- MDM
monocyte-derived macrophages
- MTT
methylthiazolyldiphenyl-tetrazolium bromide
- RCN
rat cortical neurons
References
- Adamson DC, Wildemann B, Sasaki M, Glass JD, McArthur JC, Christov VI, Dawson TM, Dawson VL. Immunologic No Synthase: Elevation in Severe AIDS Dementia and Induction by HIV-1 gp41. Science. 1996;274:1917–1926. doi: 10.1126/science.274.5294.1917. [DOI] [PubMed] [Google Scholar]
- Adle-Biassette H, Levy Y, Colombel M, Poron F, Natchev S, Keohane C, Gray F. Neuronal apoptosis in HIV infection in adults. Neuropathol App Neurobiol. 1995;21:218–227. doi: 10.1111/j.1365-2990.1995.tb01053.x. [DOI] [PubMed] [Google Scholar]
- Becker EB, Bonni A. Cell cycle regulation of neuronal apoptosis in development and disease. Prog Neurobiol. 2004;72:1–25. doi: 10.1016/j.pneurobio.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: Two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenneman DE, Westbrook GL, Fitzgerald SP, Ennist DL, Elkins KL, Ruff MR, Pert CB. Neuronal cell killing by the envelope protein of HIV and its prevention by vasoactive intestinal peptide. Nature. 1988;335:639–642. doi: 10.1038/335639a0. [DOI] [PubMed] [Google Scholar]
- Brew B, Corbeil J, Pemberton L, Evans L, Saito K, Penny R, Cooper D, Heyes M. Quinolinic acid production is related to macrophage tropic isolates of HIV-1. J Neurovirol. 1995;1:369–374. doi: 10.3109/13550289509111026. [DOI] [PubMed] [Google Scholar]
- Castilho RF, Ward MW, Nicholls DG. Oxidative stress, mitochondrial function, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurochem. 1999;72:1394–1401. doi: 10.1046/j.1471-4159.1999.721394.x. [DOI] [PubMed] [Google Scholar]
- Chen W, Sulcove J, Frank I, Jaffer S, Ozdener H, Kolson DL. Development of a human neuronal cell model for human immunodeficiency virus (HIV)-infected macrophage-induced neurotoxicity: apoptosis induced by HIV type 1 primary isolates and evidence for involvement of the Bcl-2/Bcl-xL-sensitive intrinsic apoptosis pathway. J Virol. 2002;76:9407–9419. doi: 10.1128/JVI.76.18.9407-9419.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Chai YC, Mazumder S, Jiang C, Macklis RM, Chisolm GM, Almasan A. The late increase in intracellular free radical oxygen species during apoptosis is associated with cytochrome c release, caspase activation, and mitochondrial dysfunction. Cell Death Differ. 2003;10:323–334. doi: 10.1038/sj.cdd.4401148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW, Koh JY, Peters S. Pharmacology of glutamate neurotoxicity in cortical cell culture: attenuation by NMDA antagonists. J Neurosci. 1988;8:185–196. doi: 10.1523/JNEUROSCI.08-01-00185.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryns V, Yuan J. Proteases to die for. Genes Dev. 1998;12:1551–1570. doi: 10.1101/gad.12.11.1551. [DOI] [PubMed] [Google Scholar]
- Curthoys NP, Watford M. Regulation of glutaminase activity and glutamine metabolism. Annu Rev Nutr. 1995;15:133–159. doi: 10.1146/annurev.nu.15.070195.001025. [DOI] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM, Uhl GR, Snyder SH. Human immunodeficiency virus type 1 coat protein neurotoxicity mediated by nitric oxide in primary cortical cultures. Proc Natl Acad Sci USA. 1993;90:3256–3259. doi: 10.1073/pnas.90.8.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyer EB, Kaiser PK, Offermann JT, Lipton SA. HIV-1 coat protein neurotoxicity prevented by calcium channel antagonists. Science. 1990;248:364–367. doi: 10.1126/science.2326646. [DOI] [PubMed] [Google Scholar]
- Droge W, Eck HP, Betzler M, Naher H. Elevated plasma glutamate levels in colorectal carcinoma patients and in patients with acquired immunodeficiency syndrome (AIDS) Immunobiology. 1987;174:473–479. doi: 10.1016/s0171-2985(87)80019-0. [DOI] [PubMed] [Google Scholar]
- Epstein LG, Gelbard HA. HIV-1-induced neuronal injury in the developing brain. J Leukoc Biol. 1999;65:453–457. doi: 10.1002/jlb.65.4.453. [DOI] [PubMed] [Google Scholar]
- Erdmann N, Whitney N, Zheng J. Potentiation of excitotoxicity in HIV-1-associated dementia and the significance of glutaminase. Clin Neurosci Res. 2006;6:315–328. doi: 10.1016/j.cnr.2006.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann N, Zhao J, Lopez AL, Herek S, Curthoys N, Hexum TD, Tsukamoto T, Ferraris D, Zheng J. Glutamate production by HIV-1 infected human macrophage is blocked by the inhibition of glutaminase. J Neurochem. 2007;102:539–549. doi: 10.1111/j.1471-4159.2007.04594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espey MG, Ellis RJ, Heaton RK, Basile AS. Relevance of glutamate levels in the CSF of patients with HIV-1- associated dementia complex. Neurology. 1999;53:1144–1145. doi: 10.1212/wnl.53.5.1144. [DOI] [PubMed] [Google Scholar]
- Evans TA, Raina AK, Delacourte A, Aprelikova O, Lee HG, Zhu X, Perry G, Smith MA. BRCA1 may modulate neuronal cell cycle re-entry in Alzheimer disease. Int J Med Sci. 2007;4:140–145. doi: 10.7150/ijms.4.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrarese C, Aliprandi A, Tremolizzo L, Stanzani L, De Micheli A, Dolara A, Frattola L. Increased glutamate in CSF and plasma of patients with HIV dementia. Neurology. 2001;57:671–675. doi: 10.1212/wnl.57.4.671. [DOI] [PubMed] [Google Scholar]
- Gelbard HA, James HJ, Sharer LR, Perry SW, Saito Y, Kazee AM, Blumberg BM, Epstein LG. Apoptotic neurons in brains from paediatric patients with HIV-1 encephalitis and progressive encephalopathy. Neuropathol Appl Neurobiol. 1995;21:208–217. doi: 10.1111/j.1365-2990.1995.tb01052.x. [DOI] [PubMed] [Google Scholar]
- Giardina SF, Beart PM. Kainate receptor-mediated apoptosis in primary cultures of cerebellar granule cells is attenuated by mitogen-activated protein and cyclin-dependent kinase inhibitors. Br J Pharmacol. 2002;135:1733–1742. doi: 10.1038/sj.bjp.0704636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovanni A, Wirtz-Brugger F, Keramaris E, Slack R, Park DS. Involvement of cell cycle elements, cyclin-dependent kinases, pRb, and E2F × DP, in B-amyloid-induced neuronal death. J Biol Chem. 1999;274:19011–19016. doi: 10.1074/jbc.274.27.19011. [DOI] [PubMed] [Google Scholar]
- Guaragnella N, Pereira C, Sousa MJ, Antonacci L, Passarella S, Corte-Real M, Marra E, Giannattasio S. YCA1 participates in the acetic acid induced yeast programmed cell death also in a manner unrelated to its caspase-like activity. FEBS Lett. 2006;580:6880–6884. doi: 10.1016/j.febslet.2006.11.050. [DOI] [PubMed] [Google Scholar]
- Herrup K, Busser JC. The induction of multiple cell cycle events precedes target-related neuronal death. Development. 1995;121:2385–2395. doi: 10.1242/dev.121.8.2385. [DOI] [PubMed] [Google Scholar]
- Holcomb T, Taylor L, Trohkimoinen J, Curthoys NP. Isolation, characterization and expression of a human brain mitochondrial glutaminase cDNA. Brain Res Mol Brain Res. 2000;76:56–63. doi: 10.1016/s0169-328x(99)00331-9. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- Husseman JW, Nochlin D, Vincent I. Mitotic activation: a convergent mechanism for a cohort of neurodegenerative diseases. Neurobiol Aging. 2000;21:815–828. doi: 10.1016/s0197-4580(00)00221-9. [DOI] [PubMed] [Google Scholar]
- Ino H, Chiba T. Cyclin-dependent kinase 4 and cyclin D1 are required for excitotoxin-induced neuronal cell death in vivo. J Neurosci. 2001;21:6086–6094. doi: 10.1523/JNEUROSCI.21-16-06086.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Piggee C, Heyes MP, Murphy C, Quearry B, Bauer M, Zheng J, Gendelman HE, Markey SP. Glutamate is a mediator of neurotoxicity in secretions of activated HIV- 1-infected macrophages. J Neuroimmunol. 2001;117:97–107. doi: 10.1016/s0165-5728(01)00315-0. [DOI] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–994. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- Kaul M, Zheng J, Okamoto S, Gendelman HE, Lipton SA. HIV-1 infection and AIDS: consequences for the central nervous system. Cell Death Differ. 2005;12(Suppl 1):878–892. doi: 10.1038/sj.cdd.4401623. [DOI] [PubMed] [Google Scholar]
- Krieger C, Duchen MR. Mitochondria, Ca2 + and neurodegenerative disease. Eur J Pharmacol. 2002;447:177–188. doi: 10.1016/s0014-2999(02)01842-3. [DOI] [PubMed] [Google Scholar]
- Leist M, Nicotera P. Apoptosis, excitotoxicity, and neuropathology. Exp Cell Res. 1998;239:183–201. doi: 10.1006/excr.1997.4026. [DOI] [PubMed] [Google Scholar]
- Li XF, Phillips R, LeDoux JE. NMDA and non-NMDA receptors contribute to synaptic transmission between the medial geniculate body and the lateral nucleus of the amygdala. Exp Brain Res. 1995;105:87–100. doi: 10.1007/BF00242185. [DOI] [PubMed] [Google Scholar]
- Liao XD, Tang AH, Chen Q, Jin HJ, Wu CH, Chen LY, Wang SQ. Role of Ca2 + signaling in initiation of stretchinduced apoptosis in neonatal heart cells. Biochem Biophys Res Commun. 2003;310:405–411. doi: 10.1016/j.bbrc.2003.09.023. [DOI] [PubMed] [Google Scholar]
- Lipton SA. Human immunodeficiency virus-infected macrophages, gp120, and N-methyl-D-aspartate receptor-mediated neurotoxicity. Ann Neurol. 1993;33:227–228. doi: 10.1002/ana.410330218. [DOI] [PubMed] [Google Scholar]
- Lipton S. Laboratory basis of novel therapeutic strategies to prevent HIV related neuronal injury. Res Publ Assoc Res Nerv Ment Dis. 1994;72:183–202. [PubMed] [Google Scholar]
- McShea A, Lee HG, Petersen RB, Casadesus G, Vincent I, Linford NJ, Funk JO, Shapiro RA, Smith MA. Neuronal cell cycle re-entry mediates Alzheimer disease-type changes. Biochim Biophys Acta. 2007;1772:467–472. doi: 10.1016/j.bbadis.2006.09.010. [DOI] [PubMed] [Google Scholar]
- Munsch T, Deitmer JW. Maintenance of Fura-2 fluorescence in glial cells and neurons of the leech central nervous system. J Neurosci Methods. 1995;57:195–204. doi: 10.1016/0165-0270(94)00149-b. [DOI] [PubMed] [Google Scholar]
- Nagy Z, Esiri MM, Smith AD. Expression of cell division markers in the hippocampus in Alzheimer’s disease and other neurodegenerative conditions. Acta Neuropathol (Berl) 1997;93:294–300. doi: 10.1007/s004010050617. [DOI] [PubMed] [Google Scholar]
- Newcomb R, Sun X, Taylor L, Curthoys N, Giffard RG. Increased production of extracellular glutamate by the mitochondrial glutaminase following neuronal death. J Biol Chem. 1997;272:11276–11282. doi: 10.1074/jbc.272.17.11276. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL, Castilho RF, Ward MW. Glutamate excitotoxicity and neuronal energy metabolism. Ann N Y Acad Sci. 1999;893:1–12. doi: 10.1111/j.1749-6632.1999.tb07813.x. [DOI] [PubMed] [Google Scholar]
- Nicklas WJ, Zeevalk G, Hyndman A. Interactions between neurons and glia in glutamate/glutamine compartmentation. Biochem Soc Trans. 1987;15:208–210. doi: 10.1042/bst0150208. [DOI] [PubMed] [Google Scholar]
- Nicotera P, Ankarcrona M, Bonfoco E, Orrenius S, Lipton SA. Neuronal necrosis and apoptosis: two distinct events induced by exposure to glutamate or oxidative stress. Adv Neurol. 1997;72:95–101. [PubMed] [Google Scholar]
- O’Donnell LA, Agrawal A, Jordan-Sciutto KL, Dichter MA, Lynch DR, Kolson DL. Human immunodeficiency virus (HIV)-induced neurotoxicity: roles for the NMDA receptor subtypes. J Neurosci. 2006;26:981–990. doi: 10.1523/JNEUROSCI.4617-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollenschlager G, Jansen S, Schindler J, Rasokat H, Schrappe-Bacher M, Roth E. Plasma amino acid pattern of patients with HIV infection. Clin Chem. 1988;34:1787–1789. [PubMed] [Google Scholar]
- Park DS, Levine B, Ferrari G, Greene LA. Cyclin dependent kinase inhibitors and dominant negative cyclin dependent kinase 4 and 6 promote survival of NGF-deprived sympathetic neurons. J Neurosci. 1997;17:8975–8983. doi: 10.1523/JNEUROSCI.17-23-08975.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DS, Morris EJ, Padmanabhan J, Shelanski ML, Geller HM, Greene LA. Cyclin-dependent kinases participate in death of neurons evoked by DNA-damaging agents. J Cell Biol. 1998;143:457–467. doi: 10.1083/jcb.143.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petito CK, Roberts B. Evidence of apoptotic cell death in HIV encephalitis. Am J Pathol. 1995;146:1121–1130. [PMC free article] [PubMed] [Google Scholar]
- Power C, McArthur JC, Nath A, Wehrly K, Mayne M, Nishio J, Langelier T, Johnson RT, Chesebro B. Neuronal death induced by brain-derived human immunodeficiency virus Type 1 envelope genes differs between demented and nondemented AIDS patients. J Virol. 1998;72:9045–9053. doi: 10.1128/jvi.72.11.9045-9053.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raines KW, Cao GL, Lee EK, Rosen GM, Shapiro P. Neuronal nitric oxide synthase-induced S-nitrosylation of H-Ras inhibits calcium ionophore-mediated extracellular-signal-regulated kinase activity. Biochem J. 2006;397:329–336. doi: 10.1042/BJ20052002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardieu M, Hery C, Peudenier S, Boespflug O, Montagnier L. Human immunodeficiency virus type-1 infected monocytic cells can destroy human neural cells after cell-to-cell adhesion. Ann Neurol. 1992;32:11–17. doi: 10.1002/ana.410320104. [DOI] [PubMed] [Google Scholar]
- Urushitani M, Nakamizo T, Inoue R, Sawada H, Kihara T, Honda K, Akaike A, Shimohama S. N-methyl-D-aspartate receptor-mediated mitochondrial Ca(2+) overload in acute excitotoxic motor neuron death: a mechanism distinct from chronic neurotoxicity after Ca(2+) influx. J Neurosci Res. 2001;63:377–387. doi: 10.1002/1097-4547(20010301)63:5<377::AID-JNR1032>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Valle M, Price RW, Nilsson A, Heyes M, Verotta D. CSF quinolinic acid levels are determined by local HIV infection: cross-sectional analysis and modelling of dynamics following antiretroviral therapy. Brain. 2004;127:1047–1060. doi: 10.1093/brain/awh130. [DOI] [PubMed] [Google Scholar]
- Verdaguer E, Garcia-Jorda E, Canudas AM, Dominguez E, Jimenez A, Pubill D, Escubedo E, Pallas JC, Camins A. Kainic acid-induced apoptosis in cerebellar granule neurons: an attempt at cell cycle re-entry. Neuroreport. 2002;13:413–416. doi: 10.1097/00001756-200203250-00010. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Randall RD, Thayer SA. Glutamate-induced intracellular acidification of cultured hippocampal neurons demonstrates altered energy metabolism resulting from Ca2 + loads. J Neurophysiol. 1994;72:2563–2569. doi: 10.1152/jn.1994.72.6.2563. [DOI] [PubMed] [Google Scholar]
- Ward HK, Thanki CM, Bradford HF. Glutamine and glucose as precursors of transmitter amino acids: ex vivo studies. J Neurochem. 1983;40:855–860. doi: 10.1111/j.1471-4159.1983.tb08058.x. [DOI] [PubMed] [Google Scholar]
- Wurdig S, Kugler P. Histochemistry of glutamate metabolizing enzymes in the rat cerebellar cortex. Neurosci Lett. 1991;130:165–168. doi: 10.1016/0304-3940(91)90388-a. [DOI] [PubMed] [Google Scholar]
- Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- Zhao J, Lopez AL, Erichsen D, Herek S, Cotter RL, Curthoys NP, Zheng J. Mitochondrial glutaminase enhances extracellular glutamate production in HIV-1-infected macrophages: linkage to HIV-1 associated dementia. J Neurochem. 2004;88:169–180. doi: 10.1046/j.1471-4159.2003.02146.x. [DOI] [PubMed] [Google Scholar]
- Zheng J, Thylin M, Ghorpade A, et al. Intracellular CXCR4 signaling, neuronal apoptosis and neuropathogenic mechanisms of HIV-1-associated dementia. J Neuroimmunol. 1999;98:185–200. doi: 10.1016/s0165-5728(99)00049-1. [DOI] [PubMed] [Google Scholar]
- Zheng J, Thylin MR, Cotter RL, Lopez AL, Ghorpade A, Persidsky Y, Xiong H, Leisman GB, Che MH, Gendelman HE. HIV-1 infected and immune competent mononuclear phagocytes induce quantitative alterations in neuronal dendritic arbor: relevance for HIV-1-associated dementia. Neurotoxi Res. 2001;3:443–459. doi: 10.1007/BF03033203. [DOI] [PubMed] [Google Scholar]