

Abstract
Activation of c-Jun N-terminal kinase (JNK) has been implicated as a mechanism in the development of steatohepatitis. This finding together with the reported role of JNK signaling in the development of obesity and insulin resistance, two components of the metabolic syndrome and predisposing factors for fatty liver disease, suggest that JNK may be a central mediator of the metabolic syndrome and an important therapeutic target in steatohepatitis. To define the isoform specific functions of JNK in steatohepatitis associated with obesity and insulin resistance, the effects of JNK1 or JNK2 ablation were determined in developing and established steatohepatitis induced by a high fat diet. High fat diet-fed jnk1 null mice failed to develop excessive weight gain, insulin resistance or steatohepatitis. In contrast, jnk2-/- mice fed a high fat diet were obese and insulin resistant similar to wild-type mice and had increased liver injury. In mice with established steatohepatitis, an antisense oligonucleotide knockdown of jnk1 decreased the amount of steatohepatitis in concert with a normalization of insulin sensitivity. Knockdown of jnk2 improved insulin sensitivity but had no effect on hepatic steatosis and markedly increased liver injury. A jnk2 knockdown increased hepatic expression of the pro-apoptotic Bcl-2 family members Bim and Bax and the increase in liver injury resulted in part from a Bim-dependent activation of the mitochondrial death pathway.
Conclusion:
JNK1 and JNK2 both mediate insulin resistance in high fat diet-fed mice, but the JNK isoforms have distinct effects on steatohepatitis with JNK1 promoting steatosis and hepatitis and JNK2 inhibiting hepatocyte cell death by blocking the mitochondrial death pathway.
Keywords: antisense oligonucleotides, Bim, high fat diet, metabolic syndrome, nonalcoholic fatty liver disease
The hepatocellular mechanisms underlying the development of steatosis and the progression to steatohepatitis in nonalcoholic fatty liver disease (NAFLD) remain unclear.1 A critical factor in the pathogenesis of this disease is thought to be the existence of insulin resistance, and NAFLD can be considered a component of the metabolic syndrome whose manifestations also include obesity and diabetes.2,3 Recent investigations have suggested that activation of the mitogen-activated protein kinase c-Jun N-terminal kinase (JNK) is a central mechanism for the development of obesity and insulin resistance as well as for steatohepatitis.4-7 JNK may therefore represent a unique common target for the therapy of a variety of diseases that comprise the metabolic syndrome, but whether an inhibition of JNK function will decrease or prevent further progression of established steatohepatitis is currently unknown.
Most cells including hepatocytes express two JNK genes, jnk1 and jnk2, which are alternatively spliced to yield α and β forms of both a p54 and p46 protein.8 Recent studies have demonstrated that the JNK isoforms differ in function as exemplified by the ability of JNK1 to phosphorylate and activate the transcription factor c-Jun, whereas JNK2 lacks this activity and may even oppose this action of JNK1.9 Our prior studies have implicated JNK signaling in the development of steatohepatitis. First, it was demonstrated that hepatocyte overexpression of the prooxidant enzyme cytochrome P450 2E1 as occurs in human NAFLD induced sustained JNK activation and insulin resistance.10 Second, the development of murine steatohepatitis induced by a methionine- and choline-deficient diet was associated with increased JNK activation.6 The development of steatosis and liver injury was prevented in jnk1 but not jnk2 null mice, indicating that JNK1 specifically functions in the development of this disease.6 JNK1 has also been demonstrated to mediate the development of obesity and insulin resistance in both high fat diet (HFD)-fed and genetically obese mouse models.4 JNK2 was reportedly not involved in these processes, but subsequent studies in mice haploinsufficient for jnk1 and null for jnk2 suggested that JNK2 may also promote the development of obesity and insulin resistance.7 These mice and jnk1-/- mice also failed to develop steatosis from a HFD.7 Thus, JNK1 function is clearly linked to the development of steatohepatitis, obesity and insulin resistance whereas the function of JNK2 in these conditions is less clear.
To more clearly establish the function of JNK1 and JNK2 in steatohepatitis occurring in the setting of obesity and insulin resistance, the effect of JNK1 or JNK2 ablation on the development of steatohepatitis was examined in HFD-fed mice. Although such studies are important to our understanding of the pathophysiology of NAFLD, findings derived from investigations of developing disease do not define JNK function in the progression or perpetuation of established steatohepatitis. Studies to examine the role of JNK in existing steatohepatitis have not been performed and are critical to determine whether JNK is a viable therapeutic target in this disease. The development of antisense oligonucleotides (ASO) that are effective in inhibiting JNK1 or JNK2 expression in the liver,11 led us to examine the effects of JNK inhibition in established as well as developing steatohepatitis in HFD-fed mice. The studies demonstrate that JNK1 mediates insulin resistance and both the development and progression of steatohepatitis. In contrast, although JNK2 promoted insulin resistance, this JNK isoform did not mediate the development or progression of steatohepatitis. These findings indicate that the JNK isoforms have distinct functions that differ among the various manifestations of the metabolic syndrome and have important implications for JNK as a therapeutic target in this disorder.
Materials and Methods
Animal Models
Male wild-type C57BL/6, jnk1-/-,12 jnk2-/-,13 and bim-/-14 mice (Jackson Laboratory, Bar Harbor, ME) were maintained under 12 h light/dark cycles with unlimited access to food and water. Genotyping was performed by PCR with established primers.12-14 Knockout mice had been backcrossed onto a C57BL/6 background for greater than six generations. At 4 weeks of age mice were continued on regular diet (Lab Diet, Richmond, IN, #5058) or begun on a HFD (60% kcal in fat; Research Diets, New Brunswick, NJ; #D12492). At the end of 16 weeks of HFD feeding, the mice were fasted for 6 h and then sacrificed. All studies were approved by the Animal Care and Use Committee of the Albert Einstein College of Medicine and followed the National Institutes of Health guidelines on the care and use of animals.
An acute knockdown of JNK1 and JNK2 in mice with established steatohepatitis was achieved by the administration of ASO to JNK1 and JNK2 as previously performed by Gunawan et al.11 A control oligonucleotide (Isis 141923) and ASO targeting mouse JNK1 (Isis 104492) and JNK2 (Isis 101759) were synthesized as 20 nucleotide, uniform phosphorothioate chimeric oligonucleotides and purified as previously described.15 These oligonucleotides are chimeric oligonucleotides containing 5 nuclease resistant 2′-O-methoxyethylribose-modified phosphorothioate residues on the 5′ and 3′ ends that flank a 2′-deoxyribonucleotide/phosphorothioate region that supports RNase H-based cleavage of the targeted messenger RNA. The sequences of the ASO are: control - 5′-CCTTCCCTGAAGGTTCCTCC-3′; JNK1 - 5′-TGTTGTCACGTTTACTTCTG-3′; and JNK2 -5′-GCTCAGTGGACATGGATGAG-3′. Mice fed 12 weeks of HFD were begun on twice weekly intraperitoneal injections of 25 mg/kg of ASO and received a total of 8 injections before sacrifice after 16 weeks on HFD. Some mice were similarly injected with equal volumes of phosphate-buffered saline (PBS). Studies were performed on mice two days after their last ASO injection.
Serum Assays
Serum alanine aminotransferase (ALT) was measured by commercial kit (TECO Diagnostics, Anaheim, CA). Serum glucoses were determined with an Ascensia Contour™ glucose meter (Bayer HealthCare, Mishawaka, IN). Serum insulin levels were measured by radioimmunoassay, as previously described.16 Levels of insulin resistance were determined by the homeostasis model assessment of insulin resistance, or HOMA (insulin resistance = fasting glucose mmol/L × fasting insulin mU/ml).17
Protein Isolation and Western Blotting
Total protein isolation from mouse liver and Western blotting was performed, as previously described.6 The primary antibodies were: rabbit anti-JNK, rabbit anti-c-Jun, rabbit anti-phospho-c-Jun, mouse anti-Bax (Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-phospho-JNK, rabbit anti-phospho-Bim, rabbit anti-Bad, rabbit anti-caspase 3, rabbit anti-caspase 7 (Cell Signaling, Beverly, MA), rabbit anti-Bim (EMD Biosciences, San Diego, CA), rabbit anti-Bid (kind gift of Xiao-Ming Yin, University of Pittsburgh, Pittsburgh, PA), rabbit anti-Mcl-1 (Rockland, Gilbertsville, PA) and rabbit protein disulfide isomerase (PDI; kind gift of Richard Stockert, Albert Einstein College of Medicine, Bronx, NY). Cytosolic and mitochondrial protein fractions were isolated from mouse liver, as previously described,18 and immunoblotting was performed with mouse anti-cytochrome c (PharMingen, San Diego, CA), and mouse anti-cytochrome oxidase (MitoSciences, Eugene, Oregon) antibodies in addition to the previous antibodies.
Histologic Analysis
Formalin fixed, hematoxylin and eosin stained liver sections were examined in a blinded fashion by a single pathologist and graded for steatosis and inflammation. The degree of steatosis in each specimen was determined by assessing the overall percentage of liver parenchyma containing lipid vacuoles with 0 = none, 1 = mild (<30%), 2 = moderate (30-60%), and 3 = marked (>60%). Inflammation was graded by the presence or absence of inflammatory cells with 0 = absent, 1 = minimal or focal occasional single clusters of inflammatory cells present in a few microscopic fields, 2 = mild inflammation, 3 = moderate inflammation, and 4 = marked inflammation.
Hepatic Triglyceride Content
Hepatic triglyceride (TG) content was determined by the Trig/GB Kit™ (Roche Diagnostics Indianapolis, IN), as previously described.6
Terminal Deoxynucleotide Transferase-mediated Deoxyuridine Triphosphate Nick Endlabeling (TUNEL) Assay
The TUNEL assay was employed to determine the numbers of apoptotic cells in liver sections. TUNEL was performed with a commercial kit (Promega, Madison, WI), as previously described.18 Numbers of TUNEL positive cells were counted per high powered field (400X magnification) under light microscopy in 20 random fields per liver.
Statistical analysis
Numerical results are expressed as mean ± SE that represents data from a minimum of three independent experiments. Statistical significance was determined by the Student's t test and defined as P<0.05. Calculations were made with Sigma Plot 2000 (SPSS Science, Chicago, IL).
Results
Absence of JNK1 Prevents Weight Gain and the Development of Insulin Resistance in HFD-fed Mice
To define JNK isoform specific functions in the development of steatohepatitis occurring in the setting of obesity and peripheral insulin resistance, wild-type, jnk1-/- and jnk2-/- mice were fed a HFD for 16 weeks. Wild-type mice fed the HFD had a 31% greater increase in body weight at the end of the 16 week period than mice fed regular diet (Table 1). In association with this weight gain, HFD-fed mice developed peripheral insulin resistance as demonstrated by significantly increased HOMA values (Table 1). Thus, 16 weeks of HFD feeding was sufficient to develop an overweight, insulin resistant mouse model.
Table 1.
Body and liver weights and indices of insulin resistance in regular diet (RD) and HFD fed mice.
| WT RD | WT HFD | jnk1-/- HFD | jnk2-/- HFD | |
|---|---|---|---|---|
| Body weight (g) | 31.8 ± 1.7 | 41.7 ± 1.6* | 31.2 ± 1.6† | 50.6 ± 1.1*† |
| Liver weight (g) | 1.2 ± 0.1 | 1.6 ± 0.1 | 1.2 ± 0.1† | 1.9 ± 0.1* |
| Body/liver ratio (%) | 3.9 ± 0.2 | 3.8 ± 0.2 | 3.8 ± 0.1 | 3.8 ± 0.1 |
| Glucose (mg/dl) | 120 ± 6.7 | 178 ± 10# | 131 ± 6† | 157 ± 10# |
| Insulin (ng/ml) | 2.4 ± 0.4 | 3.6 ± 0.6 | 1.2 ± 0.2#† | 4.1 ± 0.7# |
| HOMA | 16.7 ± 2.4 | 46.0 ± 9.1* | 4.6 ± 0.7#† | 80.9 ± 17.5# |
P<0.05 as compared to wild-type on RD
P<0.01 as compared to wild-type on RD
P<0.02 as compared to wild-type on HFD. Results are expressed as mean ± SE and each data point represents 6-18 animals.
JNK activation has been previously reported to mediate weight gain and the development of peripheral insulin resistance,4 two predisposing factors for human NAFLD. Consistent with this prior report, mice lacking JNK1 failed to develop the increases in total body and liver weight from the HFD feeding that occurred in wild-type mice (Table 1). HFD-induced peripheral insulin resistance also failed to develop in jnk1-/- mice. Serum glucose and insulin levels and HOMAs were significantly decreased in jnk1-/- mice as compared to wild-type mice with HFD feeding (Table 1). Insulin levels and HOMA values in HFD-fed jnk1-/- mice were even significantly lower than those of regular diet-fed wild-type mice (Table 1). In contrast, HFD-fed jnk2-/- animals had significantly greater weight gain and higher HOMA values than wild-type mice, although the differences in HOMAs did not reach statistical significance (Table 1). Thus, the excessive weight gain and insulin resistance that developed in this HFD-fed mouse model were JNK1 dependent whereas the absence of JNK2 led to increased body weight.
JNK1 but not JNK2 Null Mice Develop Decreased Steatohepatitis from a HFD
JNK activation in response to a HFD was assessed by immunoblot analysis of levels of the active, phosphorylated forms of JNK and its downstream substrate c-Jun. Protein levels of phospho-JNK and phospho-c-Jun were increased in the livers of mice fed 16 weeks of HFD (Fig. 1A). Levels of total JNK were unchanged whereas levels of total c-Jun were moderately increased (Fig. 1A). Thus, consistent with our previous findings in the methionine- and choline-deficient model of steatohepatitis,6 JNK/c-Jun activation occurred in association with the development of steatohepatitis. Jnk1-/- mice had a predominant loss of hepatic p46 JNK whereas jnk2-/- mice had decreased levels of p54 JNK (Fig. 1B and 1C), as previously reported.6 Phospho-JNK levels were decreased in both HFD-fed jnk knockout mice although jnk1-/- mice had marked decreases in both p54 and p46 phospho-JNK whereas in jnk2-/- mice the decrease was predominantly in p54 phospho-JNK (Fig. 1B and 1C). Levels of phospho-c-Jun were decreased in HFD-fed jnk1-/- mice and increased in jnk2-/- mice consistent with the known effects of the two JNK isoforms on c-Jun phosphorylation (Fig. 1B and 1C).
Fig. 1.
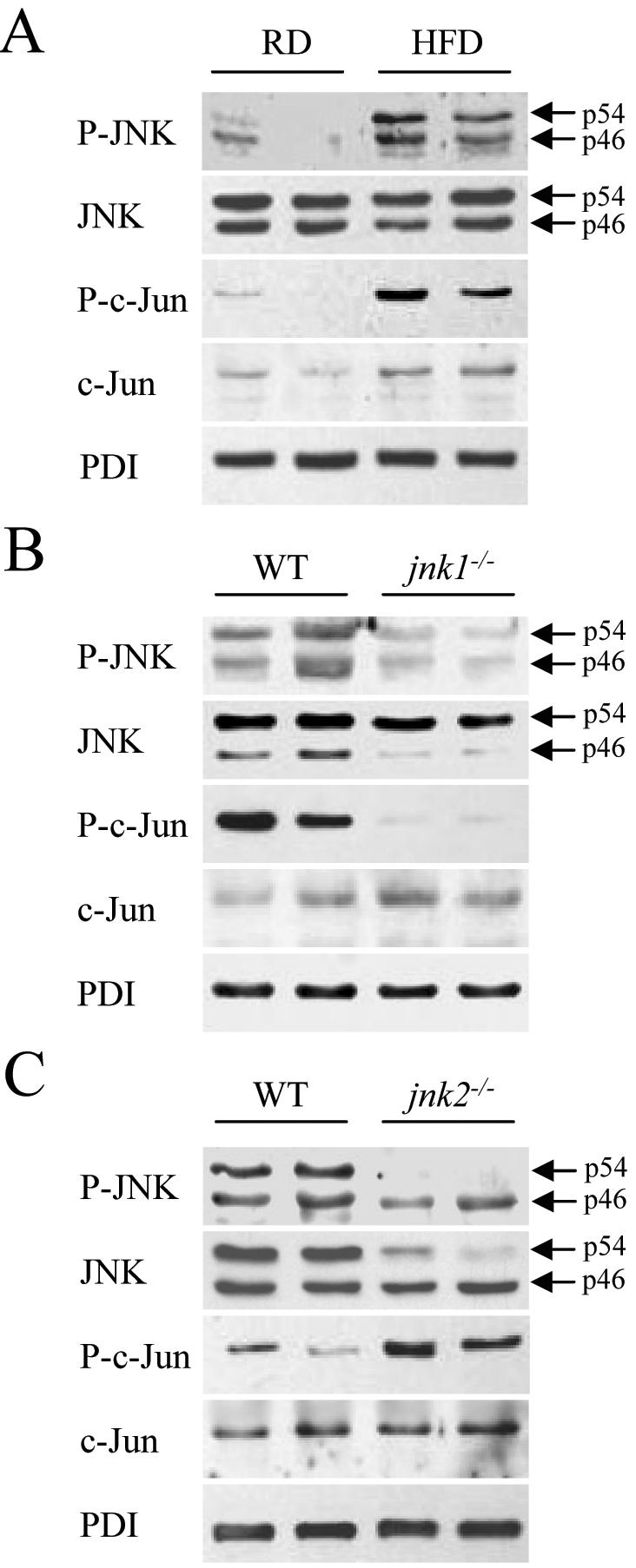
HFD induces hepatic JNK activation and ablation of jnk1 and jnk2 has differential effects on c-Jun phosphorylation. (A) Total protein was isolated from the livers of regular diet (RD)- and HFD-fed mice and immunoblotted with antibodies for phospho-JNK (P-JNK), total JNK, phospho-c-Jun (P-c-Jun) and total c-Jun. Stripped blots were reprobed for PDI. (B) Protein isolated from HFD-fed wild-type (WT) and jnk1-/- mice was immunoblotted with the same antibodies. (C) Immunoblots of protein from HFD-fed wild-type and jnk2-/- mice. The p54 and p46 forms of JNK are indicated with arrows. Findings are representative of those from 3 independent experiments.
By histology wild-type mice developed significant steatosis and hepatitis with a HFD (Fig. 2A and 2B). Blinded histological grading of the degree of steatosis revealed increased hepatic fat accumulation in wild-type mice fed the HFD (Fig. 3A). In contrast, steatosis did not develop in jnk1-/- mice (Fig. 2C and 3A). Histological evidence of steatosis in jnk2-/- mice was equivalent to that in wild-type mice (Fig. 2D and 3A). The protective effect of JNK1 ablation on the development of steatosis was confirmed by measures of hepatic TG levels. A significant increase in TG content occurred with HFD feeding in wild-type and jnk2-/- mice, but jnk1-/- mice had levels equivalent to regular diet-fed wild-type mice (Fig. 3B). The development of hepatic steatosis in response to a HFD was therefore JNK1 but not JNK2 dependent.
Fig. 2.
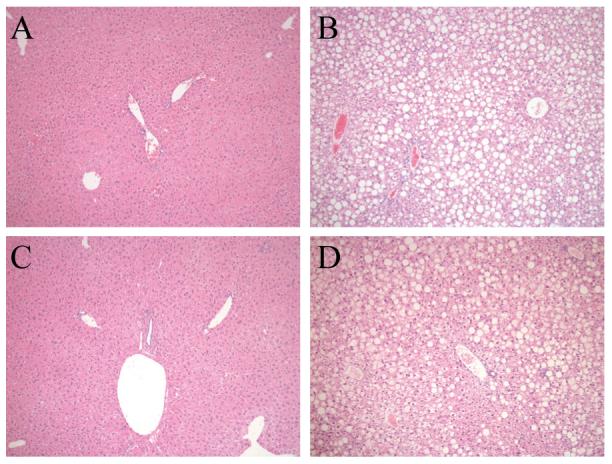
HFD-fed jnk1-/- mice have improved histology. Hematoxylin and eosin stained sections of (A) wild-type mice fed a regular diet, (B) wild-type mice fed 16 weeks of a HFD, (C) jnk1-/- mice fed a HFD, and (D) HFD-fed jnk2-/- mice (magnification 100X).
Fig. 3.

HFD-induced steatohepatitis is decreased in jnk1-/- mice whereas liver injury increased in jnk2-/- mice. (A) Histological steatosis grade in HFD-fed wild-type (WT), jnk1-/- and jnk2-/- mice (n=7-12; *P<0.0001 compared to wild-type or jnk2-/- mice). (B) TG content in the livers of regular diet (RD)- and HFD-fed wild-type, jnk1-/- and jnk2-/- mice (n=6-15; *P<0.01 compared to RD-fed wild-type mice; **P<0.0003 compared to jnk1-/- or jnk2-/- HFD-fed mice). (C) Serum ALT levels from the same animals (n=5-13; *P<0.001 as compared to RD-fed wild-type mice; **P<0.0001 as compared to HFD-fed wild-type mice; §P<0.01 when compared to HFD-fed wild-type or jnk1-/- mice). (D) Percentage of TUNEL positive cells in the same livers (n=5-9; *P<0.0001 compared to RD-fed wild-type mice; **P<0.00001 compared to wild-type or jnk2-/- HFD-fed mice; §P<0.005 compared to HFD-fed wild-type mice). (E) Histological inflammation grade in HFD-fed wild-type, jnk1-/- and jnk2-/- mice (n=7-12; *P<0.02 compared to wild-type or jnk2-/- mice).
Livers were also examined for the extent of injury and inflammation. In parallel with an absence of steatosis, jnk1-/- mice had reduced liver injury as reflected in decreased serum ALT levels and numbers of TUNEL positive cells (Fig. 3C and 3D). Histological evidence of inflammation was also significantly decreased in HFD-fed jnk1-/- mice (Fig. 3E). In contrast, HFD-fed jnk2-/- mice had significantly higher levels of serum ALT and TUNEL staining as compared to wild-type mice indicating that liver injury was increased in the absence of JNK2 (Fig. 3C and 3D). Thus, JNK1 function was critical for the development of steatosis and hepatitis in response to a HFD. In contrast, although lipid accumulation was unaffected by loss of JNK2, JNK2 functioned to protect against hepatocyte injury in the setting of a fatty liver.
Knockdowns of JNK1 and JNK2 both Reverse Insulin Resistance but have Opposing Effects on Established Steatohepatitis in HFD-fed Animals
Although initial studies established that the development of HFD-induced murine steatohepatitis was differentially regulated by JNK1 and JNK2, these investigations did not address the function of these factors in established liver disease. In an effort to define whether JNK1 could be a therapeutic target in NAFLD, it was necessary to investigate the role of the JNK isoforms in promoting or perpetuating established steatohepatitis. To test the effect of JNK inhibition on existing steatohepatitis, and to obtain further evidence of the differential effects of JNK1 and JNK2 in the pathophysiology of this disease, we employed ASO to acutely inhibit JNK function. In addition to allowing the initiation of JNK inhibition late in the progression of the disease, this alternative approach to knocking down JNK function had the potential advantage of avoiding the confounding variable of compensatory changes that may develop in knockout mice and affect their biological responses.
Within 12 weeks of being started on a HFD, wild-type mice developed significant insulin resistance (HOMA values of 41.9 ± 7.6 in HFD versus 16.5 ± 4.5 with regular diet), steatosis (triglyceride levels of 95.3 ± 16.8 and 22.1 ± 7.3 ug/mg protein in HFD- and regular diet-fed mice, respectively) and steatohepatitis (ALT levels of 95.3 ± 16.8 IU/ml in HFD-fed and 22.1 ± 7.3 IU/ml in regular diet-fed mice). Mice fed 12 weeks of the HFD were randomly assigned to twice weekly injections of a control oligonucleotide or the JNK1 or JNK2 ASO. Injections were continued for 4 weeks during which time the animals were continued on a HFD. The effectiveness and specificity of the ASO treatment protocol were assessed by immunoblots for total JNK levels as compared to PBS and control ASO-injected animals. Administration of the JNK1 ASO significantly decreased levels of p46 JNK without affecting those for p54 JNK (Fig. 4). Conversely JNK2 ASO treatment reduced levels of p54 but not p46 JNK (Fig. 4). The effects of the JNK ASO on levels of phospho- and total c-Jun (Fig. 4) were also similar to those in the knockout mice (Fig. 1B and 1C).
Fig. 4.
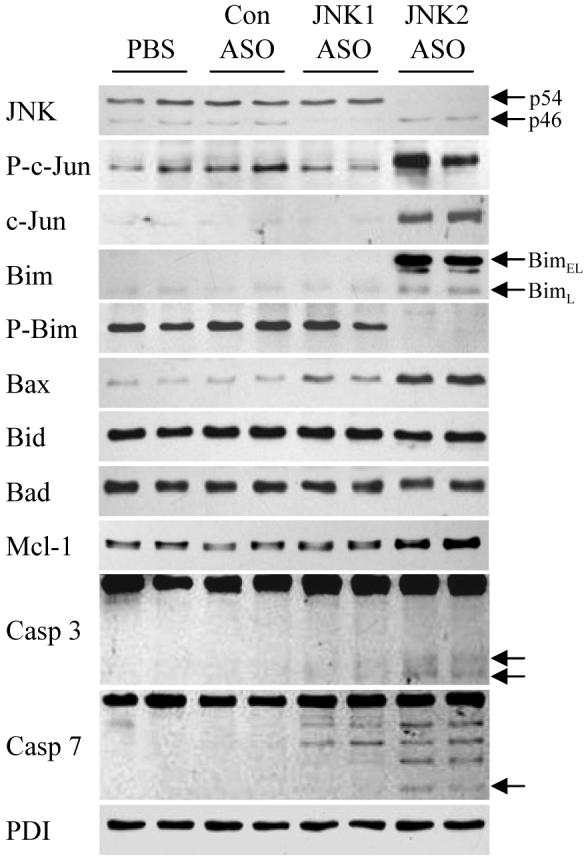
JNK2 ASO treatment of HFD-fed mice results in increased Bim and Bax expression. Total protein isolated from the livers of HFD-fed mice treated with PBS, or control (Con), JNK1 or JNK2 ASO was immunoblotted for total JNK, Bim, Ser65 phospho-Bim (P-Bim), Bax, Bad, Mcl-1, caspase 3 (Casp 3), caspase 7 (Casp 7) or PDI. Arrows indicate the p54 and p46 forms of JNK, BimEL and BimL and the cleaved fragments of caspase 3 and 7. Results are representative of three independent experiments.
ASO-treated animals were sacrificed after 16 weeks of HFD to determine the effect of 4 weeks of JNK inhibition on established steatohepatitis. Injection of the JNK1 ASO led to a complete reversal of insulin resistance as these animals had significantly reduced serum levels of glucose (Fig. 5A) and insulin (Fig. 5B) and HOMA (Fig. 5C). Surprisingly JNK2 ASO-injected animals had an even more profound decrease in serum glucose levels as well as a reduction in insulin levels (Fig. 5A and 5B). HOMA values were decreased to a greater extent in these animals than in JNK1 ASO-injected mice (Fig. 5C).
Fig. 5.

JNK1 and JNK2 ASO treatment of HFD-fed mice has differential effects on insulin resistance, steatosis and liver injury. (A) Fasting serum glucose levels in HFD-fed mice treated with the control (Con), JNK1 or JNK2 ASO (n=7-9; *P<0.0002 compared to control ASO treated; **P<0.00001 as compared to control or JNK1 ASO treated). (B) Fasting serum insulin levels in the same mice (n=7-9; *P<0.02 compared to control ASO-injected mice). (C) HOMA values in the animals (n=7-9; *P<0.01 compared to control ASO treated; **P<0.01 as compared to control or JNK1 ASO treated). (D) Hepatic TG levels (n=7-8; *P<0.04 as compared to wild-type and P<0.004 when compared to JNK2 mice). (E) Percentage of TUNEL positive cells in the same livers along with those from regular diet (RD)-fed mice who did not receive ASO treatment (∅) (n=3-5; *P<0.01 compared to control ASO injected; **P<0.01 compared to control or JNK1 ASO-treated mice).
Control ASO-injected mice had significant steatohepatitis by histology (Fig. 6A). In contrast, JNK1 ASO-injected mice had markedly decreased steatosis and hepatitis (Fig. 6B). JNK2 ASO-injected mice had marked liver injury with lobular disarray, increased inflammation and dying hepatocytes (Fig. 6C and 6D). These histological impressions were confirmed by biochemical analyses that revealed significantly decreased TG content, serum ALT levels and TUNEL positivity in JNK1 ASO-injected mice as compared to control ASO-injected mice (Fig. 5D, 5E and 5F). There was some nonspecific effect of ASO treatment as control ASO-injected animals had lower ALT levels, but similar TUNEL positivity, than uninjected animals (Fig. 3C and 3D versus Fig. 5E and 5F). TG levels were unaffected by the JNK2 ASO but levels of ALT and TUNEL positivity were markedly increased (Fig. 5D, 5E and 5F). Thus, similar to the effects of genetic JNK1 and JNK2 ablation on the development of steatohepatitis, knockdown of JNK1 reduced established steatohepatitis whereas a JNK2 knockdown worsened hepatitis without affecting the degree of steatosis. However, in the case of established steatohepatitis the effect of a loss of JNK2 on liver injury was far more pronounced.
Fig. 6.
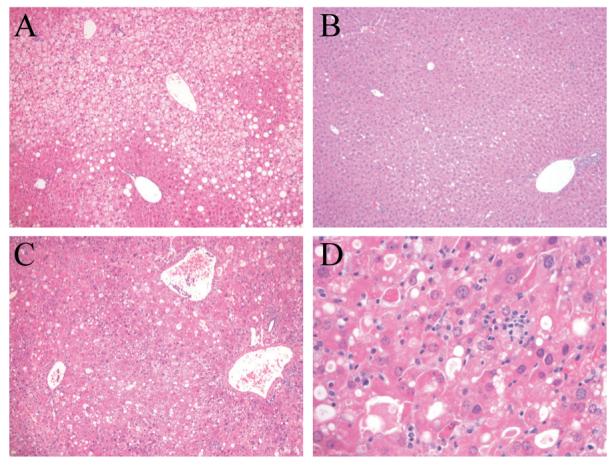
JNK1 ASO treatment decreases steatohepatitis in HFD-fed mice whereas JNK2 ASO treatment worsens liver injury. Hematoxylin and eosin stained sections of HFD-fed mice treated with the control (A), JNK1 (B) or JNK2 (C) ASO (magnification 100X). (D) Higher magnification (400X) of (C).
Acute JNK2 Knockdown Results in Bim Overexpression in HFD-fed Mice
Inhibition of JNK2 function not only failed to block the development of steatohepatitis or decrease the extent of preexistent disease but also worsened liver injury in both models. The fact that loss of JNK2 function did not affect the amount of steatosis suggested that increased injury resulted not from greater fat accumulation but rather from the induction of a cellular injury/death pathway. JNK is known to regulate the function of several Bcl-2 family proteins suggesting that an effect on one of these proteins may mediate the pro-apoptotic effect of the JNK2 knockdown. Investigations in cultured hepatocytes by Malhi et al.,19 previously demonstrated that increased levels of fatty acids induce JNK-dependent injury mediated by the pro-apoptotic Bcl-2 family member Bim. To investigate the possibility that loss of JNK2 may increase liver injury by altering Bim expression, the effects of JNK knockdowns on levels of the pro-apoptotic Bcl-2 family members were examined in HFD-fed mice. Levels of BimEL were markedly increased with ASO-mediated JNK2 inhibition (Fig. 4). BimL levels were also increased and BimS was undetectable (Fig. 4). Bim is regulated at the post-translational level by phosphorylation at Ser65 which triggers Bim degradation.20 In parallel with the increase in total Bim, levels of Ser65 phosphorylated Bim were decreased (Fig. 4). Levels of total and phospho-Bim were unaffected by JNK1 knockdown. In addition to the changes in Bim, levels of Bax were also significantly increased in JNK2 ASO-treated animals and to a lesser extent in mice with a knockdown of JNK1 (Fig. 4). Levels of Bid and Bad were equivalent in all animals (Fig. 4). The anti-apoptotic Bcl-2 family member Mcl-1 is also regulated by JNK phosphorylation which acts to decrease Mcl-1's anti-apoptotic effect.21 JNK2 ASO treatment led to a slight increase in Mcl-1 levels but no shift in mobility to indicate an alteration in Mcl-1 phosphorylation and therefore Mcl-1 function.
The function of pro-apoptotic Bcl-2 family members is also regulated by their release from cytoplasmic binding partners and translocation to the mitochondria where they activate the mitochondrial death pathway.22 Bim and Bax translocated to the mitochondria in HFD-fed, JNK2 ASO-treated mice but not in control ASO-injected mice (Fig. 7A). Similar to the findings for total protein, mitochondrial Bid and Bad levels were unaffected by ASO treatment (Fig. 7A). Low levels of mitochondrial Bim and Bax were detected in JNK1 ASO-treated animals (Fig. 7A). Bim/Bax mitochondrial translocation led to activation of the mitochondrial death pathway as demonstrated by the presence of cytoplasmic cytochrome c only in HFD-fed mice treated with the JNK2 ASO (Fig. 7B). Interestingly, despite mitochondrial release of cytochrome c, JNK2 ASO-treated mice had increased levels of mitochondrial cytochrome c whereas JNK1 ASO-treated mice had decreased levels (Fig. 7A), suggesting that JNK functioned to regulate mitochondrial cytochrome c levels exclusive of the effects of apoptosis. Purity of the mitochondrial and cytoplasmic isolates along with equivalency of protein loading was confirmed by probing stripped membranes for cytochrome oxidase and protein disulfide isomerase, respectively (Fig. 7A and 7B). In parallel with cytochrome c release, activation of the effector caspases 3 and 7 was detectable only in JNK2 ASO-treated mice fed a HFD as indicated by the presence of the active, cleaved caspase forms in these animals (Fig. 4). These data demonstrate that JNK2 inhibition in the setting of steatohepatitis led to increased levels of Bim/Bax, translocation of these proteins to the mitochondria and activation of the mitochondrial death pathway and downstream effector caspases.
Fig. 7.
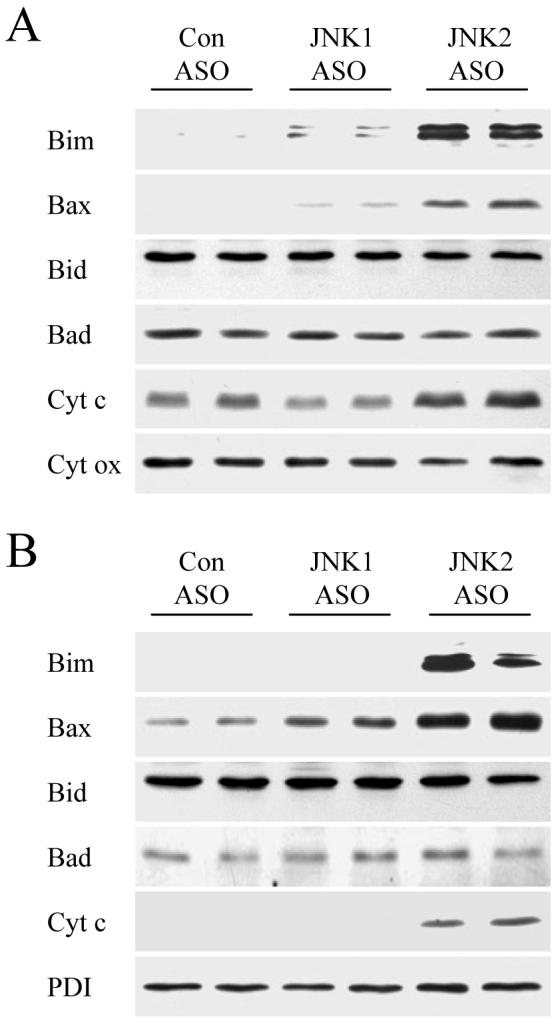
Hepatic mitochondrial Bim and Bax translocation and cytochrome c release occur in JNK2 ASO-treated HFD-fed mice. Mitochondrial (A) and cytoplasmic (B) protein fractions were isolated from HFD-fed mice administered a control (Con), JNK1 or JNK2 ASO. Proteins were immunoblotted with antibodies for Bim, Bax, Bid, Bad and cytochrome c (cyt c). Stripped blots were reprobed for cytochrome oxidase (cyt ox) and PDI as indicated.
Bim Null Mice Are Protected Against the Increase in Steatohepatitis Induced by a JNK2 Knockdown
To determine whether increased Bim expression was a mechanism of the increased hepatic injury that resulted from JNK2 inhibition, liver injury was compared in HFD-fed wild-type and bim-/- mice treated with the JNK2 ASO. JNK2 ASO-injected bim null mice had significant decreases in serum ALT levels (Fig. 8A), and the numbers of TUNEL positive cells (Fig. 8B), as compared to JNK2 ASO-treated wild-type mice. Thus, the increase in Bim expression that resulted from JNK2 inhibition mediated the increase in liver injury in HFD-fed mice.
Fig. 8.
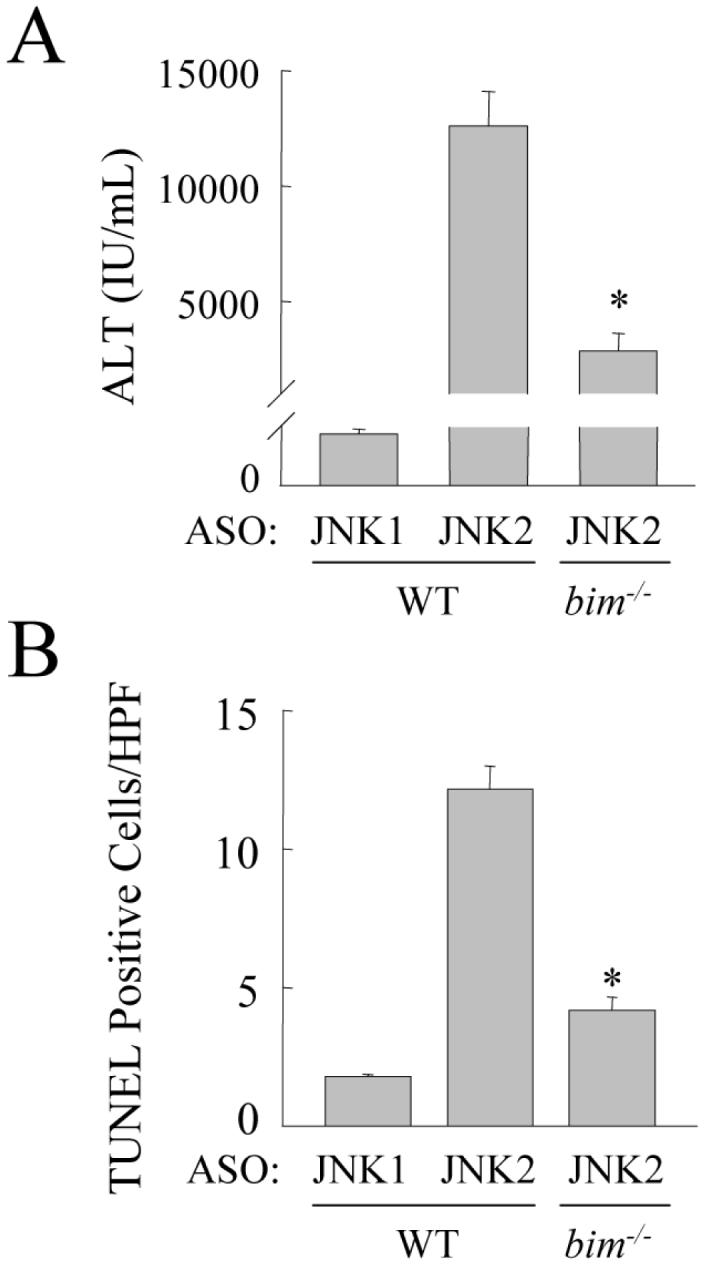
Bim null mice are protected from the increase in liver injury and cell death induced by a JNK2 knockdown. (A) Serum ALT levels in HFD-fed wild-type mice (WT) treated with the JNK1 or JNK2 ASO and bim-/- mice treated with JNK2 ASO (n=5-12; P<0.0001 as compared to wild-type mice treated with the JNK2 ASO). (B) Numbers of TUNEL positive cells in the same mice (n=6; P<0.00001 as compared to wild-type mice treated with the JNK2 ASO).
Discussion
Activation of the mitogen-activated protein kinase JNK has been increasingly implicated as a central mechanism of hepatic injury.6,11,18,19,23-26 Our previous studies demonstrated that JNK was activated during the development of murine steatohepatitis induced by a methionine- and choline-deficient diet, and that ablation of JNK1 but not JNK2 decreased steatosis and hepatitis.6 However, this dietary model lacks the human NAFLD features of obesity and peripheral insulin resistance,27 although the mice do develop hepatic insulin resistance.10 The fact that activation of JNK contributes to both obesity and insulin resistance,4,5,7,28 suggested that JNK may play an even more important role in steatohepatitis occurring in the setting of these two NAFLD risk factors. The present studies employed a HFD-fed mice model with excessive weight gain and insulin resistance to demonstrate that JNK1 promotes the development and persistence of steatohepatitis, whereas JNK2 performs the protective function of down regulating cell death.
The studies of JNK1 null mice and NAFLD development and JNK1 ASO-treated mice and established NAFLD demonstrated the same function for JNK1 - promotion of steatosis and liver injury. JNK1 inhibition by either method dramatically reduced hepatic TG content in concert with decreased liver injury and cell death. In particular, JNK1 was critical in steatosis development as HFD-fed jnk1-/- mice had hepatic TG levels equivalent to regular diet-fed wild-type mice. The ability of JNK1 to mediate both steatosis and cellular injury may reflect one or multiple functions of this kinase. JNK1 may simply promote hepatic lipid accumulation, and the dramatic reduction in steatosis from JNK1 inhibition decreases liver injury as well. Alternatively, JNK1 may promote steatosis and hepatitis by distinct mechanisms. The mechanism by which JNK1 promotes steatosis remains to be defined and may involve hepatic and/or peripheral effects. The prevention or reversal of steatosis may have resulted simply from the dramatic effect of JNK inhibition on insulin sensitivity. JNK1 is known to induce insulin resistance by phosphorylating inhibitory serine residues on insulin receptor substrate 1 and mediating obesity-induced inflammation.29 Against the reduction in hepatic lipid being merely secondary to effects on insulin resistance is our finding that JNK2 ASO treatment restored insulin sensitivity but did not affect steatosis. Thus, direct effects of JNK1 on lipid metabolism may underlie the development and maintenance of hepatic steatosis. Further studies will examine the regulation of components of lipid metabolism as JNK targets in these pathways have not yet been reported.30
The studies demonstrate a different function for JNK2 in HFD-induced steatohepatitis. In both developing and established steatohepatitis, JNK2 inhibition did not affect the amount of steatosis and worsened liver injury. As discussed above, JNK2 ASO administration had no effect on steatosis despite a reversal of insulin resistance. This effect was different from findings in the HFD-fed JNK2 null animals which had no improvement in insulin sensitivity and even a trend to greater resistance. This difference may have been secondary to compensatory changes in jnk2 null mice that did not occur in ASO-treated mice, or to disparate functions of JNK2 in developing versus established insulin resistance. A similar explanation likely underlies the differences between our findings and those of Tuncman et al.7 who reported that JNK1 and JNK2 both contribute to steatosis based on studies of jnk2 null mice haploinsufficient for jnk1. Alternatively, findings in the ASO-treated mice may have differed because ASO treatment resulted in a more liver specific effect of JNK inhibition as the liver has one of the highest concentrations of delivered ASO among tissues.31 In addition, the findings with JNK2 inhibition suggest that improved insulin sensitivity by itself is not sufficient to reverse established steatosis. Other recent studies have found that steatosis can persist despite reversal of insulin resistance,28 or suggested that insulin resistance may not mediate the development of this disease.32
The function of JNK2 in liver injury also differed from that of JNK1. In contrast to the improvement in liver injury in mice lacking JNK1 function, knockout or knockdown of JNK2 significantly worsened liver injury and apoptosis. The absence of an increase in steatosis with JNK2 inhibition suggested that JNK2 functioned specifically to block cell death pathways. Based on the reported ability of JNK to regulate Bim expression,33-35 and findings that Bim modulated hepatocyte toxicity from fatty acids in vitro,19 the effect of a knockdown of JNK2 on Bim was examined. Loss of JNK2 led to increased levels of Bim and to a lesser extent of Bax. Bim regulation is complex with steady-state protein levels reflecting changes in transcription, phosphorylation-mediated degradation and inhibitory protein binding and translocation. A number of kinases including JNK, extracellular signal-regulated kinase 1/2 and Akt have been reported to phosphorylate Bim in different cell types.20,33,36 In the livers of HFD-fed mice, JNK2 function was required for Bim phosphorylation at Ser65, a site whose phosphorylation triggers Bim degradation.20 Lack of Ser65 phosphorylation was associated with increased steady-state levels of total Bim in JNK2 knockdown mice. Our results are consistent with recent findings in a leukemia cell line in which JNK promoted chemoresistance by phosphorylating Bim at Ser65 and causing its degradation,33 and demonstrate that JNK can act to down regulate Bim-dependent apoptosis as well as to activate it. The effects of a JNK2 knockdown on Bim/Bax led to activation of the mitochondrial death pathway and apoptosis as indicated by mitochondrial cytochrome c release, cleavage of the effector caspases 3 and 7 and increased TUNEL staining in JNK2 ASO-treated, HFD-fed mice. In contrast to findings with Bim and Bax, in response to a JNK2 knockdown levels of Bid and Bad were unchanged, Bid cleavage was not detected and neither protein translocated to the mitochondria, indicating that they were unaffected by the loss of JNK2 and uninvolved in the increased cell death. Bim clearly mediated the increase in liver injury from JNK2 inhibition as bim null mice had a significant, but partial, reduction in liver injury and cell death. The partial nature of the effect may have been secondary to increased Bax translocation that promoted mitochondrial death pathway activation independent of Bim. JNK2-dependent promotion of liver injury in HFD-fed mice by activation of the mitochondrial death pathway is in direct opposition to our findings in a model of toxin-induced liver injury in which JNK2 functioned to inhibit this pathway.18 These differences point to the complexity of JNK function in the liver and likely reflect the effects of different types of injury, the time course of the injury, crosstalk with other signaling pathways and possible extrahepatic effects of JNK.
The findings from the ASO-treated animals demonstrate that HFD-induced steatohepatitis in mice is quickly reversible. Despite the fact that approximately two weeks of injections were required to achieve a JNK knockdown, a dramatic decrease in steatohepatitis was achieved with only four weeks of therapy. The rapidity of this effect is further evidence of the critical nature of JNK1 activation in steatohepatitis and the potential efficacy of anti-JNK therapy in the treatment of this disease.
The findings demonstrate that the JNK isoforms have differential functions in insulin resistance and steatohepatitis. Both JNK forms contributed to insulin resistance, however, JNK1 promoted both hepatic fat accumulation and injury whereas JNK2 was uninvolved in the steatosis and inhibited liver injury. Significantly the findings demonstrate that anti-JNK therapy can reverse chronic steatohepatitis in the absence of any reduction in the stimulus for the disease, a high fat diet. Kinase inhibitors are already employed in the treatment of human diseases,37 and JNK inhibitors have successfully reversed diabetes in animals.5 Our findings suggest that steatohepatitis can also be treated with this approach but that therapy will have to be targeted specifically to the JNK1 isoform. Inhibiting JNK2 may be effective in increasing insulin sensitivity but have detrimental effects on the associated liver disease.
Acknowledgements
We thank Neil Kaplowitz for his helpful suggestions, Xiao-Ming Yin for the Bid antibody and Richard Stockert for the PDI antibody.
Supported by NIH grants DK061498 (MJC) and DK020541 and an American Liver Foundation Postdoctoral Research Fellowship Award (RS).
Abbreviations
- ASO
antisense oligonucleotide
- HFD
high fat diet
- HOMA
homeostasis model assessment of insulin resistance
- JNK
c-Jun N-terminal kinase
- NAFLD
nonalcoholic fatty liver disease
- PBS
phosphate-buffered saline
- PDI
protein disulfide isomerase
- TG
triglyceride
References
- 1.Adams LA, Lindor KD. Nonalcoholic fatty liver disease. Ann Epidemiol. 2007;17:863–869. doi: 10.1016/j.annepidem.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 2.Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M, et al. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes. 2001;50:1844–1850. doi: 10.2337/diabetes.50.8.1844. [DOI] [PubMed] [Google Scholar]
- 3.Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183–1192. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 4.Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 5.Kaneto H, Nakatani Y, Miyatsuka T, Kawamori D, Matsuoka TA, Matsuhisa M, et al. Possible novel therapy for diabetes with cell-permeable JNK-inhibitory peptide. Nat Med. 2004;10:1128–1132. doi: 10.1038/nm1111. [DOI] [PubMed] [Google Scholar]
- 6.Schattenberg JM, Singh R, Wang Y, Lefkowitch JH, Rigoli RM, Scherer PE, et al. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology. 2006;43:163–172. doi: 10.1002/hep.20999. [DOI] [PubMed] [Google Scholar]
- 7.Tuncman G, Hirosumi J, Solinas G, Chang L, Karin M, Hotamisligil GS. Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proc Natl Acad Sci USA. 2006;103:10741–10746. doi: 10.1073/pnas.0603509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 9.Sabapathy K, Hochedlinger K, Nam SY, Bauer A, Karin M, Wagner EF. Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Mol Cell. 2004;15:713–725. doi: 10.1016/j.molcel.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 10.Schattenberg JM, Wang Y, Singh R, Rigoli RM, Czaja MJ. Hepatocyte CYP2E1 overexpression and steatohepatitis lead to impaired hepatic insulin signaling. J Biol Chem. 2005;280:9887–9894. doi: 10.1074/jbc.M410310200. [DOI] [PubMed] [Google Scholar]
- 11.Gunawan BK, Liu ZX, Han D, Hanawa N, Gaarde WA, Kaplowitz N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology. 2006;131:165–178. doi: 10.1053/j.gastro.2006.03.045. [DOI] [PubMed] [Google Scholar]
- 12.Dong C, Yang DD, Wysk M, Whitmarsh AJ, Davis RJ, Flavell RA. Defective T cell differentiation in the absence of Jnk1. Science. 1998;282:2092–2095. doi: 10.1126/science.282.5396.2092. [DOI] [PubMed] [Google Scholar]
- 13.Yang DD, Conze D, Whitmarsh AJ, Barrett T, Davis RJ, Rincon M, et al. Differentiation of CD4+ T cells to Th1 cells requires MAP kinase JNK2. Immunity. 1998;9:575–585. doi: 10.1016/s1074-7613(00)80640-8. [DOI] [PubMed] [Google Scholar]
- 14.Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–1738. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- 15.Baker BF, Lot SS, Condon TP, Cheng-Flournoy S, Lesnik EA, Sasmor HM, et al. 2′-O-(2-Methoxy)ethyl-modified anti-intercellular adhesion molecule 1 (ICAM-1) oligonucleotides selectively increase the ICAM-1 mRNA level and inhibit formation of the ICAM-1 translation initiation complex in human umbilical vein endothelial cells. J Biol Chem. 1997;272:11994–12000. doi: 10.1074/jbc.272.18.11994. [DOI] [PubMed] [Google Scholar]
- 16.D'Ambra R, Surana M, Efrat S, Starr RG, Fleischer N. Regulation of insulin secretion from beta-cell lines derived from transgenic mice insulinomas resembles that of normal beta-cells. Endocrinology. 1990;126:2815–2822. doi: 10.1210/endo-126-6-2815. [DOI] [PubMed] [Google Scholar]
- 17.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Singh R, Lefkowitch JH, Rigoli RM, Czaja MJ. Tumor necrosis factor-induced toxic liver injury results from JNK2-dependent activation of caspase-8 and the mitochondrial death pathway. J Biol Chem. 2006;281:15258–15267. doi: 10.1074/jbc.M512953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281:12093–12101. doi: 10.1074/jbc.M510660200. [DOI] [PubMed] [Google Scholar]
- 20.Ley R, Ewings KE, Hadfield K, Howes E, Balmanno K, Cook SJ. Extracellular signal-regulated kinases 1/2 are serum-stimulated “BimEL kinases” that bind to the BH3-only protein BimEL causing its phosphorylation and turnover. J Biol Chem. 2004;279:8837–8847. doi: 10.1074/jbc.M311578200. [DOI] [PubMed] [Google Scholar]
- 21.Inoshita S, Takeda K, Hatai T, Terada Y, Sano M, Hata J, et al. Phosphorylation and inactivation of myeloid cell leukemia 1 by JNK in response to oxidative stress. J Biol Chem. 2002;277:43730–43734. doi: 10.1074/jbc.M207951200. [DOI] [PubMed] [Google Scholar]
- 22.Baskin-Bey ES, Gores GJ. Death by association: BH3 domain-only proteins and liver injury. Am J Physiol Gastrointest Liver Physiol. 2005;289:G987–G990. doi: 10.1152/ajpgi.00371.2005. [DOI] [PubMed] [Google Scholar]
- 23.Corazza N, Jakob S, Schaer C, Frese S, Keogh A, Stroka D, et al. TRAIL receptor-mediated JNK activation and Bim phosphorylation critically regulate Fas-mediated liver damage and lethality. J Clin Invest. 2006;116:2493–2499. doi: 10.1172/JCI27726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwabe RF, Uchinami H, Qian T, Bennett BL, Lemasters JJ, Brenner DA. Differential requirement for c-Jun NH2-terminal kinase in TNFα- and Fas-mediated apoptosis in hepatocytes. FASEB J. 2004;18:720–722. doi: 10.1096/fj.03-0771fje. [DOI] [PubMed] [Google Scholar]
- 25.Uehara T, Bennett B, Sakata ST, Satoh Y, Bilter GK, Westwick JK, et al. JNK mediates hepatic ischemia reperfusion injury. J Hepatol. 2005;42:850–859. doi: 10.1016/j.jhep.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 26.Qiao L, Han SI, Fang Y, Park JS, Gupta S, Gilfor D, et al. Bile acid regulation of C/EBPβ, CREB, and c-Jun function, via the extracellular signal-regulated kinase and c-Jun NH2-terminal kinase pathways, modulates the apoptotic response of hepatocytes. Mol Cell Biol. 2003;23:3052–3066. doi: 10.1128/MCB.23.9.3052-3066.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rinella ME, Green RM. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J Hepatol. 2004;40:47–51. doi: 10.1016/j.jhep.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 28.Solinas G, Naugler W, Galimi F, Lee MS, Karin M. Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proc Natl Acad Sci USA. 2006;103:16454–16459. doi: 10.1073/pnas.0607626103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Solinas G, Vilcu C, Neels JG, Bandyopadhyay GK, Luo JL, Naugler W, et al. JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metab. 2007;6:386–397. doi: 10.1016/j.cmet.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 30.Bogoyevitch MA, Kobe B. Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol Mol Biol Rev. 2006;70:1061–1095. doi: 10.1128/MMBR.00025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geary RS, Yu RZ, Watanabe T, Henry SP, Hardee GE, Chappell A, et al. Pharmacokinetics of a tumor necrosis factor-α phosphorothioate 2′-O-(2-methoxyethyl) modified antisense oligonucleotide: comparison across species. Drug Metab Dispos. 2003;31:1419–1428. doi: 10.1124/dmd.31.11.1419. [DOI] [PubMed] [Google Scholar]
- 32.Fishman S, Muzumdar RH, Atzmon G, Ma X, Yang X, Einstein FH, et al. Resistance to leptin action is the major determinant of hepatic triglyceride accumulation in vivo. FASEB J. 2007;21:53–60. doi: 10.1096/fj.06-6557com. [DOI] [PubMed] [Google Scholar]
- 33.Leung KT, Li KK, Sun SS, Chan PK, Ooi VE, Chiu LC. Activation of the JNK pathway promotes phosphorylation and degradation of BimEL-a novel mechanism of chemoresistance in T-cell acute lymphoblastic leukemia. Carcinogenesis. 2008;29:544–551. doi: 10.1093/carcin/bgm294. [DOI] [PubMed] [Google Scholar]
- 34.Putcha GV, Moulder KL, Golden JP, Bouillet P, Adams JA, Strasser A, et al. Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron. 2001;29:615–628. doi: 10.1016/s0896-6273(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 35.Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J. Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron. 2001;29:629–643. doi: 10.1016/s0896-6273(01)00239-2. [DOI] [PubMed] [Google Scholar]
- 36.Qi XJ, Wildey GM, Howe PH. Evidence that Ser87 of BimEL is phosphorylated by Akt and regulates BimEL apoptotic function. J Biol Chem. 2006;281:813–823. doi: 10.1074/jbc.M505546200. [DOI] [PubMed] [Google Scholar]
- 37.Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]