

Abstract
Ischemia-reperfusion injury (IRI) is one of the major causes of acute kidney injury (AKI) and evidence supporting the involvement of both innate and adaptive immunity in renal IRI has accumulated in recent years. In addition to leukocytes, kidney endothelial cells promote inflammation after IRI by increasing adhesion molecule expression and vascular permeability. Kidney tubular epithelial cells increase complement binding and upregulate toll-like receptors, both of which lead to cytokine/chemokine production in IRI. Activation of kidney resident dendritic cells, interferon-γ-producing neutrophils, infiltrating macrophages, CD4+ T cells, B cells and invariant natural killer T cells are all implicated in the pathogenesis of AKI. The complex interplay between innate and adaptive immunity in renal IRI is still not completely understood, but major advances have been made. This review summarizes these recent advances to further our understanding of the immune mechanisms of acute kidney injury.
Keywords: Innate immunity, Adaptive immunity, Leukocytes, Acute renal failure
Introduction
Acute kidney injury (AKI) is associated with a high degree of morbidity and mortality and the incidence remains unacceptably high. Ischemia-reperfusion injury (IRI) is one of the major causes of AKI. Currently, no pharmacological agents have proven to prevent AKI and the mortality rate of patients with severe AKI has not declined in recent decades [1].
Ischemia and/or reperfusion initiate changes in vascular endothelial cells, tubular epithelial cells and leukocytes that result in the loss of immune system homeostasis in the kidney [2-6]. The ensuing inflammation leads to kidney parenchymal cell death and in severe cases AKI. The inflammatory response can be mediated by two different, but related, arms of the immune system: innate and adaptive immunity. The innate immune system is activated very early in infectious or inflammatory states in a non-antigen-specific fashion and is comprised of neutrophils, monocytes/macrophages, dendritic cells (DCs), natural killer (NK) cells and natural killer T (NKT) cells. In contrast, the adaptive immune system becomes responsive to specific antigens (from pathogens or dead self cells) over the course of several days and includes DC maturation and antigen presentation, CD4 and CD8 T lymphocyte proliferation and activation, and T to B lymphocyte interactions. Leukocytes such as DCs and macrophages play key roles in both types of immunity by producing pro-inflammatory cytokines and presenting antigen to lymphocytes. Evidence supporting the involvement of both innate and adaptive immunity in renal IRI has accumulated in recent years. This review will highlight some of the new concepts in the immunologic mechanisms of ischemia-induced AKI.
Renal Vascular Endothelium
One of the early events in renal IRI is activation of the endothelium leading to an increase in vascular permeability [7] which promotes extravasation of leukocytes into the kidney. Brodsky et al. [8] showed that after renal IRI, there was loss of endothelial cells from afferent arterioles and interruption of endothelial cell contacts, an effect reversed through transfer of endothelial cells [8] or through treatment with a sphingosine-1-phosphate analog prodrug, FTY-720 [9]. In addition to changes in the integrity of the endothelial cell layer of the renal vasculature, IRI upregulates the expression of adhesion molecules that facilitate leukocyte-endothelial cell interactions. The expression of intracellular adhesion molecule 1 (ICAM-1) increases in the kidney by 1 h after IRI and mice lacking ICAM-1 are protected from renal IRI [4]. Leukocyte adhesion to endothelial cells leads to inflammation and extension of cellular injury. Additionally, renal endothelial cells upregulate the expression of CX3CL1 (fractalkine), a ligand for the CX3CR1 receptor highly expressed on macrophages that mediates macrophage recruitment in the inflamed kidney, and pretreatment with a neutralizing CX3CR1 mAb reduced the severity of AKI [10]. Therefore, the endothelium plays an important early role in the inflammatory response to kidney damage by promoting the accumulation of leukocytes.
Renal Tubular Epithelium
Several studies have demonstrated that tubular epithelial cells (TECs) play a pro-inflammatory role in kidney IRI. Normally, the epithelial cells lining the proximal tubules of the kidney express the complement inhibitor Crry preferentially on the basolateral membrane [2]. After renal IRI, Crry is redistributed away from the basolateral surface of the cell, which permits the deposition of C3 on the tubular epithelium [2]. In support of a protective role for proximal tubular Crry expression, mice deficient in Crry are more susceptible to kidney IRI [2]. Complement activation, by the alternative pathway, is required for the production of the pro-inflammatory chemokines macrophage inflammatory factor-2 (MIP-2) and keratinocyte-derived chemokine (KC) by the renal tubular epithelium after IRI [11]. These chemokines attract neutrophils and macrophages to the injured kidney. Another recent study demonstrated that toll-like receptor 4 (TLR4) is upregulated in TECs after IRI and deficiency of TLR4 on kidney parenchymal cells was more effective at preventing kidney IRI than TLR4 deficiency on bone marrow-derived cells [12]. TLRs are a family of pattern recognition receptors that detect motifs of pathogens and host material released during injury that are important for activation of innate immunity. TLR4 deficiency blunted the IRI-induced production of pro-inflammatory cytokines and chemokines and inhibited macrophage and neutrophil accumulation [12]. A similar study showed, using bone marrow chimeras, that lack of TLR2 expression on kidney parenchymal cells also inhibited renal IRI and kidney pro-inflammatory cytokine production was reduced in TLR2−/− mice compared to wild-type controls [13]. Molecules such as high-mobility group B1 (HMGB1), heat shock proteins, hyaluronan and biglycan released from damaged tissues activate TLRs and lead to downstream activation of transcription factors that regulate the expression of survival genes or proinflammatory cytokines and chemokines. TLRs expressed on endothelial cells and epithelial cells are involved in kidney IRI via both MyD88-dependent and independent pathways [14]. These studies highlight the important role for renal endothelial and epithelial cells in the inflammation of AKI.
Neutrophils
Neutrophils rapidly respond to injury and are important mediators of innate immunity. Adherence of neutrophils to the vascular endothelium is a crucial early process in the initiation of damage to ischemic tissues. Neutrophils respond to invading pathogens either by phagocytosis or releasing granules containing proteases and other enzymes, which generate reactive oxygen species. In inflammatory states, neutrophil degranulation can lead to the destruction of normal self cells in the inflamed tissue. One of the hallmarks of renal IRI, in mouse models, is neutrophil accumulation in the post-ischemic kidney [3, 4, 12] and depletion of neutrophils prevents AKI [4]. Our laboratory has demonstrated that blocking upstream activation of invariant NKT (iNKT) cells (see below) prevents renal accumulation of IFN-γ-producing neutrophils and kidney dysfunction after IRI in mice [3]. These studies suggest the involvement of neutrophils in the pathogenesis of kidney dysfunction in the widely used murine model of IRI-induced AKI. Furthermore, neutrophil activation and infiltration may be governed by other leukocytes, such as iNKT cells. In contrast, studies in other species (rabbit and rat) have not reported extensive neutrophil accumulation or protective effects of neutrophil depletion in mild or severe renal IRI [15].
Macrophages
Macrophages are derived from monocytes in the blood and are named for their role as phagocytes. In addition to phagocytosis, macrophages produce pro-inflammatory cytokines that can stimulate the activity of other leukocytes [Li and Okusa, unpubl. data; 16 ]. Macrophages infiltrate the injured kidney shortly after neutrophils (within 1 h of reperfusion), and this infiltration is mediated by CCR2 [Li and Okusa, unpubl. data] and CX3CR1 signaling pathways [10]. These macrophages have a distinct F4/80lowLy6ChighGR-1+CX3CR1low ‘inflamed’ phenotype [Li and Okusa, unpubl. data; 16]. Depletion of kidney and spleen macrophages, using liposomal clodronate, prior to renal IRI prevented AKI and adoptive transfer of macrophages reconstituted AKI [5]. Intracellular cytokine staining of kidney infiltrating macrophages by flow cytometry demonstrated that these leukocytes are significant producers of the cytokines IL-1α, IL-6, IL-12p40/70 and TNF-α [Li and Okusa, unpubl. data]. Another study identified IL-6 expression in renal outer medulla interstitial macrophages by in situ hybridization 4 h after IRI [16]. The increased abundance of IFN-γ from iNKT cells and neutrophils provides potent stimulation for macrophage activation early in IRI.
Dendritic Cells
DCs are an important link between innate and adaptive immunity and their role in AKI is not completely understood. CD11c+ MHC class II+ DCs are the most abundant leukocyte subset in the normal mouse kidney suggesting an important role in renal immunity and inflammation. Upon stimulation, DCs can convert to a mature cell type characterized by high levels of class II major histocompatibility complex (MCH class II) and co-stimulatory molecules and low phagocytic capacity. Mature DCs are specialized in T cell activation. However, DCs are also important in the innate immune response by releasing pro-inflammatory factors, interacting with NKT cells via CD40–CD40L and presenting glycolipids via the CD1d molecule to activate iNKT cells. Dong et al. [17] demonstrated that after IRI renal DCs produce the pro-inflammatory cytokines/chemokines TNF, IL-6, MCP-1 and RANTES, and that depletion of DCs prior to IRI significantly reduced the kidney levels of TNF produced after IRI. IL-12 and its new family member IL-23 are mainly produced from activated DCs and macrophages, and their downstream cytokines IFN-γ and IL-17, associated with macrophage activation and neutrophil recruitment, may amplify the immune response following kidney reperfusion. These results suggest a role for the innate response of DCs in AKI. In a separate study, DCs were shown to traffic to the renal draining lymph nodes after IRI and induce T cell proliferation in an antigen-specific fashion, implicating renal DCs in the adaptive immune response to IRI [18]. While these studies strongly suggest that DCs play an important role in ischemia-induced AKI, additional studies are needed to determine the effect of specific-DC depletion in IRI-induced AKI. The use of a genetically engineered mouse in which the DC-specific surface protein CD11c is conjugated to the human diphtheria toxin receptor (CD11c-DTR mouse) should facilitate DC depletion studies and offer more insight into the role of renal DCs in IRI.
Lymphocytes
Lymphocytes are the major mediators of adaptive immunity. Antigen presentation by APCs, in the presence of sufficient co-stimulation, causes expansion and activation of T cells with a T cell receptor (TCR) specific for the presented antigen. B cells do not require antigen presentation; rather, they recognize soluble antigens that they engulf and process to present to T cells with a TCR specific for the same antigen. The interaction of the B and T cell stimulates the B cell to generate antibodies specific for the antigen. Other antigens can induce antibody production in the absence of T cell participation. A role for T cells in the pathogenesis of kidney IRI has been established in different mouse models lacking certain types of lymphocytes [6, 19]. In nu/nu mice (which lack CD4 and CD8 T cells), IRI measured by serum creatinine levels and renal histology was significantly reduced compared to wild-type controls [19]. Reconstitution of nu/nu mice with CD4+ T cells alone and not CD8+ T cells alone restores kidney injury after IRI [19]. Additionally, RAG-1−/− mice (lacking both B and T cells) are also protected from IRI and adoptive transfer of CD4+ T cells from wild-type mice reconstitutes injury [6]. Importantly, transfer of CD4+ T cells from IFN-γ −/− mice failed to re-establish injury in this model [6]. These results suggest that CD4+ T cells, and specifically IFN-γ produced by these cells, mediate the early phase of IRI.
Mice deficient in B cells (μMT mice) are also protected from IRI [20]. The adoptive transfer of purified B cells back into these mice, however, does not restore kidney injury after ischemia [20]. On the other hand, transfer of serum from wild-type mice does result in higher serum creatinine values after IRI compared to the μMT mice without serum transfer [20]. The authors suggest that lack of a circulating factor, possibly an immunoglobulin, may be responsible for the protection observed in B cell-deficient mice.
Other investigators have reported a lack of protection from IRI in RAG-1−/− mice [21, 22]. Burne-Taney et al. [22] reported that while RAG-1−/− mice were not protected from IRI, RAG-1−/− mice reconstituted with either T or B cells alone were protected. The reasons for the discrepancy between laboratories in results using the RAG-1−/− mice are unclear at present and cannot be explained by strain differences [21, 22]. It is possible that in some models, combined T and B cell deficiency leads to increased innate immune responses [22].
Invariant Natural Killer T Cells
Several studies have demonstrated that CD4+ T cells are involved in renal IRI (see above). However, conventional CD4+ T cells are thought to play a role in antigen-specific, adaptive immunity that requires 2–4 days for T cell processing, a time course that cannot explain the rapid, innate immune response following IRI. NKT cells are a unique subset of T lymphocytes with surface receptors and functional properties shared with conventional T cells and NK cells. Invariant NKT cells posses a conserved invariant TCR (Vα14/Jα18 and Vβ8.2,Vβ2 or Vβ7) together with the NK cell marker NK1.1. In contrast to conventional T cells, the NKT cell TCR does not interact with peptide antigen presented by classical MHC-class I or II, rather it recognizes glycolipids presented by the class I-like molecule, CD1d. A glycolipid, α-galactosylceramide, is the most efficient activator for iNKT cells. The most remarkable property of iNKT cells is their ability to rapidly produce large amounts of cytokines, including Th1-type (IFN-γ, TNF) and Th2-type (IL-4, IL-13) at the same time within 1–2 h. The rapid response by iNKT cells following activation can amplify and regulate the function of DCs, regulatory T cells, NK and B cells, as well as conventional T cells, and thus links innate and adaptive immunity. A recent finding from our laboratory is that early IRI (30 min following reperfusion) leads to an increase in activated CD4+CD69+ cells and the number of IFN-γ-producing iNKT cells in the kidney is significantly increased by 3 h of reperfusion compared to sham-operated mice [3]. At this time point, there is also a significant increase in IFN-γ + neutrophil recruitment in the IRI kidney. Blockade of NKT cell activation with the anti-CD1d mAb, NKT cell depletion with an anti-NK1.1 mAb in wild-type mice, or use of iNKT cell-deficient mice (Jα18−/−) inhibited the accumulation of IFN-γ-producing neutrophils after IRI and prevented AKI [3]. Given that (1) there is a major disconnect between the timing of the protection seen in CD4+ T cell-deficient mice and the timing of conventional T cell activation, (2) IFN-γ −/− CD4+ T cells do not reconstitute injury in RAG-1−/− mice, and (3) the mouse CD4+ T cell population contains iNKT cells that can be activated within hours, the current findings suggest that iNKT cells are the major early-acting CD4+ cell type in renal IRI. CD1d-restricted NKT cells include type I NKT (iNKT) cells and type II NKT cells; the role of type II NKT cells in kidney IRI has not been examined.
Conclusions
Over the past decade many new concepts in the role of inflammation in AKI have emerged (fig. 1). Among them are pro-inflammatory changes in the endothelial and epithelial cells of the kidney. Additionally, complement, TLRs and numerous cytokines and chemokines are clearly involved in amplifying the immune response to kidney injury. The complex interplay between innate and adaptive immunity in renal IRI is still not completely understood but advances have been made in this area. Critical early roles for neutrophils, macrophages, and T and B and NKT cells have been established in mouse models of AKI. Finally, these new concepts may lead to new targets for development of clinically relevant treatment strategies for AKI.
Fig. 1.
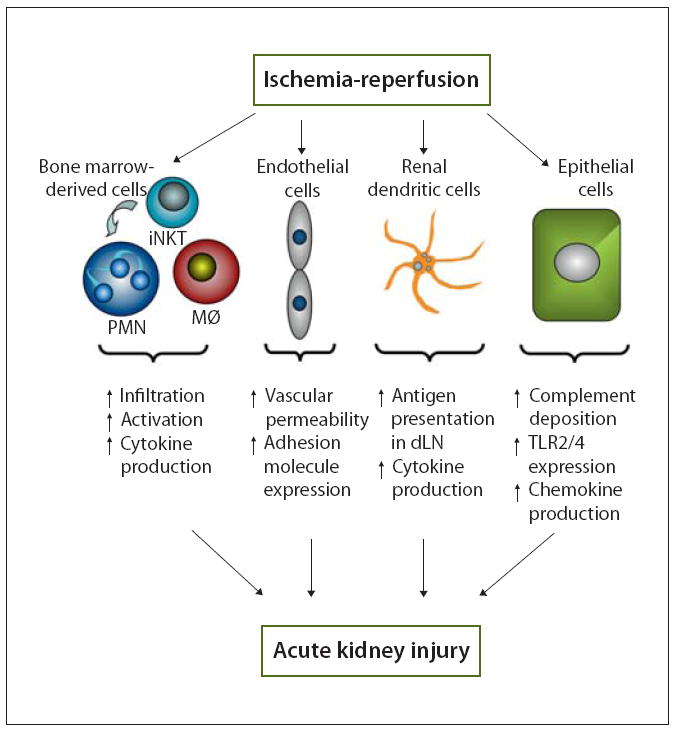
Inflammatory role of bone marrow-derived and kidney cells in AKI. Ischemia-reperfusion induces changes in leukocytes, endothelial cells and tubular epithelial cells that result in kidney inflammation and mediate AKI. Bone marrow derived cells such as iNKT cells [3], neutrophils (PMN [3, 4, 12]) and macrophages (MØ [16]) accumulate in the kidney, are activated and produce pro-inflammatory cytokines (i.e. IFN-γ production by iNKT cells and PMNs [3]). Endothelial cells are damaged by IRI leading to increased vascular permeability [8, 9] and expression of adhesion molecules such as ICAM-1 [4] and fractalkine [10]. These changes facilitate the accumulation of leukocytes in the kidney. Renal dendritic cells produce cytokines and chemokines [17] and traffic to the renal draining lymph node and present antigens to T cells [18]. Tubular epithelial cells exhibit increased complement deposition [2] and upregulate the expression of toll-like receptors (TLRs [12, 13]), both of which mediate chemokine and cytokine production in the injured kidney [11-13]. Changes in each cell type directly or indirectly influence the other cells involved to promote inflammation after renal IRI. These interactions between kidney and bone marrow derived cells and between innate and adaptive immunity demonstrate the complex nature of the inflammation associated with AKI.
Acknowledgments
This work was supported by grants from the National Institutes of Health RO1 DK56223, RO1 DK62324, and RO1 DK06595.
References
- 1.Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med. 1996;334:1448–1460. doi: 10.1056/NEJM199605303342207. [DOI] [PubMed] [Google Scholar]
- 2.Thurman JM, Ljubanovic D, Royer PA, Kraus DM, Molina H, Barry NP, Proctor G, Levi M, Holers VM. Altered renal tubular expression of the complement inhibitor crry permits complement activation after ischemia/reperfusion. J Clin Invest. 2006;116:357–368. doi: 10.1172/JCI24521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li L, Huang L, Sung SS, Lobo PI, Brown MG, Gregg RK, Engelhard VH, Okusa MD. NKT cell activation mediates neutrophil IFN-gamma production and renal ischemia-reperfusion injury. J Immunol. 2007;178:5899–5911. doi: 10.4049/jimmunol.178.9.5899. [DOI] [PubMed] [Google Scholar]
- 4.Kelly KJ, Williams WW, Jr, Colvin RB, Meehan SM, Springer TA, Gutierrez-Ramos JC, Bonventre JV. Intercellular adhesion molecule-1-deficient mice are protected against ischemic renal injury. J Clin Invest. 1996;97:1056–1063. doi: 10.1172/JCI118498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Day YJ, Huang L, Ye H, Linden J, Okusa MD. Renal ischemia-reperfusion injury and adenosine 2a receptor-mediated tissue protection: role of macrophages. Am J Physiol Renal Physiol. 2005;288:F722–F731. doi: 10.1152/ajprenal.00378.2004. [DOI] [PubMed] [Google Scholar]
- 6.Day YJ, Huang L, Ye H, Li L, Linden J, Okusa MD. Renal ischemia-reperfusion injury and adenosine 2a receptor-mediated tissue protection: the role of CD4+ T cells and IFN-gamma. J Immunol. 2006;176:3108–3114. doi: 10.4049/jimmunol.176.5.3108. [DOI] [PubMed] [Google Scholar]
- 7.Sutton TA, Mang HE, Campos SB, Sandoval RM, Yoder MC, Molitoris BA. Injury of the renal microvascular endothelium alters barrier function after ischemia. Am J Physiol Renal Physiol. 2003;285:F191–F198. doi: 10.1152/ajprenal.00042.2003. [DOI] [PubMed] [Google Scholar]
- 8.Brodsky SV, Yamamoto T, Tada T, Kim B, Chen J, Kajiya F, Goligorsky MS. Endothelial dysfunction in ischemic acute renal failure: rescue by transplanted endothelial cells. Am J Physiol Renal Physiol. 2002;282:F1140–F1149. doi: 10.1152/ajprenal.00329.2001. [DOI] [PubMed] [Google Scholar]
- 9.Awad AS, Ye H, Huang L, Li L, Foss FW, Jr, Macdonald TL, Lynch KR, Okusa MD. Selective sphingosine 1-phosphate 1 receptor activation reduces ischemia-reperfusion injury in mouse kidney. Am J Physiol Renal Physiol. 2006;290:F1516–F1524. doi: 10.1152/ajprenal.00311.2005. [DOI] [PubMed] [Google Scholar]
- 10.Oh DJ, Dursun B, He Z, Lu L, Hoke TS, Ljubanovic D, Faubel S, Edelstein CL. Fractalkine receptor (CX3CR1) inhibition is protective against ischemic acute renal failure in mice. Am J Physiol Renal Physiol. 2008;294:F264–F271. doi: 10.1152/ajprenal.00204.2007. [DOI] [PubMed] [Google Scholar]
- 11.Thurman JM, Lenderink AM, Royer PA, Coleman KE, Zhou J, Lambris JD, Nemenoff RA, Quigg RJ, Holers VM. C3a is required for the production of CXC chemokines by tubular epithelial cells after renal ishemia/reperfusion. J Immunol. 2007;178:1819–1828. doi: 10.4049/jimmunol.178.3.1819. [DOI] [PubMed] [Google Scholar]
- 12.Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, Alexander SI, Sharland AF, Chadban SJ. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117:2847–2859. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leemans JC, Stokman G, Claessen N, Rouschop KM, Teske GJ, Kirschning CJ, Akira S, van der Poll T, Weening JJ, Florquin S. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J Clin Invest. 2005;115:2894–2903. doi: 10.1172/JCI22832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shigeoka AA, Holscher TD, King AJ, Hall FW, Kiosses WB, Tobias PS, Mackman N, McKay DB. TLR2 is constitutively expressed within the kidney and participates in ischemic renal injury through both MyD88-dependent and -independent pathways. J Immunol. 2007;178:6252–6258. doi: 10.4049/jimmunol.178.10.6252. [DOI] [PubMed] [Google Scholar]
- 15.Thornton MA, Winn R, Alpers CE, Zager RA. An evaluation of the neutrophil as a mediator of in vivo renal ischemic-reperfusion injury. Am J Pathol. 1989;135:509–515. [PMC free article] [PubMed] [Google Scholar]
- 16.Kielar ML, John R, Bennett M, Richardson JA, Shelton JM, Chen L, Jeyarajah DR, Zhou XJ, Zhou H, Chiquett B, Nagami GT, Lu CY. Maladaptive role of IL-6 in ischemic acute renal failure. J Am Soc Nephrol. 2005;16:3315–3325. doi: 10.1681/ASN.2003090757. [DOI] [PubMed] [Google Scholar]
- 17.Dong X, Swaminathan S, Bachman LA, Croatt AJ, Nath KA, Griffin MD. Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia-reperfusion injury. Kidney Int. 2007;71:619–628. doi: 10.1038/sj.ki.5002132. [DOI] [PubMed] [Google Scholar]
- 18.Dong X, Swaminathan S, Bachman LA, Croatt AJ, Nath KA, Griffin MD. Antigen presentation by dendritic cells in renal lymph nodes is linked to systemic and local injury to the kidney. Kidney Int. 2005;68:1096–1108. doi: 10.1111/j.1523-1755.2005.00502.x. [DOI] [PubMed] [Google Scholar]
- 19.Burne MJ, Daniels F, El Ghandour A, Mauiyyedi S, Colvin RB, O’Donnell MP, Rabb H. Identification of the CD4(+) T cell as a major pathogenic factor in ischemic acute renal failure. J Clin Invest. 2001;108:1283–1290. doi: 10.1172/JCI12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burne-Taney MJ, Ascon DB, Daniels F, Racusen L, Baldwin W, Rabb H. B cell deficiency confers protection from renal ischemia reperfusion injury. J Immunol. 2003;171:3210–3215. doi: 10.4049/jimmunol.171.6.3210. [DOI] [PubMed] [Google Scholar]
- 21.Park P, Haas M, Cunningham PN, Bao L, Alexander JJ, Quigg RJ. Injury in renal ischemia-reperfusion is independent from immunoglobulins and T lymphocytes. Am J Physiol Renal Physiol. 2002;282:F352–F357. doi: 10.1152/ajprenal.00160.2001. [DOI] [PubMed] [Google Scholar]
- 22.Burne-Taney MJ, Yokota-Ikeda N, Rabb H. Effects of combined T- and B-cell deficiency on murine ischemia reperfusion injury. Am J Transplant. 2005;5:1186–1193. doi: 10.1111/j.1600-6143.2005.00815.x. [DOI] [PubMed] [Google Scholar]