

Abstract
Mouse fibroblast growth factor 15 (FGF15) and human ortholog FGF19 have been identified as the bile acid-induced intestinal factors that mediate bile acid feedback inhibition of cholesterol 7α-hydroxylase gene transcription in mouse liver. The mechanism underlying FGF15/FGF19 inhibition of bile acid synthesis in hepatocytes remains unclear. Chenodeoxycholic acid (CDCA) and a farnesoid X receptor (FXR)-specific agonist GW4064 strongly induced FGF19 but inhibited CYP7A1 mRNA levels in primary human hepatocytes. FGF19 strongly and rapidly repressed CYP7A1 but not small heterodimer partner (SHP) mRNA levels. Kinase inhibition and phosphorylation assays revealed that the MAPK/ERK1/2 pathway played a major role in mediating FGF19 inhibition of CYP7A1. However, siRNA knockdown of SHP did not affect FGF19 inhibition of CYP7A1. Interestingly, CDCA stimulated tyrosine phosphorylation of the FGF receptor 4 (FGFR4) in hepatocytes. FGF19 antibody and siRNA specific to FGFR4 abrogated GW4064 inhibition of CYP7A1. These results suggest that bile acid-activated FXR is able to induce FGF19 in hepatocytes to inhibit CYP7A1 by an autocrine/paracrine mechanism. We conclude that the hepatic FGF19/FGFR4/ERK1/2 pathway may inhibit CYP7A1 independent of SHP. In addition to inducing FGF19 in the intestine, bile acids in hepatocytes may activate the liver FGF19/FGFR4 signaling pathway to inhibit bile acid synthesis and prevent accumulation of toxic bile acid in human livers.
Keywords: CYP7A1, gene regulation, cytokines, bile acid synthesis, human hepatocytes
Introduction
Hepatic bile acid synthesis is feedback inhibited by bile acids returning to the liver via enterohepatic circulation (1). It has been known for more than fifty years that the rate of bile acid synthesis is stimulated by bile fistula and inhibited by intraduodenal infusion of taurine-conjugated bile acids (2, 3). More recent in vivo studies have shown that bile acids exert their negative feedback regulation at the first and rate-limiting enzyme of the pathway, CYP7A1 (4, 5). Intriguingly intraduodenal infusion, but not intravenous infusion of taurocholate markedly reduced CYP7A1 expression in bile fistula rats (6). We suggest that a putative intestinal factor, released or absorbed in the presence of bile acids in the intestine lumen, may play a role in the regulation of bile acid synthesis (6).
Bile acid-activated receptor, farnesoid X receptor (FXR) is known to induce a negative nuclear receptor, SHP, which interacts with liver receptor homolog-1 (LRH-1) and inhibits CYP7A1 gene expression (7, 8). Targeted deletion of the FXR gene in mice impaired bile acid and lipid homeostasis supporting the critical role of FXR in bile acid and lipid metabolism (9). However, ablation of the SHP gene in mice impaired but did not eliminate bile acid negative feedback inhibition of bile acid synthesis suggesting SHP-independent mechanisms exist (10, 11). These include bile acid-induced inflammatory cytokines, FGF receptor 4 (FGFR4) signaling, JNK/c-Jun, and pregnane X receptor (PXR) (10, 12-14).
Several recent studies have shown that the bile acid-activated FXR binds to a response element located in the second intron of the mouse FGF15, human FGF19 and rat FGF15 genes (15, 16). Adenovirus-mediated overexpression of FGF15 inhibits CYP7A1 gene expression (17). These investigators suggest that intestine FGF15 is transported to the liver to activate FGFR4 signaling to inhibit CYP7A1 gene transcription. However, these investigators were unable to identify FGF15 in the mouse sera and livers, and reported that feeding a synthetic FXR agonist GW4064 or cholic acid did not induce FGF15 in the mouse livers (17). Therefore, it is not clear as how the intestine FGF15 is transported to the liver to activate the FGFR4 and how FGFR4 signal inhibits CYP7A1 gene transcription. The FGF family of mitogenic cytokine consists of more than 20 small secreted-peptides involved in cell growth, development and migration (18, 19). FGF15 and FGF19 have been shown to increase metabolic rate, reverse diet-induced diabetes and decrease adiposity (20). FGF19 binds and activates FGFR4 in human and mouse livers (18). FGFR4 receptor tyrosine kinase activates several signaling pathways including JNK and ERK1/2 MAP kinases to exert its biological effects (15, 21, 22). FGF15 inhibition of CYP7A1 is partially abolished in SHP-/- mice suggesting that SHP-independent pathway may be involved in mediating FGFR signaling (17). Furthermore, FGF15 does not induce SHP in mouse and human hepatocytes and the expression of SHP is significantly decreased in FGFR4 transgenic mice expressing the constitutively active human FGFR4 (15, 22). Therefore the pathway that mediates FGF19 signaling in the liver remains to be identified. We studied bile acid induction of FGF19 mRNA and protein expression in primary human hepatocytes, and the role of FGF19 and FGFR4 signaling in mediating bile acid repression of CYP7A1 in the liver.
Materials and methods
Cell culture
HepG2 cells were obtained from ATCC (Manassas, VA). Primary human hepatocytes were isolated from human donors and were obtained from the Liver Tissue Procurement and Distribution System of National Institute of Health (S. Strom, University of Pittsburgh, PA). Cells were maintained as described previously (23).
Reagents
The reagents were obtained from the following sources: PD98059, SB203580 and SP600125 were from CalBiochem; U0126 was from Upstate Biotec (Lake Placid, NY). Recombinant FGF19 was from R&D systems (Minneapolis, MN). GW4064 was a generous gift from Dr. C. Kremoser (Phenex Pharma AG, Ludwigshafen, Germany).
RNA isolation and Quantitative Real-time PCR (Q-PCR)
Total RNA was isolated using Tri-reagent (Sigma, St. Louis, MO) according to the manufacturer's instruction. Reverse-transcription and Q-PCR were performed to detect relative mRNA expression using Taqman probes (ABI) as described previously (23).
Immunoblotting analysis
Cell lysate preparation and Immunoblotting analysis were performed as described previously (23). Anti-FGFR4 and anti-SHP antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA); anti-FGF19 antibody was from Upstate (Lake Placid, NY); antibodies against ERK1/2, phospho-ERK1/2, JNK, phospho-JNK, p38, phospho-p38, and anti-phosphotyrosine antibodies were from Cell Signaling Technology (Beverly, MA). The Immunoblots were visualized by ECL chemiluminescence detection system (GE Healthcare, UK).
Enzyme-linked immunosorbent assay (ELISA)
A sandwich ELISA kit was used for colorimetric detection of FGF19 in cell culture media (FGF19 Quantikine ELISA kit, R&D Systems) following the manufacturer's instructions. All cell culture media samples from three primary human hepatocytes were assayed in duplicate, the coefficient of variation was 2%.
Small Interference RNA (siRNA) assays
The SMART pool siRNAs for human FGFR4 and SHP were purchased from Dharmacon Research (Lafayette, Co) and transfected into HepG2 cells using LipofectAMINE 2000 reagent (Life Technologies, Inc., Gaithersburg, MD). Forty-eight hours after transfection, cells were extracted and analyzed the mRNA levels of FGFR4 and CYP7A1 and SHP by Q-PCR.
Statistical analyses
All experimental data are shown as the means ± SD. Multiple groups were tested by one-way ANOVA followed by Dunnett's test to determine which groups are significantly different from the control group. A p value < 0.05 was considered to be significant. *, p < 0.05; **, p < 0.001
Results
Activation of FXR strongly induces FGF19 in primary human hepatocytes
To study the role of FGF19 in the regulation of CYP7A1 in hepatocytes, we treated primary human hepatocytes with CDCA (50 μM), a natural ligand of FXR or GW4064 (1 μM), an efficacious and highly specific FXR agonist, and assayed the mRNA expression levels of FGF19, CYP7A1, SHP, and hepatocyte nuclear factor 4α (HNF4α) by real time PCR. Basal FGF19 mRNA levels in primary human hepatocytes were very low as indicated by high Ct values of 30 ±2.0 (n=6). Both CDCA and GW4064 dramatically increased FGF19 mRNA levels with time, reaching a maximum level of over 100-fold higher than the basal level in 24 h (Fig. 1A). CDCA and GW4064 rapidly and strongly repressed CYP7A1 mRNA expression in 3 to 6 h (Fig. 1B). CDCA and GW4064 induced SHP mRNA expression rapidly by about 5-fold within 1 h. However, SHP expression levels returned to low basal levels after 24 h. (Fig. 1C). CDCA and GW4064 had little effect on HNF4α mRNA levels (Fig. 1D). We also investigated the effect of cholic acid, a weaker endogenous FXR agonist on FGF19 and CYP7A1 mRNA expression. Treatment of human primary hepatocytes with 50 μM cholic acid for 24 hrs only induced FGF19 mRNA expression by about 10-fold and repressed CYP7A1 mRNA expression by 50% (data not shown). These effects are much weaker than CDCA (Fig. 1A and Fig. 1B). These results suggest that FGF19 is highly induced by CDCA or GW4064-activated FXR in human hepatocytes. FGF19 expression levels are inversely correlated to CYP7A1 mRNA expression.
Fig. 1.
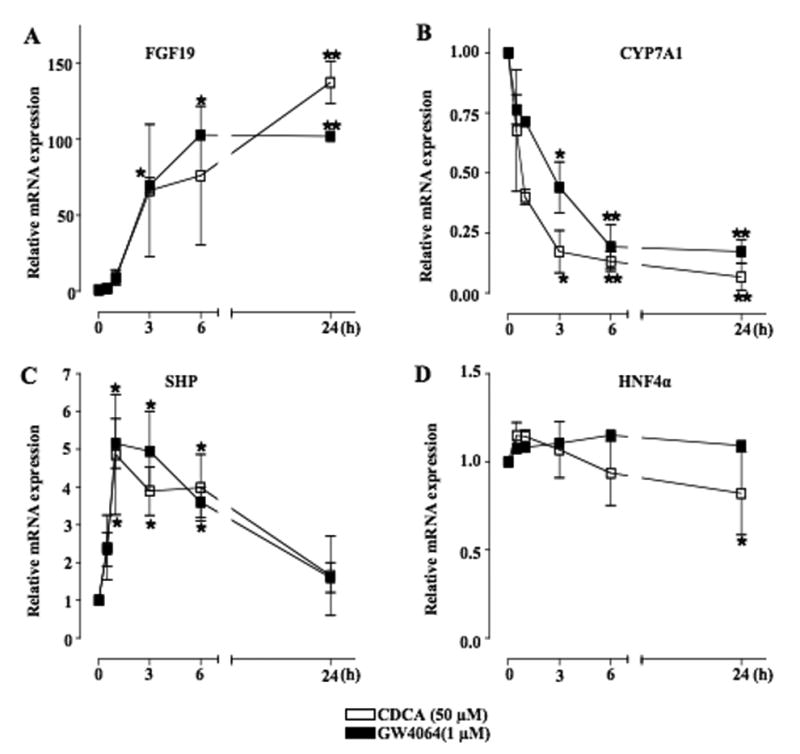
CDCA and GW4064 increased FGF19 and repressed CYP7A1 mRNA expression in primary human hepatocytes. Primary human hepatocytes were treated with CDCA (50 μM) or GW4064 (1 μM) for a period of time indicated. Total RNA was isolated for real-time quantitative PCR analysis of relative mRNA expression of FGF19 (A), CYP7A1 (B), SHP (C) and HNF4α (D) in hepatocytes. Data represent the mean ± SD of at least three different donor human primary hepatocytes.
To assay FGF19 protein expression, we performed immunoblot analysis with the FGF19-specific antibody. CDCA (50 μM) induced FGF19 protein expression within 3 h of treatment and gradually increasing in 24 to 48 h (Fig. 2A). Since FGFs contain a signal sequence for secretion, an ELISA assay was used to measure FGF19 levels in cell culture media. Without treatment, FGF19 was not detectable in cell culture media. CDCA (50 μM) time-dependently induced FGF19 excretion to 1,300 pg/ml in cultured media at 48 h (Fig. 2B). Moreover, CDCA dose-dependently induced FGF19 excretion (Fig. 2C). At physiological concentrations of CDCA (10 to 25 μM), CDCA strongly induced FGF19 secretion. These results suggest that FGF19 is highly induced by CDCA and GW4064 in human hepatocytes.
Fig. 2.
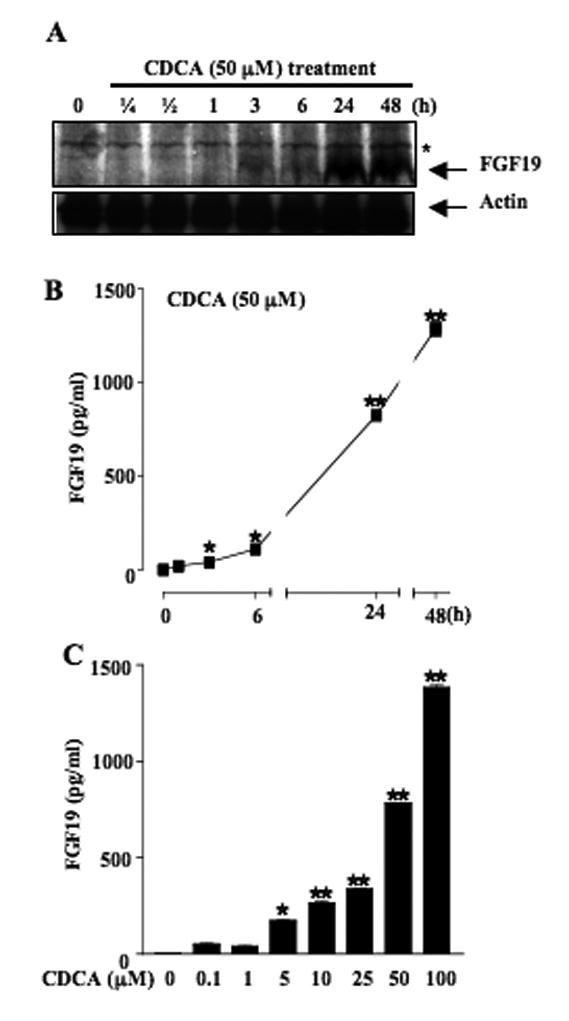
CDCA induced FGF19 protein expression and secretion in primary human hepatocytes. (A) Western Blot analysis of FGF19 protein expression in primary human hepatocytes by CDCA. Primary human hepatocytes were treated with CDCA (50 μM) for a period of time indicated and whole cell extracts (50 μg/lane) were then analyzed by immunoblotting with FGF19 and actin antibodies. Data are representative of two independently performed experiments. An asterisk indicates non-specific binding. (B) Effects of CDCA on FGF19 secretion to culture media. Primary human hepatocytes were treated with CDCA for a period of time indicated and collected for analysis by ELISA assay. (C) CDCA dose-responses on FGF19 secretion. Primary human hepatocytes were treated with CDCA for 24 h in the concentrations indicated. FGF19 expression was determined by ELISA assay.
FGF19 inhibits CYP7A1 mRNA expression in primary human hepatocytes
We then studied the effect of FGF19 on CYP7A1 mRNA expression in primary human hepatocytes. Fig. 3A shows that FGF19 (40 ng/ml) rapidly inhibits CYP7A1 mRNA expression in 3-6 h, persisting for at least 48 h. FGF19 dose-dependently inhibited CYP7A1 mRNA expression in primary human hepatocytes (Fig. 3B). To address whether FGF19 regulates SHP, we assayed SHP mRNA levels. In contrast to CDCA and GW4064, FGF19 did not have a significant effect on SHP mRNA expression in primary human hepatocytes (Fig. 3C and 3D). These results suggested that FGF19 repression of CYP7A1 might not require SHP induction.
Fig. 3.
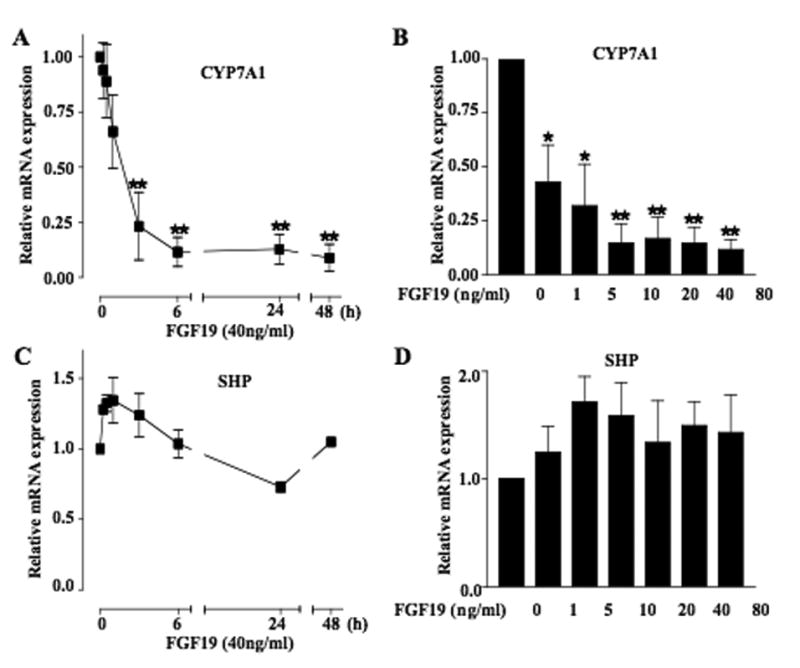
FGF19 repressed CYP7A1 mRNA expression in primary human hepatocytes. Primary human hepatocytes were treated with FGF19 (40 ng/ml) for a period of time or treated for 24 h with different concentrations of FGF19. Total RNAs were isolated for real-time quantitative PCR analysis of relative mRNA expression levels of CYP7A1 (A and B) and SHP (C and D). Data represent the mean ± SD of at least three donors primary human hepatocytes.
Knockdown of SHP expression did not abolish FGF19 repression on CYP7A1
To further confirm the role of SHP in FGF19-mediated repression of CYP7A1 gene expression, we examined the CYP7A1 mRNA expression levels in HepG2 cells after siRNA knockdown of SHP expression. Immunoblot analysis showed that siRNA to SHP efficiently knocked down SHP protein expression in HepG2 (Fig. 4A), and SHP mRNA expression by 50% with or without FGF19 (Fig. 4B). Knockdown of SHP mRNA by siRNA increased CYP7A1 mRNA levels by 50% in the absence of FGF19, but failed to block FGF19 repression of CYP7A1 mRNA (Fig. 4C). These data support the hypothesis that SHP may not be required for FGF19 inhibition of CYP7A1.
Fig. 4.
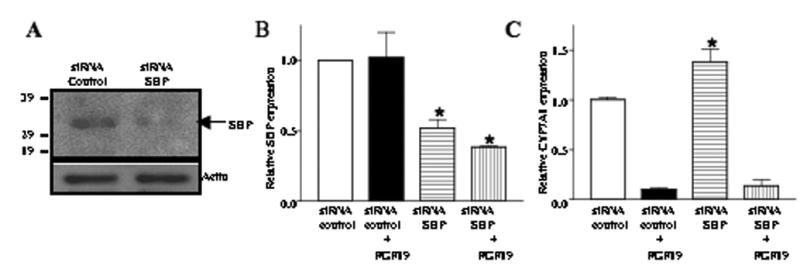
siRNA knockdown of SHP did not abolish FGF19 repression of CYP7A1 expression. (A) The effects of siRNA-SHP on the SHP protein expression were measured by Western blot analysis Actin expression was used as a loading control. Data represent one of three separate experiments. (B) HepG2 cells were transfected with the SMART Pool of siRNA-SHP (200 pmol) and control siRNA (cyclophilin B, 200 pmol) and treated with or without FGF19 (40 ng/ml) for 6 h. Total RNA was isolated for Q-PCR analysis of SHP ( A B) and CYP7A1 (C) mRNA levels. Data show relative mRNA expression of siRNA-SHP (200 pmol) treated to the control siRNA (200 pmol) treated samples. Data represent the mean ± SD of at least three individual experiments.
The ERK1/2 MAPK pathway s plays a major role in mediating FGF19 inhibition of CYP7A1
To identify signaling pathways that may mediate FGF19 inhibition of CYP7A1 gene transcription in primary human hepatocytes, we used several specific inhibitors of MAP kinases to study their effect on FGF19 inhibition of CYP7A1 mRNA expression. Fig. 5A shows that U0126 (10 μM), an inhibitor of MEK in the ERK1/2 pathway, strongly stimulates CYP7A1 mRNA expression by 7.5-fold in the absence of FGF19 (open bars). This indicates that CYP7A1 is under the negative regulation by the ERK1/2 signaling pathway in human hepatocytes. Furthermore, FGF19 reduced the stimulatory effect of U1026 on CYP7A1 by 50% (closed bar). On the other hand, a JNK inhibitor, SP600125 (25 μM) stimulated basal levels of CYP7A1 mRNA expression by only 50% and FGF19 reduced SP600125 stimulatory effect by about 40%. Moreover, combination of U1026 and SP600125 induced CYP7A1 mRNA by 12-fold and almost completely blocked FGF19 inhibition on CYP7A1. A p38 kinase inhibitor, SB203580 (25 μM) had no effect on basal or FGF19 inhibition of CYP7A1 mRNA expression suggesting that the p38 kinase is not involved in FGF19 inhibition of CYP7A1 (data not shown). These results suggest that FGF19 inhibition of CYP7A1 in human hepatocytes is at least partially mediated through activation of the ERK1/2MAPK pathway.
Fig. 5.
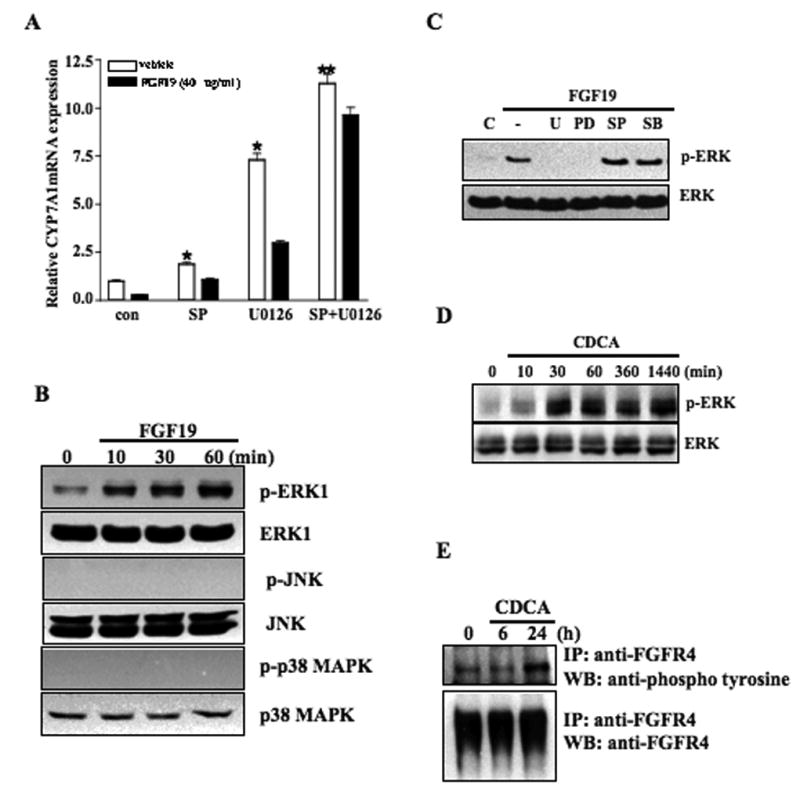
CDCA, FGF19, and MAP kinase inhibitors stimulate CYP7A1 mRNA expression and activate MAP kinases in primary human hepatocytes. (A) Primary human hepatocytes were treated with specific MAP kinase inhibitors: ERK1/2:U0126 (10 μM) and/or JNK: SP600125 (SP, 25 μM) for 1 h, and then with FGF19 (40 ng/ml) for 6 h. Total RNAs were isolated for real-time quantitative PCR analysis of relative mRNA expression levels of CYP7A1 in primary human hepatocytes. Data represent the mean ± SD of at least three donor hepatocytes independently performed. (B) Primary human hepatocytes were treated with FGF19 (40 ng/ml) for a period of time indicated and whole cell extracts (50 μg/lane) were then analyzed by immunoblotting with antibodies against ERK1/2, phospho-ERK1/2, JNK, phospho-JNK, p38, or phospho-p38 as indicated. (C) Primary human hepatocytes were treated with specific MAP kinase inhibitors: ERK1/2:U0126 (U, 10 μM), PD98059 (PD, 25 μM) and/or JNK: SP600125 (SP, 25 μM) for 1 h, and then with FGF19 (40 ng/ml) for 30 min. Whole cell extracts (50 μg/lane) were then analyzed by immunoblotting with ERK1/2 or phospho-ERK1/2 antibodies. Data are representative of two donor hepatocytes independently performed. (D) Primary human hepatocytes were treated with CDCA (50 μM) for a period of time indicated. Cell lysates were isolated for western blots using antibodies against ERK or phospho-ERK. (E) Primary human hepatocytes were treated with CDCA (50 μM) for a period of time indicated and cell extracts were immunoprecipitated with anti-FGFR4 antibody. Western blot analysis was performed using anti-phosphor-tyrosine antibody. Western blot analysis for FGFR4 using goat anti-FGFR4 antibody was performed to confirm the equal amounts of FGFR4 precipitant.
We next investigated the effects of FGF19 on activation of MAP kinases by kinase phosphorylation assays. Primary human hepatocytes were treated with 40 ng/ml FGF19 for 10, 30 and 60 min. Lysates were analyzed by mmunoblotting using specific antibodies to each kinase and their phospho-specific antibodies. As shown in Fig. 5B, ERK was phosphorylated without FGF19 treatment. This is consistent to the results from kinase inhibition assays that ERK1/2 is active in inhibiting CYP7A1 and the ERK1/2 inhibitor stimulated CYP7A1 mRNA expression in human hepatocytes (Fig. 5A). FGF19 (40 ng/ml) time-dependently stimulated phosphorylation of ERK1/2 in primary human hepatocytes (Fig. 5B). FGF19 did not activate p38 kinase. Surprisingly, FGF19 did not activate JNK in this assay. This result does not support a key role of JNK in mediating FGF19 inhibition of CYP7A1. To further confirm that FGF19 activates the ERK1/2 pathway, primary human hepatocytes were pretreated with FGF19 and ERK1/2 inhibitors for 30 min, and ERK and phospho-ERK in cell lysates were detected by immunoblot analysis. As shown in Fig. 5C, pretreatment of an ERK1/2 inhibitor PD98059 or U0126 blocked FGF19 phosphorylation of ERK1/2. As negative controls, SP600125 (JNK inhibitor) and SB203580 (p38 inhibitor) did not affect FGF19 phosphorylation of ERK. Interestingly, CDCA (50 μM) strongly phosphorylated ERK1/2 in 30 min and lasted for 24 h (Fig. 5D). We conclude that FGF19 may specifically activate the ERK1/2 pathway, which may play a major role in mediating FGF19 inhibition of CYP7A1.
It is well established that FGF19 specifically activates a receptor tyrosine kinase FGFR4, which is mainly expressed in the liver (24). To determine whether CDCA activates FGFR4 by phosphorylation, we treated human primary hepatocytes with CDCA and isolated cell lysates for analysis of FGFR4 protein phosphorylation using an anti-phosphotyrosine antibody. Fig. 5E shows that immunoprecipitation with anti-FGFR4 antibodies detected abundant FGFR4 in non-treated primary human hepatocytes. CDCA treatment (50 μM) did not alter total FGFR4 protein levels. However, an anti-phosphotyrosine antibody detected increased expression of phosphorylated FGFR4 in primary human hepatocytes after CDCA treatment for 24 h (Fig. 5E). Since CDCA is not an activator of FGFR4, this result suggests that a FXR/FGF19/FGFR4 auto regulatory loop may exist in human hepatocytes.
Inhibition of FGF19 and FGFR4 abrogated FGF19 inhibition of CYP7A1 in primary human hepatocytes
To study the involvement of FGF19/FGFR4 signaling in inhibition of CYP7A1, we used a FGF19 neutralizing antibody and siRNA probes to FGFR4 to block FGF19/FGFR4 signaling. As shown in Fig. 6A (open bars), GW4064 treatment (2 μg/ml) strongly repressed CYP7A1 mRNA levels in primary human hepatocytes. Anti-FGF19 antibody partially abrogated the inhibitory effect of GW4064 on CYP7A1 mRNA expression (filled bars). Consistently, siRNA knockdown of FGFR4 (siFGFR4) reduced FGFR4 protein and mRNA expression in HepG2 cells (Fig. 6B and Fig. 6C), and resulted in 50% reduction of GW4064-mediated repression of CYP7A1 mRNA expression compared to control siRNA (Fig. 6D). Therefore, these results suggest that FXR-mediated induction of FGF19 contributes to the repression of CYP7A1 expression and the FGF19/FGFR4 an autocrine/paracrine pathway may regulate CYP7A1 in human hepatocytes.
Fig. 6.
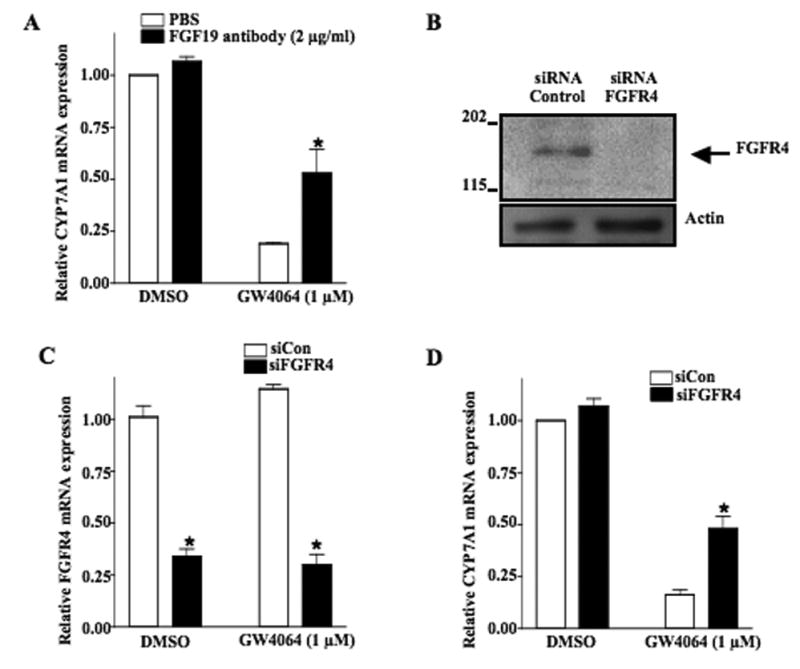
Inhibition of FGF19 and FGFR4 activity abrogates inhibition of CYP7A1 by FXR agonist GW4064 in human hepatocytes. (A) Primary human hepatocytes were treated with anti-FGF19 antibody (closed bar) with or without GW4064. Total RNAs were isolated for Q-PCR analysis of relative CYP7A1 mRNA expression levels in hepatocytes. (B) Western blot analysis of the effect of siRNA-FGFR4 on the FGFR4 protein expression. Actin expression was used as a loading control. Data represent one of three separate experiments. (C) HepG2 cells were transfected with the SMART Pool of siRNA to FGFR4 (siFGFR4, 200 pmol, closed bar) or control siRNA (siCon, 200 pmol, open bar) and treated with GW4064 for 24 h. Total RNAs were isolated for Q-PCR analysis of relative mRNA expression levels of FGFR4. (D) mRNAs isolated in (C) were used to assay relative mRNA expression levels of CYP7A1.
Discussion
Recent studies from Kliewer's laboratory have identified FGF15 as a bile acid/FXR induced intestinal hormone that inhibits Cyp7A1 gene transcription in the liver (17). FGF15 also stimulates gallbladder refilling in mice, thus serving as a feed forward signal for biliary bile acid excretion and storage (25). These investigators suggest that FGF15 in the intestine is secreted into circulation, transported to the liver to activate the FGFR4 signaling pathway and inhibit CYP7A1. This endocrine regulation of hepatic CYP7A1 by intestinal FGF15 is based on the fact that FGF15 is exclusively expressed in mouse jejunum and ileum but not in liver, while FGF15-specific receptor FGFR4 is highly expressed in mouse liver (17, 18). However, these investigators have not been able to detect FGF15 in mouse sera and livers (17, 26). A recent study of conditional liver- and intestine-FXR knockout mice has clearly demonstrated that knockout of the liver FXR did not affect GW4064 inhibition of CYP7A1, whereas knockout of the intestine FXR abrogated GW4064 inhibition of CYP7A1 (27). This study provides the first direct and compelling evidence that intestinal FXR, but not liver FXR is required for bile acid feedback inhibition of CYP7A1 and bile acid synthesis in the liver. The bile acid-induced FGF15 appears to be the “intestine factor” we proposed in 1995 (6).
It has been shown that FGF19 inhibits CYP7A1 in primary human hepatocytes and HepG2 cells (15), and FGF19 is detected in human patient sera (28). Interestingly the serum FGF19 levels show diurnal variation similar to serum 7α-hydroxy-4-cholesten-3-one, a product of CYP7A1 and a marker of the rate of bile acid synthesis and CYP7A1 activity in humans. These investigators suggest that serum FGF19 is derived from the intestine and is transported via blood circulation to hepatocytes to inhibit CYP7A1 in human liver. Here we show that FGF19 mRNA and protein are expressed at very low levels in human hepatocytes. An endogenous FXR ligand, CDCA and a specific FXR agonist, GW4064 strongly induced FGF19 mRNA and protein expression by more than one hundred-fold to inhibit CYP7A1 mRNA expression in primary human hepatocytes. Moreover, neutralizing FGF19 by a FGF19 antibody or siRNA knockdown of FGFR4 abrogated FGF19 inhibition of CYP7A1. FGF19 is readily detectable after CDCA treatment in human hepatocytes indicating that the human liver may contribute significant amounts of circulating FGF19 in humans. Our results also suggest that FGF19 produced in the liver may be a paracrine factor that inhibits CYP7A1 expression in hepatocytes by activating FGFR4 signaling in surrounding hepatocytes or an autocrine factor that directly activates intracellular signaling pathways in hepatocytes to inhibit CYP7A1 and bile acid synthesis in human livers. Thus bile acid-activated FXR may induce the production and secretion of FGF19 in both intestine and liver in humans. This is in contrast to the mouse that only the intestine produces and secretes FGF15. We also show that cholic acid, a weaker FXR ligand is less effective in inducing FGF19 and inhibiting CYP7A1. It should be mentioned that the bile acid pool in mice is highly hydrophilic consisting mostly cholic acid and muricholic acids, which are synthesized in mouse liver and are poor FXR ligands. This may explain lack of induction of FGF15 in the mouse liver by bile acids. In humans, the bile acid pool is highly hydrophobic consisting mostly cholic acid, chenodeoxycholic acid and deoxycholic acid, which are potent FXR ligands in human livers. Our current findings suggest that the liver-produced FGF19 may be a feed forward signal for biliary secretion and refilling of bile acids in the gallbladder, and a feedback signal for bile acid inhibition of bile acid synthesis. Like bile acid induction of inflammatory cytokines, bile acid activation of FGF19 signaling may be an rapid response to cholestatic liver injury to protect the liver against bile acid toxicity (29).
Despite a plethora of studies of the nuclear receptor regulation of CYP7A1 in recent years, the molecular mechanism of bile acid feedback inhibition of CYP7A1 gene transcription remains unclear. It is generally recognized that the FXR/SHP cascade mechanism mediates bile acid feedback inhibition of CYP7A1 in the liver (7, 8). SHP is a negative factor that interacts with many nuclear receptors including LRH-1. SHP inhibits the trans-activating activity of LRH-1, which binds to the CYP7A1 gene. It has been proposed that LRH-1 is required for CYP7A1 expression and is a competence factor of the liver orphan receptor α (LXRα) that binds to and stimulates CYP7A1 gene in mice (8). However, the human CYP7A1 gene does not bind LXRα (30) and LRH-1 may compete with HNF4α for binding to the overlapping sequence and inhibits the human CYP7A1 gene (31, 32). A recent study of conditional liver LRH-1 knockout mice revealed that mRNA expression of sterol 12α-hydroxylase (CYP8B1) involved in cholic acid synthesis was markedly suppressed, but surprisingly CYP7A1 mRNA expression remained unchanged (33, 34). These investigators suggest that LRH-1 plays a key role in regulating CYP8B1 and bile acid composition but is not required for bile acid inhibition of CYP7A1 gene transcription (33, 34). Since LRH-1 is the major target gene of SHP, the physiological significance of the cascade FXR/SHP/LRH-1 pathway mediating bile acid feedback inhibition of CYP7A1 needs to be re-evaluated.
It has been suggested that FGF19/FGFR4 signaling activates JNK/c-Jun, which cooperates with SHP to inhibit CYP7A1 mRNA expression (15). However, study of human FGFR4 transgenic mice reveals that SHP mRNA expression levels are markedly decreased in FGFR4 transgenic mouse livers, but not increased in FGFR4 null mice. These investigators suggested that bile acid inhibition of bile acid synthesis and FGFR4 activation of c-Jun/JNK pathway are independent of each other, and JNK- and SHP-independent pathways for bile acid feedback inhibition may exist (22). It should be emphasized that induction of SHP mRNA expression by CDCA or GW4064 is rapid and transient, in contrast to a sustained induction of FGF19 and inhibition of CYP7A1 by CDCA and GW4064. This may be because CDCA activates several signaling pathways in addition to activation of FXR (35). Our current study reveals that FGF19 does not induce SHP expression in primary human hepatocytes. Furthermore, FGF19 strongly inhibits CYP7A1 expression even when expression of SHP was knock-downed. These results suggest that SHP may not be required for FGF19 to inhibit CYP7A1 at least in human hepatocytes.
Several recent studies have confirmed that FGFR4 activity requires a co-receptor β-Klotho, which is highly expressed in hepatocytes (24, 36, 37). β-Klotho induces ERK1/2 phosphorylation in response to FGF19 and is required for FGF19 binding to FGFR4 and inhibition of CYP7A1 (24, 36, 37). Thus our results on kinase inhibition and activation assays are consistent with the ERK1/2 pathway being a major MAP kinase involved in mediating FGF19 inhibition of CYP7A1. This is also consistent to a recent report that activation of FXR by CDCA or GW4064 increases ERK1/2 phosphorylation and knockout of the Fxr gene in mice reduced ERK1/2 phosphorylation (38). It appears that the FXR is required for ERK1/2 phosphorylation and FGF19 probably is the major factor mediating FXR activation of the ERK1/2/MAPK pathway. The major downstream targets of ERK1/2 signaling are c-Fos and early growth responsive-1 (EGR-1), the latter is highly induced by FGF19 in the liver (37). In summary, our present study provides the direct evidence that in addition to inducing FGF19 in the intestine, bile acids in hepatocytes may activate the liver FGF19/FGFR4 signaling pathway to inhibit bile acid synthesis and prevent accumulation of toxic bile acid in human livers.
Acknowledgments
This research was supported by NIH grants DK44442 and DK58379 to JYLC, and DK92310 to SS (LTPADS)
Abbreviations used
- BARE
bile acid response element
- CDCA
chenodeoxycholic acid
- CYP7A1
cholesterol 7α-hydroxylase
- CYP8B1
sterol 12α-hydroxylase
- FGF
fibroblast growth factor
- FGFR4
FGF receptor 4
- FXR
farnesoid X receptor
- LRH
liver receptor homolog
- SHP
small heterodimer partner
Footnotes
Potential conflict of interest: nothing to report.
References
- 1.Myant NB, Mitropoulos KA. Cholesterol 7 alpha-hydroxylase. J Lipid Res. 1977;18:135–153. [PubMed] [Google Scholar]
- 2.Eriksson S. Biliary excretion of bile acids and cholesterol in bile fistula rats; bile acids and steroids. Proc Soc Exp Biol Med. 1957;94:578–582. doi: 10.3181/00379727-94-23018. [DOI] [PubMed] [Google Scholar]
- 3.Bergstrom S, Danielsson H. On the regulation of bile acid formation in the rat liver. Acta Physiol Scand. 1958;43:1–7. doi: 10.1111/j.1748-1716.1958.tb01572.x. [DOI] [PubMed] [Google Scholar]
- 4.Shefer S, Hauser S, Bekersky I, Mosbach EH. Biochemical site of regulation of bile acid biosynthesis in the rat. J Lipid Res. 1970;11:404–411. [PubMed] [Google Scholar]
- 5.Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–174. doi: 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]
- 6.Pandak WM, Heuman DM, Hylemon PB, Chiang JY, Vlahcevic ZR. Failure of intravenous infusion of taurocholate to down-regulate cholesterol 7 alpha-hydroxylase in rats with biliary fistulas. Gastroenterology. 1995;108:533–544. doi: 10.1016/0016-5085(95)90083-7. [DOI] [PubMed] [Google Scholar]
- 7.Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6:517–526. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 8.Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, Mangelsdorf DJ. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell. 2000;6:507–515. doi: 10.1016/s1097-2765(00)00050-2. [DOI] [PubMed] [Google Scholar]
- 9.Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–744. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 10.Wang L, Lee YK, Bundman D, Han Y, Thevananther S, Kim CS, Chua SS, et al. Redundant pathways for negative feedback regulation of bile acid production. Dev Cell. 2002;2:721–731. doi: 10.1016/s1534-5807(02)00187-9. [DOI] [PubMed] [Google Scholar]
- 11.Kerr TA, Saeki S, Schneider M, Schaefer K, Berdy S, Redder T, Shan B, et al. Loss of nuclear receptor SHP impairs but does not eliminate negative feedback regulation of bile acid synthesis. Dev Cell. 2002;2:713–720. doi: 10.1016/s1534-5807(02)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyake JH, Wang SL, Davis RA. Bile acid induction of cytokine expression by macrophages correlates with repression of hepatic cholesterol 7alpha-hydroxylase. J Biol Chem. 2000;275:21805–21808. doi: 10.1074/jbc.C000275200. [DOI] [PubMed] [Google Scholar]
- 13.Yu C, Wang F, Kan M, Jin C, Jones RB, Weinstein M, Deng CX, et al. Elevated cholesterol metabolism and bile acid synthesis in mice lacking membrane tyrosine kinase receptor FGFR4. J Biol Chem. 2000;275:15482–15489. doi: 10.1074/jbc.275.20.15482. [DOI] [PubMed] [Google Scholar]
- 14.Li T, Chiang JY. Mechanism of rifampicin and pregnane X receptor inhibition of human cholesterol 7 alpha-hydroxylase gene transcription. Am J Physiol Gastrointest Liver Physiol. 2005;288:G74–84. doi: 10.1152/ajpgi.00258.2004. [DOI] [PubMed] [Google Scholar]
- 15.Holt JA, Luo G, Billin AN, Bisi J, McNeill YY, Kozarsky KF, Donahee M, et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003;17:1581–1591. doi: 10.1101/gad.1083503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J, Pircher PC, Schulman IG, Westin SK. Regulation of complement C3 expression by the bile acid receptor FXR. J Biol Chem. 2005;280:7427–7434. doi: 10.1074/jbc.M411473200. [DOI] [PubMed] [Google Scholar]
- 17.Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 18.Xie MH, Holcomb I, Deuel B, Dowd P, Huang A, Vagts A, Foster J, et al. FGF-19, a novel fibroblast growth factor with unique specificity for FGFR4. Cytokine. 1999;11:729–735. doi: 10.1006/cyto.1999.0485. [DOI] [PubMed] [Google Scholar]
- 19.Fu L, John LM, Adams SH, Yu XX, Tomlinson E, Renz M, Williams PM, et al. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology. 2004;145:2594–2603. doi: 10.1210/en.2003-1671. [DOI] [PubMed] [Google Scholar]
- 20.Tomlinson E, Fu L, John L, Hultgren B, Huang X, Renz M, Stephan JP, et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology. 2002;143:1741–1747. doi: 10.1210/endo.143.5.8850. [DOI] [PubMed] [Google Scholar]
- 21.Yu C, Wang F, Jin C, Wu X, Chan WK, McKeehan WL. Increased carbon tetrachloride-induced liver injury and fibrosis in FGFR4-deficient mice. Am J Pathol. 2002;161:2003–2010. doi: 10.1016/S0002-9440(10)64478-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu C, Wang F, Jin C, Huang X, McKeehan WL. Independent repression of bile acid synthesis and activation of c-Jun N-terminal kinase (JNK) by activated hepatocyte fibroblast growth factor receptor 4 (FGFR4) and bile acids. J Biol Chem. 2005;280:17707–17714. doi: 10.1074/jbc.M411771200. [DOI] [PubMed] [Google Scholar]
- 23.Song KH, Chiang JY. Glucagon and cAMP inhibit cholesterol 7alpha-hydroxylase (CYP7A1) gene expression in human hepatocytes: discordant regulation of bile acid synthesis and gluconeogenesis. Hepatology. 2006;43:117–125. doi: 10.1002/hep.20919. [DOI] [PubMed] [Google Scholar]
- 24.Kurosu H, Choi M, Ogawa Y, Dickson AS, Goetz R, Eliseenkova AV, Mohammadi M, et al. Tissue-specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J Biol Chem. 2007;282:26687–26695. doi: 10.1074/jbc.M704165200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi M, Moschetta A, Bookout AL, Peng L, Umetani M, Holmstrom SR, Suino-Powell K, et al. Identification of a hormonal basis for gallbladder filling. Nat Med. 2006;12:1253–1255. doi: 10.1038/nm1501. [DOI] [PubMed] [Google Scholar]
- 26.Nishimura T, Utsunomiya Y, Hoshikawa M, Ohuchi H, Itoh N. Structure and expression of a novel human FGF, FGF-19, expressed in the fetal brain. Biochim Biophys Acta. 1999;1444:148–151. doi: 10.1016/s0167-4781(98)00255-3. [DOI] [PubMed] [Google Scholar]
- 27.Kim I, Ahn SH, Inagaki T, Choi M, Ito S, Guo GL, Kliewer SA, et al. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J Lipid Res. 2007;48:2664–2672. doi: 10.1194/jlr.M700330-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Lundasen T, Galman C, Angelin B, Rudling M. Circulating intestinal fibroblast growth factor 19 has a pronounced diurnal variation and modulates hepatic bile acid synthesis in man. J Intern Med. 2006;260:530–536. doi: 10.1111/j.1365-2796.2006.01731.x. [DOI] [PubMed] [Google Scholar]
- 29.Li T, Jahan A, Chiang JY. Bile acids and cytokines inhibit the human cholesterol 7 alpha-hydroxylase gene via the JNK/c-jun pathway in human liver cells. Hepatology. 2006;43:1202–1210. doi: 10.1002/hep.21183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chiang JY, Kimmel R, Stroup D. Regulation of cholesterol 7alpha-hydroxylase gene (CYP7A1) transcription by the liver orphan receptor (LXRalpha) Gene. 2001;262:257–265. doi: 10.1016/s0378-1119(00)00518-7. [DOI] [PubMed] [Google Scholar]
- 31.Chiang JY, Stroup D. Identification and characterization of a putative bile acid-responsive element in cholesterol 7 alpha-hydroxylase gene promoter. J Biol Chem. 1994;269:17502–17507. [PubMed] [Google Scholar]
- 32.Stroup D, Crestani M, Chiang JY. Identification of a bile acid response element in the cholesterol 7 alpha-hydroxylase gene CYP7A. Am J Physiol. 1997;273:G508–517. doi: 10.1152/ajpgi.1997.273.2.G508. [DOI] [PubMed] [Google Scholar]
- 33.Mataki C, Magnier BC, Houten SM, Annicotte JS, Argmann C, Thomas C, Overmars H, et al. Compromised intestinal lipid absorption in mice with a liver-specific deficiency of liver receptor homolog 1. Mol Cell Biol. 2007;27:8330–8339. doi: 10.1128/MCB.00852-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee YK, Schmidt DR, Cummins CL, Choi M, Peng L, Zhang Y, Goodwin B, et al. Liver Receptor Homolog-1 Regulates Bile Acid Homeostasis But Is Not Essential for Feedback Regulation of Bile Acid Synthesis. Mol Endocrinol. 2008;22:1345–1356. doi: 10.1210/me.2007-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chiang JY. Regulation of bile acid synthesis: pathways, nuclear receptors, and mechanisms. J Hepatol. 2004;40:539–551. doi: 10.1016/j.jhep.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 36.Wu X, Ge H, Gupte J, Weiszmann J, Shimamoto G, Stevens J, Hawkins N, et al. Co-receptor requirements for fibroblast growth factor-19 signaling. J Biol Chem. 2007;282:29069–29072. doi: 10.1074/jbc.C700130200. [DOI] [PubMed] [Google Scholar]
- 37.Lin BC, Wang M, Blackmore C, Desnoyers LR. Liver-specific activities of FGF19 require Klotho beta. J Biol Chem. 2007;282:27277–27284. doi: 10.1074/jbc.M704244200. [DOI] [PubMed] [Google Scholar]
- 38.Wang YD, Yang F, Chen WD, Huang X, Lai L, Forman BM, Huang W. Farnesoid X receptor protects liver cells from apoptosis induced by serum deprivation in vitro and fasting in vivo. Mol Endocrinol. 2008;22:1622–1632. doi: 10.1210/me.2007-0527. [DOI] [PMC free article] [PubMed] [Google Scholar]