

Summary
Most solid tumors are aneuploid and many frequently mis-segregate chromosomes. This chromosomal instability is commonly caused by persistent maloriented attachment of chromosomes to spindle microtubules. Chromosome segregation requires stable microtubule attachment at kinetochores, yet those attachments must be sufficiently dynamic to permit correction of malorientations. How this balance is achieved is unknown, and the permissible boundaries of attachment stability versus dynamics essential for genome stability remain poorly understood. Here we show that two microtubule-depolymerizing kinesins, Kif2b and MCAK, stimulate kinetochore-microtubule dynamics during distinct phases of mitosis to correct malorientations. Few-fold reductions in kinetochore-microtubule turnover, particularly in early mitosis, induce severe chromosome segregation defects. In addition, we show that stimulation of microtubule dynamics at kinetochores restores chromosome stability to chromosomally unstable tumor cell lines, establishing a causal relationship between deregulation of kinetochore-microtubule dynamics and chromosomal instability. Thus, temporal control of microtubule attachment to chromosomes during mitosis is central to genome stability in human cells.
Most solid tumors have aneuploid karyotypes ranging from 40 to 90 or more chromosomes1. Aneuploidy is the irreversible consequence of chromosome mis-segregation during mitosis and is a state where the cellular karyotype deviates from multiples of the haploid (n) number of chromosomes2. Some aneuploid tumor cells remain genetically stable by faithfully segregating their improper chromosome number3. However, many aneuploid tumors are genetically unstable and mis-segregate whole chromosomes at high rates throughout consecutive generations4. This chromosomal instability (CIN) indicates an underlying persistent defect in the fidelity of chromosome segregation during mitosis. CIN produces continuous changes in gene expression patterns that can confer growth advantage and complicate chemotherapeutic strategies5 explaining why it positively correlates with propensity for metastasis and poor patient prognosis6,7. Despite this clinical importance, little is known about the molecular mechanisms underlying CIN.
Chromosome segregation in mitosis is mediated by a microtubule-based structure called the spindle. Microtubules attach to chromosomes at kinetochores, and in human cells each kinetochore binds 20-25 microtubules8. Proper segregation occurs when kinetochores of sister chromatids achieve bioriented attachment to spindle microtubules. In principle, centromere geometry provides an intrinsic bias for sister chromatid biorientation9,10. However, due to stochastic interactions between kinetochores and microtubules at early stages of mitosis (prometaphase), many chromosomes fail to initially biorient and kinetochores do not immediately attain full microtubule occupancy11. Consequently, some chromosomes mono-orient with one or both kinetochores attached to microtubules emanating from one spindle pole, whereas others have kinetochores attached to spindle microtubules oriented to both poles forming merotelic attachments12. The spindle assembly checkpoint (SAC) delays anaphase onset to permit mono-oriented chromosomes sufficient time to achieve biorientation13,14, and mutations in genes encoding SAC proteins have been reported in a few aneuploid tumor cells with CIN15. It has also been reported that CIN may arise from defects in sister chromatid cohesion16. However, those studies explain the cause of CIN in a minority of tumor cells and recent evidence suggests that persistent merotely is a more widespread cause of CIN17. Unlike other malorientations, merotely evades SAC detection18. Instead, merotelic correction is regulated by the Aurora B kinase to orchestrate release of inappropriately oriented microtubules permitting replacement by properly oriented microtubules14,19-21. This correction mechanism underscores the importance of the dynamic attachment/detachment of microtubules to kinetochores, but little is known about how these events are regulated or how they relate to CIN. Here, we examine mechanisms governing kinetochore-microtubule (kMT) dynamics and their relationship to correction of chromosome malorientation using high-resolution quantitative microscopy and fluorescence dissipation after photoactivation (FDAPA) techniques. We also utilize clonal cell analyses to test if manipulating kMT dynamics influences CIN in human tumor cell lines.
Two kinesin-13s cooperate to promote mitotic fidelity
Precedent for a role of kinesin-13 catalyzed microtubule depolymerization in the correction of chromosome malorientation has been established through work on MCAK19-23. The kinesin-13 protein Kif2b localizes to kinetochores during early mitosis24 suggesting it may also play a role in correcting attachment errors25. To directly test this, we scored lagging chromosomes in Kif2b-deficient anaphase cells (Fig. 1). Kif2b expression in human U2OS cells was suppressed using RNA interference and suppression was confirmed by immunoblot analysis in GFP-Kif2b expressing cells due to the low expression levels of the endogenous protein (Supp. Fig. 1a, ref. 24). Whereas some Kif2b-deficient cells build bipolar spindles and progress into anaphase, others form monopolar spindles24. Live cell analysis shows these monopolar spindles are transient and can resolve to bipolar spindles that subsequently undergo anaphase (Supp. Fig. 1b and c), demonstrating that Kif2b-deficiency does not irreversibly block bipolar spindle assembly. Once bipolar, spindles in Kif2b-deficient cells display no dramatic defects in either spindle length or spindle multipolarity (Supp. Fig. 2a and b). Spindle length decreases by ∼10% when Kif2b is either depleted or overexpressed. Markedly, more than 80% of Kif2b-deficient cells display lagging chromosomes in anaphase with an average of 3 to 7 lagging chromosomes per cell (Fig. 1c and d). Kinetochores on these lagging chromosomes are distended and display microtubule attachments oriented toward both spindle poles consistent with persistent merotelic attachment (Fig. 1a, arrow). For comparison, only ∼45% of MCAK-depleted cells show lagging chromosomes at anaphase with an average of 1 to 2 lagging chromosomes per anaphase. It is possible that the extended prometaphase in Kif2b-deficient cells contributes to elevating the frequencies of lagging chromosomes in anaphase. However, simultaneous suppression of MCAK and Kif2b restores a normal mitotic index, results in primarily bipolar spindles, and does not alleviate the segregation defects observed in the absence of Kif2b alone. Moreover, defects in chromosome segregation in Kif2b-deficient cells are not a consequence of a defective SAC since cells depleted of Kif2b, MCAK or both arrest as efficiently as control cells if mitotic progression is blocked using nocodazole (Supp. Fig. 1d).
Figure 1. Lagging chromosomes in the absence of Kif2b and MCAK.
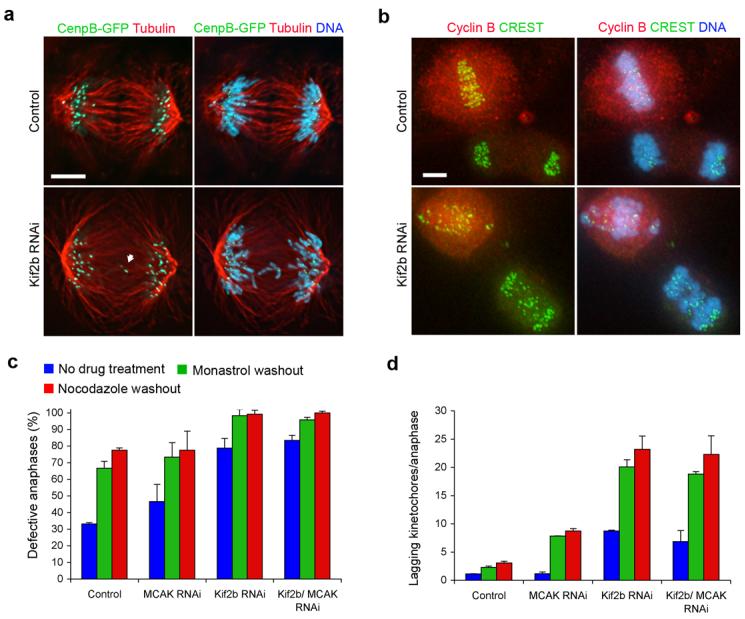
a, Anaphase spindles of both untreated and Kif2b-deficient U2OS cells containing CenpB-GFP to indicate kinetochores and stained for microtubules and DNA. White arrow highlights merotelic kinetochores. For clarity, a cell with only 2 lagging chromosomes was selected for this panel. b, Anaphase and pre-anaphase cells of both untreated and Kif2b-deficient RPE1 cells following recovery from nocodazole treatment stained for kinetochores with CREST, cyclin B, and DNA. c, d, Number of anaphase U2OS cells with lagging kinetochores (c) and the average number of laggards per anaphase (d) in untreated cells (blue) or cells following recovery from monastrol (green) or nocodazole (red) treatment. Bars represent mean ± s.e.m, n = 150 cells, 3 experiments. Scale bars, 5 μm.
Centromere and spindle geometries can bias sister chromatid bi-orientation reducing the incidence of malattachment9,10. Thus, perturbing such geometries would highlight the requirement for Kif2b-mediated correction of malorientation. We measured the frequency of lagging chromosomes in anaphase cells that were allowed to recover from spindle disruption induced by treatment with either nocodazole- or monastrol. Nearly 100% of Kif2b-deficient U2OS cells exhibit defective anaphases when the incidence of chromosome malorientation is artificially increased by recovery from either drug treatment. These cells display an average of 8 to 20 lagging chromosomes per anaphase representing 10-25% of the total number of chromosomes in each cell (Fig. 1 c and d). Similar defects are also observed in diploid RPE1 cells depleted of Kif2b confirming that these effects are not limited to transformed cells. The exceedingly high numbers of lagging chromosomes in these cells, such as those following recovery from nocodazole, could be mis-construed as mis-aligned chromosomes in prometaphase. However, diminution of cyclin B levels confirms that they are in anaphase (Fig. 1b). Whereas a subset of these lagging chromosomes might result from delayed sister chromatid separation or slowed anaphase movement of individual chromosomes, our observations that most sister kinetochores have separated by anaphase, single kinetochores on lagging chromosomes are distended toward both spindle poles, and the absence of kinetochore localization of Kif2b during anaphase24 make those possibilities unlikely.
The strikingly high incidence of lagging chromosomes in Kif2b-deficient cells recovering from nocodazole or monastrol treatments implicates Kif2b in the correction of other types of errors such as chromosome mono-orientation. To test this, we treated U2OS cells with monastrol to induce monopolar spindles replete with mono-oriented chromosomes, and then scored chromosome alignment on bipolar spindles that form after monastrol washout26. Monastrol washout did not induce drastic changes in spindle multipolarity or length (Supp. Fig. 2a and b). 45-50 minutes after monastrol removal, control and MCAK-deficient cells efficiently form bipolar spindles and most align all chromosomes (Fig 2a and b). Many Kif2b-deficient cells also efficiently build bipolar spindles after monastrol removal, but over 50% of these cells fail to align all chromosomes at the spindle equator and numerous chromosomes remain adjacent to spindle poles.
Figure 2. Kif2b is required for correction of attachment errors.
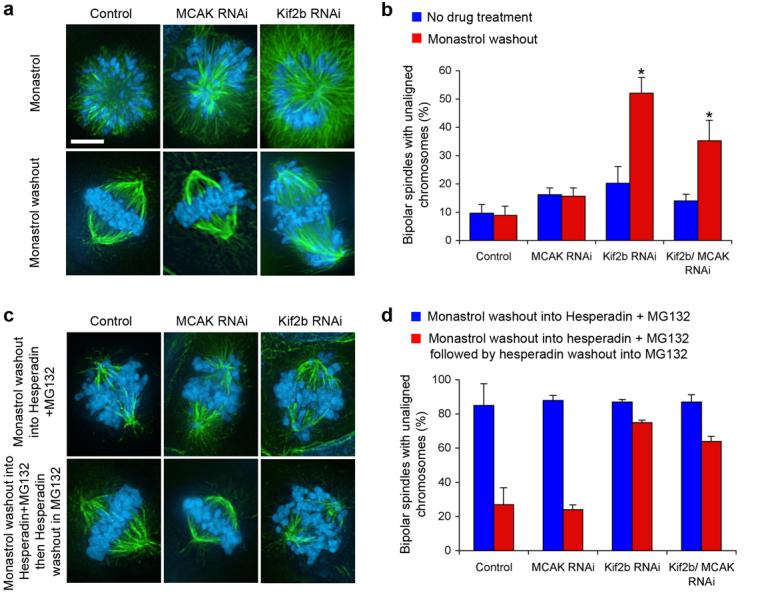
a, Spindles of untreated (Control), MCAK-deficient (MCAK RNAi), and Kif2b-deficient (Kif2b RNAi) U2OS cells in the presence of monastrol (top panels) or after recovery from monastrol treatment (bottom panels). b, Number of cells with bipolar spindles that failed to align chromosomes with no treatment (blue) or following recovery from monastrol treatment (red). c, Spindles of untreated (Control), MCAK-deficient (MCAK RNAi), and Kif2b-deficient (Kif2b RNAi) cells following recovery from monastrol treatment in the presence of hesperadin (top panels) and then followed by recovery from hesperadin treatment (bottom panels). d, Number of bipolar cells that failed to align chromosomes after monastrol washout in the presence of hesperadin (blue) and then after recovery from hesperadin treatment (red). Bars in b and d represent mean ± s.e.m, n = 600, 3 experiments cells for b, 100 cells, 2 experiments for d. Scale bars, 5 μm. *, p < 0.0002, t-test.
Aurora B kinase regulates various kinetochore proteins to control correction of mono-oriented chromosomes by mediating release of mal-oriented microtubules14,26. Accordingly, over 80% of control, MCAK- and Kif2b-deficient cells that recover from monastrol treatment in the presence of the Aurora kinase inhibitor, hesperadin27, form bipolar spindles with numerous unaligned chromosomes (Fig 2c and d). Subsequent removal of hesperadin restores Aurora kinase activity which in turn permits efficient chromosome alignment in most control and MCAK-deficient cells. In contrast, almost 80% of Kif2b-deficient spindles fail to align chromosomes upon restoration of Aurora kinase activity functionally placing Kif2b downstream of Aurora B. Consistent with this conclusion, we find that kinetochore localization of GFP-Kif2b on monopolar spindles is sensitive to Aurora inhibition (Fig. 3) and is reversibly restored following recovery from hesperadin treatment. It remains to be determined whether or not this regulation is direct. Taken together, these data demonstrate that in human cells both Kif2b and MCAK are essential for the correction of merotelic orientations and potential other segregation defects, and that Kif2b's role is unique in correcting mono-oriented attachments that typically arise during the initial attachment of chromosomes to spindle microtubules.
Figure 3. Kinetochore localization of GFP-Kif2b is sensitive to the Aurora-inhibitor, Hesperadin.
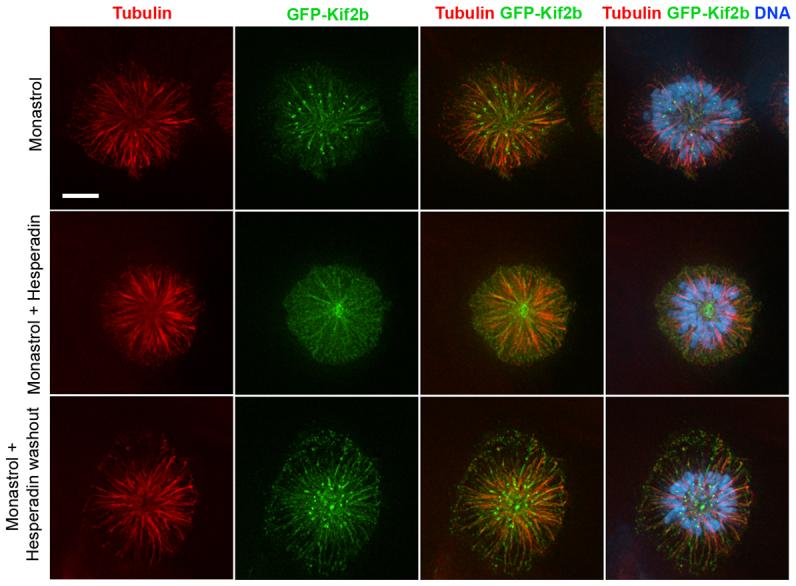
Monopolar spindles induced with monastrol showing microtubules (red), GFP-Kif2b (green), and DNA (blue) in the presence of hesperadin and following removal of hesperadin. Scale bar, 5 μm.
Kif2b and MCAK influence kMT dynamics
Release of inappropriately oriented microtubules from kinetochores is the rate-limiting step in correction of maloriented chromosomes28. Thus, the dynamic attachment/detachment of kinetochore-microtubules is critical to ensure error-free mitosis, and the microtubule depolymerizing activity of the kinesin-13 proteins may be required for efficient microtubule detachment from kinetochores. To directly test if Kif2b and MCAK promote error correction by modulating the dynamics of kMT attachments, we used FDAPA to examine spindle microtubule dynamics in live mitotic cells. GFP fluorescence was activated in human U2OS cells expressing photoactivatable GFP-tubulin using several pulses from a diffraction-limited 405nm laser (Fig. 4a). Average pixel intensities within the activated ∼2 μm2 region were obtained at various times, background subtracted, and then corrected for photobleaching as previously described29-31. Fluorescence intensities fit a double exponential decay curve (R2>0.99) whereby the slow-decaying fluorescence corresponds to the less dynamic kMT population (Fig. 4b, ref 31). Consistently, this population of slow-decaying fluorescent microtubules is significantly reduced in cells depleted of Nuf2, an essential protein for kMT attachment (not shown). In control cells, this kMT population exhibits an average half-life of approximately 3 and 7 min. in prometaphase and metaphase, respectively, closely matching previously reported values29-31.
Figure 4. Temporal regulation of kinetochore-microtubule dynamics.

a, Differential interference contrast (D.I.C.) and time-lapse fluorescent images of prometaphase spindles in untreated (Control) and Kif2b-deficient (Kif2b RNAi) U2OS cells before (Pre-PA) and at the indicated times (sec) after activation (Post-PA) of GFP-tubulin fluorescence. Scale bar, 5 μm. b, Normalized fluorescence intensity over time after photoactivation of spindles in untreated (filled squares), MCAK-deficient (filled circles), and Kif2b-deficient (white squares) prometaphase and metaphase cells. Datapoints represent mean ± s.e.m, n = 5 to 11 cells. c, Calculated kMT half-life under different conditions. Error bars represent S.E. from the regression analysis in b.
Interestingly, both Kif2b and MCAK depletions affect kMT turnover albeit at distinct phases of mitosis (Fig. 4c). Kif2b-deficient cells have a four-fold increase in kMT half-life in prometaphase whereas MCAK-deficient cells are indistinguishable from prometaphase control cells. On the other hand, Kif2b-deficiency does not significantly alter kMT dynamics at metaphase whereas MCAK-deficient cells have a two-fold increase in half-life. Conversely, overexpression of mCherry-Kif2b or RFP-MCAK significantly increases kMT turnover. This effect is limited to metaphase with mCherry-Kif2b and RFP-MCAK overexpressing cells displaying kMT half-life's of 3.5 ± 0.2 (n=6 cells) and 5.2 ± 0.9 (n=5 cells), respectively. Moreover, Kif2b-deficient mitotic cells show delayed kMT depolymerization in the presence of high doses of nocodazole whereas overexpression of GFP-Kif2b increases nocodazole sensitivity indicating reduced kMT stability (Supp. Fig. 3). Thus, stabilization of kMT attachments at either prometaphase or metaphase by depletion of Kif2b or MCAK prevents release of maloriented microtubule attachments leading to defective chromosome segregation. Collectively, these data show that Kif2b and MCAK have non-overlapping roles in ensuring accurate chromosome segregation.
Suppression of chromosomal instability
We recently demonstrated that a common cause of CIN in aneuploid tumor cells is persistent merotely17. Because merotelic attachment is a widespread outcome of functional disruption of a variety of proteins and pathways impacting the formation or maintenance of properly bioriented kMT attachment, we postulated that promoting kMT detachment would allow for correction of merotely in CIN tumor cells regardless of the specific upstream underlying cause. Based on increased kMT stability in Kif2b- or MCAK-deficient mitotic cells (Fig. 4), the reduced kMT stability in GFP-Kif2b overexpressing cells (Supp. Fig. 3), and the reduced kMT stability in mCherry-Kif2b or RFP-MCAK overexpressing cells, we tested the impact of overexpression of GFP-Kif2b and GFP-MCAK on chromosome segregation fidelity in CIN cells. As expected, GFP-Kif2b and GFP-MCAK target to kinetochores and centromeres, respectively, in U2OS cells (Supp. Fig. 4) and overexpression of each kinesin significantly suppresses the incidence of lagging chromosomes in both U2OS and MCF-7 cell lines (Fig. 5a and Supp. Fig. 5a). Importantly, overexpression of neither GFP-tubulin nor GFP-Kif2a, the third kinesin-13 protein encoded in the human genome which does not target to kinetochores, suppresses the frequency of lagging chromosomes in U2OS cells (Fig. 5a). This indicates that suppression of lagging chromosomes is specific to kinetochore-associated microtubule depolymerizing activity of Kif2b and MCAK. Overexpression of either GFP-Kif2b or GFP-MCAK also suppresses frequencies of lagging chromosomes induced when spindle geometry is perturbed during recovery from either monastrol or nocodazole treatment in U2OS cells (Supp. Fig. 5b). In addition, overexpression of each kinesin significantly suppresses lagging chromosomes in RPE1 cells recovering from either drug treatment, confirming that these results are not limited to transformed cells (Supp. Fig. 5c). We also found that overexpression of GFP-Kif2b or GFP-MCAK suppresses lagging chromosomes resulting from MCAK or Kif2b-depletion, respectively (Fig. 5b). Given that overexpression of either kinesin significantly reduces lagging chromosomes in two chromosomally unstable cell lines (U2OS & MCF-7) and a chromosomally stable diploid cell line (RPE1) where segregation errors were artificially induced, these data demonstrate that kMT release is likely the furthest downstream target in the correction of merotely regardless of its origin.
Figure 5. Suppression of chromosome mis-segregation in cancer cell lines.
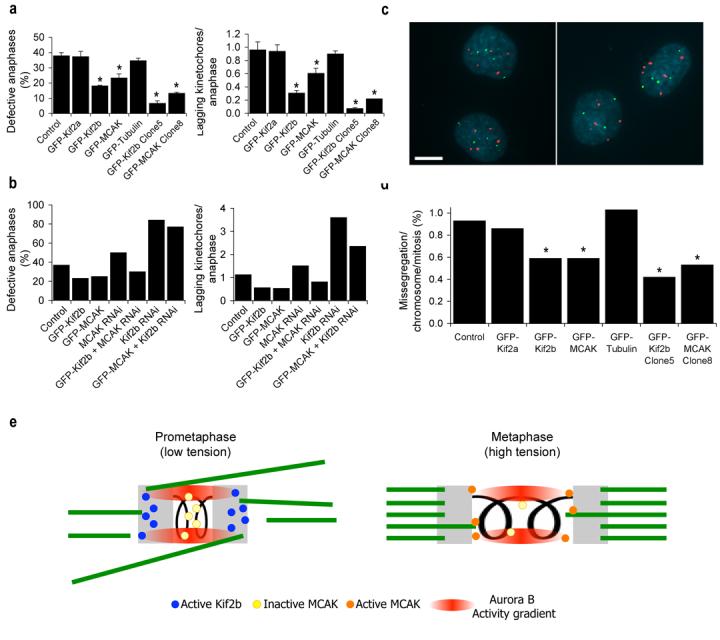
a, b, Percent of anaphase cells with lagging kinetochores and average numbers of lagging chromosomes in anaphase of U2OS cells from representative experiments. Clone 5 and clone 8 are from Table 1. Bars represent mean ± s.e.m, n = 150 cells, 3 experiments (a) and bars represent values from 100 cells, 2 experiements (b), *, p < 0.05, t-test. c, Examples of a proper chromosome segregation in U2OS cells expressing GFP-MCAK (left panel) and mis-segregation event in U2OS cells expressing GFP-Kif2a (right panel) using FISH with probes for chromosomes 2 (green) and 3 (red) and DNA stained with DAPI (blue). Scale bars, 10 μm. d, Mis-segregation rates per chromosome per mitosis in untreated U2OS cells and U2OS cells overexpressing GFP-Kif2a, GFP-Kif2b, GFP-MCAK, or GFP-tubulin as indicated. Clone 5 and clone 8 are from Table 1. n > 660 cells. *, p < 0.05, Chi-square test. e, Model for temporal regulation of kMT dynamics. At low inter-kinetochore tension during prometaphase, the Aurora B kinase activity gradient recruits Kif2b to kinetochores and inhibits centromeric MCAK. The tension generated upon biorientation causes sister kinetochores to exceed the boundaries of the Aurora B kinase activity gradient releasing Kif2b from kinetochores and activating a subset of MCAK in the outer centromere. Kinetochores are shown in grey and microtubules are shown in green.
Next, we tested if chromosome mis-segregation rates decrease in accordance with reductions in lagging chromatid frequencies in overexpressing cells. We measured chromosome mis-segregation directly using fluorescence in situ hybridization (FISH) with chromosome-specific α-satellite DNA probes following single mitotic events (Fig. 5c, ref. 17). The mis-segregation rate in untreated U2OS cells is ∼1% per chromosome, corresponding to two chromosome mis-segregation events every three divisions when corrected for the modal chromosome number in this cell line. Overexpression of either GFP-Kif2b or GFP-MCAK significantly reduces chromosome mis-segregation rates in these cells (Fig. 5d). In contrast, overexpression of either of GFP-Kif2a or GFP-tubulin fails to reduce chromosome mis-segregation rates demonstrating the specificity of Kif2b and MCAK in correcting chromosome malorientations that directly lead to chromosome mis-segregation.
Elevated frequencies of chromosome mis-segregation are a direct indicator of CIN4,17. Since overexpression of GFP-Kif2b or GFP-MCAK significantly reduces chromosome mis-segregation rates in U2OS cells we postulated that they might suppress CIN in this unstable aneuploid tumor cell line. To assess CIN in untreated, GFP-Kif2b, or GFP-MCAK overexpressing U2OS cells, we determined the modal chromosome number and scored the percentage of cells that deviate from that mode in isolated colonies that visibly express GFP-Kif2b or GFP-MCAK using FISH probes for chromosomes 2, 3, 7, and 15. Colonies were grown for ∼27 generations and growth times were adjusted accordingly to ensure equivalent generations were reached for each colony. Untreated U2OS cells display deviations from the mode ranging from 16% to as high as 54% for each chromosome measured befitting a CIN cell line (Table 1). In each clone examined for cells overexpressing either GFP-Kif2b or GFP-MCAK there is significant reduction in the deviation from the modal chromosome number for two or more of the four chromosomes examined. In contrast, no significant reductions in chromosome deviations are detected in cells from colonies expressing GFP-Kif2a (not shown). Notably, the deviation from the modal chromosome number for some chromosomes in cells expressing GFP-Kif2b or GFP-MCAK (i.e. clone 5 overexpressing GFP-Kif2b) is reduced to ∼4-6%, comparable to previously reported values for chromosomally stable diploid cell lines4,17. This low deviation is striking because these aneuploid cells possess higher modal chromosome numbers than stable diploid cells. Moreover, these reduced deviations are maintained when these clones are carried for 50 generations indicating lasting reductions in chromosome mis-segregation (not shown). Immnoblotting with a GFP-specific antibody reveals that both clones 5 and 8 contain higher levels of GFP-Kif2b and GFP-MCAK than populations with mixed GFP expression (Supp. Fig. 4e). Accordingly, the frequency of lagging chromosomes in anaphase and chromosome mis-segregation rates are further reduced in these clonal populations, demonstrating that the relative level of overexpression of the GFP-constructs is proportional to the magnitude of suppression of segregation errors (Fig. 5a and d). For example, GFP-Kif2b clone 5 displays 6.7% of anaphase cells with lagging chromosomes and a mis-segregation rate of ∼0.42% per chromosome, and GFP-MCAK clone 8 shows 13.3% of anaphase cells with lagging chromosomes and a mis-segregation rate of ∼0.53% per chromosome. GFP-Kif2b and GFP-MCAK overexpression also suppresses CIN in MCF-7 breast cancer cell line demonstrating that this mechanism for suppression of CIN is not limited to a single tumor cell line (Table 2). Thus, chromosomal stability can be restored to otherwise chromosomally unstable aneuploid tumor cell lines by overexpression of kinesin-13 proteins that act by promoting kMT turnover to release inappropriate attachments.
Table 1.
Modal Chromosome Numbers in U2OS Cell Clones (% cells that deviate from mode)
| Chromosome | ||||
|---|---|---|---|---|
|
Cell line |
2 mode (% deviation) |
3 mode (% deviation) |
7 mode (% deviation) |
15 mode (% deviation) |
| U2OS (clone 1) | 4 (20.3) < | 4 (27.7) | 4 (18.7) | 3 (20.7) |
| U2OS (clone 2) | 4 (25.7) | 5 (54.3) | 3 (17.0) | 2 (20.0)< |
| U2OS (clone 3) | 4 (26.3) | 4 (23.3) < | 4 (16.0) < | 3 (23.3) |
| U2OS GFP-Kif2b (clone 4) | 4 (11.3)* | 4 (14.3)* | 4 (10.3)* | 3 (15.7) |
| U2OS GFP-Kif2b (clone 5) | 4 (4.0)* | 5 (17.0) | 4 (6.0)* | 2 (5.3)* |
| U2OS GFP-Kif2b (clone 6) | 4 (6.0)* | 4 (18.0) | 4 (12.0) | 2 (7.3)* |
| U2OS GFP-MCAK (clone 7) | 4 (9.0)* | 4 (15.3)* | 4 (9.3)* | 3 (13.7) * |
| U2OS GFP-MCAK (clone 8) | 4 (7.3)* | 4 (13.3)* | 4 (7.7)* | 2 (15.7) |
| U2OS GFP-MCAK (clone 9) | 4 (6.3)* | 4 (12.3)* | 4 (8.3)* | 2 (12.7)* |
Control cell clone used for statistical comparison for each chromosome.
Chi-square test, p < 0.05.
Table 2.
Modal Chromosome Numbers in MCF-7 Cell Clones (% cells that deviate from mode)
| Chromosome | ||||
|---|---|---|---|---|
|
Cell line |
2 mode (% deviation) |
3 mode (% deviation) |
7 mode (% deviation) |
15 mode (% deviation) |
| MCF-7 (clone 1) | 3 (16.0) | 5 (28.7) | 4 (18.7) | 3 (24.7) |
| MCF-7 (clone 2) | 3 (13.0) < | 5 (18.7) < | 4 (11.3) < | 3 (27.7) |
| MCF-7 (clone 3) | 3 (14.3) | 5 (23.3) | 4 (17.0) | 3 (21.0) < |
| MCF-7 GFP-Kif2b (clone 4) | 3 (6.3)* | 4 (9.0)* | 4 (8.7) | 3 (11.3)* |
| MCF-7 GFP-Kif2b (clone 5) | 3 (5.3)* | 5 (10.3)* | 4 (9.0) | 2 (11.7)* |
| MCF-7 GFP-Kif2b (clone 6) | 2 (12.0) | 4 (16.3) | 4 (13.0) | 3 (11.0)* |
| MCF-7 GFP-MCAK (clone 7) | 3 (8.7) | 5 (17.7) | 4 (10.3) | 3 (8.7)* |
| MCF-7 GFP-MCAK (clone 8) | 3 (7.3)* | 5 (23.0) | 4 (15.0) | 3 (11.0)* |
| MCF-7 GFP-MCAK (clone 9) | 3 (6.0)* | 5 (15.7) | 4 (11.3) | 3 (14.3)* |
Control cell clone used for statistical comparison for each chromosome.
Chi-square test, p < 0.05.
Discussion
Genome stability requires faithful chromosome segregation during mitosis and is ensured by two cooperating mechanisms. First, centromere geometry inherently biases bi-oriented attachment of chromosomes to spindle microtubules thereby constituting a prevention mechanism against malorientation9,10. However, this geometrical constraint is insufficient to guarantee error-free mitosis because the stochastic interaction of microtubules with kinetochores often results in improper attachments12,25. Thus, a second corrective mechanism acts to release these improper attachments and we show that kinetochore-associated microtubule depolymerizing activities of Kif2b and MCAK fulfill this critical function. Kif2b localizes to kinetochores exclusively during prometaphase24 where it destabilizes kMTs to allow for the correction of both mono-oriented chromosomes and merotelic kinetochores that frequently occur during early mitosis25. On the other hand, MCAK localizes to centromeres and corrects the remaining errors through maintenance of kMT dynamic behavior during metaphase19-23.
Kinetochore microtubules become progressively stabilized as cells progress from prometaphase to metaphase29,30, and it has been shown that tension contributes to stabilizing kinetochore-microtubule attachments28. Aurora B kinase forms spatial activity gradients in mitosis32 and it has been postulated that tension influences the Aurora B kinase activity gradient across centromeres14,19-21. Whereas Aurora B kinase may inhibit Kif2b microtubule deploymerase activity akin to its regulation of MCAK, we propose a model (Fig. 5e) where low tension in prometaphase allows Aurora B kinase activity to simultaneously suppress centromeric MCAK activity19-21 and recruit Kif2b to outer kinetochores to promote microtubule destabilization (Fig. 3). Tension generated upon bi-orientation in metaphase would limit the extent of the Aurora B kinase activity gradient resulting in release of Kif2b from outer kinetochores and activation of a subset of MCAK19-21 at the outer centromere. Thus, tension-dependent changes in Aurora B kinase activity gradient at centromeres would temporally regulate kMT dynamics through the activities of Kif2b and MCAK ultimately leading to the correction of maloriented kinetochore-microtubule attachments. We cannot discount models invoking Aurora B-mediated targeting of correction machinery specifically to maloriented attachments21, our data support the view that temporally controlled global shifts in the dynamics of kMT attachments during mitotic progression promotes biorientation. Given that kinetochores have a bias to capture microtubules emanating from the spindle pole they face28, promoting indiscriminate release of both proper and improper kMT attachments would probabilistically favor the accumulation of properly attached microtubules without the need to target the correction machinery specifically to maloriented kinetochores.
Despite the benefit of promoting kMT release, the dynamic range of microtubule attachment to kinetochores cannot be infinitely large. kMT dynamics must be intricately controlled to prevent undesirable consequences resulting from excessively stable or unstable attachments. Hypostable attachment would prevent kinetochores from reaching adequate microtubule occupancy required to silence the SAC13. Thus, mutations causing excess kMT detachment would be rarely observed as they would likely lead to inviability. Indeed, only modest levels of GFP-Kif2b or GFP-MCAK overexpression were tolerated (Supp. Fig. 4) while increasingly high levels reduced cell viability. This provides a potential explanation for the extremely low cellular abundance of Kif2b whose activity must be temporally and spatially regulated to prevent perpetual destabilization of kMT attachment. Conversely, mutations causing even modest hyperstability of kMT attachment would disrupt correction of malorientations predisposing cells to chromosome mis-segregation and CIN. Not surprisingly, multiple gene mutations such as APC33,34, hCDC435, and BRCA136 have been reported to increase lagging chromosomes in anaphase. Nevertheless our data show that overexpression of enzymes required for the latest stages of malorientation correction bypass upstream defects in chromosome segregation and hence suppress CIN irrespective of its origin. These results open the possibility to directly investigate the contribution of CIN toward tumorigenicity, and provide additional means to enhance tumor cell response to chemotherapeutic agents by reducing the incidence of drug resistance arising from genetic instability.
Methods
GFP-Tubulin photoactivation
Mitotic cells were identified by D.I.C. microscopy and several pulses from a 405 nm diffraction-limited laser (Photonic Instruments, St Charles, IL) were used to photoactivate and area of < 2.5 μm2 of GFP within the spindle. Images were acquired with a Hamamatsu Orca II camera binned 2 × 2 with a 63X, 1.4 NA objective on a Zeiss Axioplan 2 microscope. 3 × 1-μm stacks of fluorescent images were collected <1s before and after photoactivation. Subsequently, images were collected every 30 s. D.I.C. microscopy was then used to verify that cells did not undergo anaphase.
Photoactivation analysis
FDAPA analysis was performed primarily as described previously29-31. Briefly, pixel intensities were measured within a 2 μm2 rectangular area surrounding the region with the brightest fluorescence and pixel intensities from an equal area from the opposite half-spindle were subtracted. Correction for photobleaching was made by normalizing to values obtained from photoactivated taxol-stabilized spindles where the photoactivatable region clearly did not dissipate. Bleaching-induced decrease in average fluorescence after 30-captured images was 35%. For each cell, fluorescence values were normalized to the first time-point after photoactivation. Normalized fluorescence was then averaged for multiple cells at each time-point. A double exponential regression analysis was used to fit the data to the following equation: A1*exp(−k1*t)+A2*exp(−k2*t), where A1 and A2 represent less (non-kMT) and more (kMT) stable microtubule populations with decay rates of k1 and k2, respectively.
Chromosome mis-segregation assay
Cells were grown in flasks, isolated by mitotic shake-off, plated at low density on glass slides, and incubated for 16 hours to allow completion of mitosis. Cells were fixed and fluorescence in situ hybridization (FISH) was performed as described17 using α-satellite probes specific for chromosomes 2, 3, and 7 (Cytocell). Chromosome signals in pairs of daughter nuclei were recorded using previously defined criteria37.
Chromosomal instability assay
U2OS cells were grown in flasks, isolated by shake-off and plated at very low density. Single clones were isolated at 15-20 generations using cloning rings. Isolated clones were maintained until they reached 27 generations. Cells were fixed and FISH was performed on clonal populations as described17 with probes for chromosomes 2, 3, 7, and 15 (Cytocell). 300 nuclei were counted per chromosome per cell clone.
Cell culture
Cells were maintained at 37 °C in a 5% CO2 atmosphere in Dulbecco's modified medium (DMEM) or McCoy's medium (PA-GFP-tubulin cells) containing 10% fetal bovine serum, 50 IU/ml penicillin, and 50 μg/ml streptomycin. For plasmid selection cells were maintained in 0.5-1.0 mg/ml of G418 (geneticin).
Antibodies
Kif2b-specific antibody24, MCAK-specific antibody38, Tubulin-specific mAb DM1α (Sigma-Aldrich, St. Louis, MO), CREST antibody (provided by Kevin Sullivan), Hec1-specific mAb (Novus Biologicals, Littleton, CO)
RNA interference and plasmid transfection
Published sequences were used to deplete MCAK39 and Kif2b24. 70-200 nM dsRNA (Ambion) were transfected into cells using Oligofectamine reagent (Invitrogen) as previously described40 and cells were analyzed 70 hours after transfection. Plasmids encoding GFP-tagged Kif2a, Kif2b and MCAK were provided by Linda Wordeman (U. Washington). Cells were transfected with plasmid DNA using the lipofectamine reagent (Invitrogen).
Indirect immunofluorescence microscopy
Cell fixation and antibody staining were performed as previously described24. Cells were fixed with ice-cold methanol when staining for microtubules, CREST, Cyclin B, DNA. Cells were fixed with 1% gluteraldehyde when staining for microtubules, DNA and for visualization of GFP-constructs. Images were acquired with a Hamamatsu Orca II CCD camera mounted on a Zeiss Axioplan 2 microscope with 63X and 100X, 1.4 NA objectives or with a Hamamatsu Orca ER cooled CCD camera mounted on a Nikon TE-2000E microscope with a 60x, 1.4 NA objective. Image series in the z-axis were obtained using 0.2- or 0.25-μm optical sections. Image deconvolution and contrast enhancement was performed using Openlab and Phylum live software (Improvision, Lexington, MA). Final images represent selected overlaid planes.
Fluorescence time-lapse microscopy
Glass coverslips were mounted on a stainless steel modified Rose chamber containing appropriate growth medium and sealed with VALAP. All time-lapse images were collected at 37°C on the Zeiss or Nikon TE-2000E microscopes as described above.
Drug treatments
Drug concentrations used were the following: 100 μM monastrol, 0.1 μg/ml of nocodazole, 50 nM hesperadin (Tarun Kapoor, the Rockefeller University) and 5 μM of MG132. Drug washouts were performed by either 3 washes with fresh growth medium or two washes with PBS followed by fresh growth medium.
Nocodazole sensitivity assays
In nocodazole sensitivity assay, 100 μg/ml nocodazole and 100 μM of monastrol were added to cells for various periods of time and cells were fixed with gluteraldehyde. Total microtubule fluorescence was quantified with background subtraction while also subtracting mean fluorescence of spindles exposed to nocodazole for 120min. Then fluorescence values were normalized to the first time point.
Supplementary Material
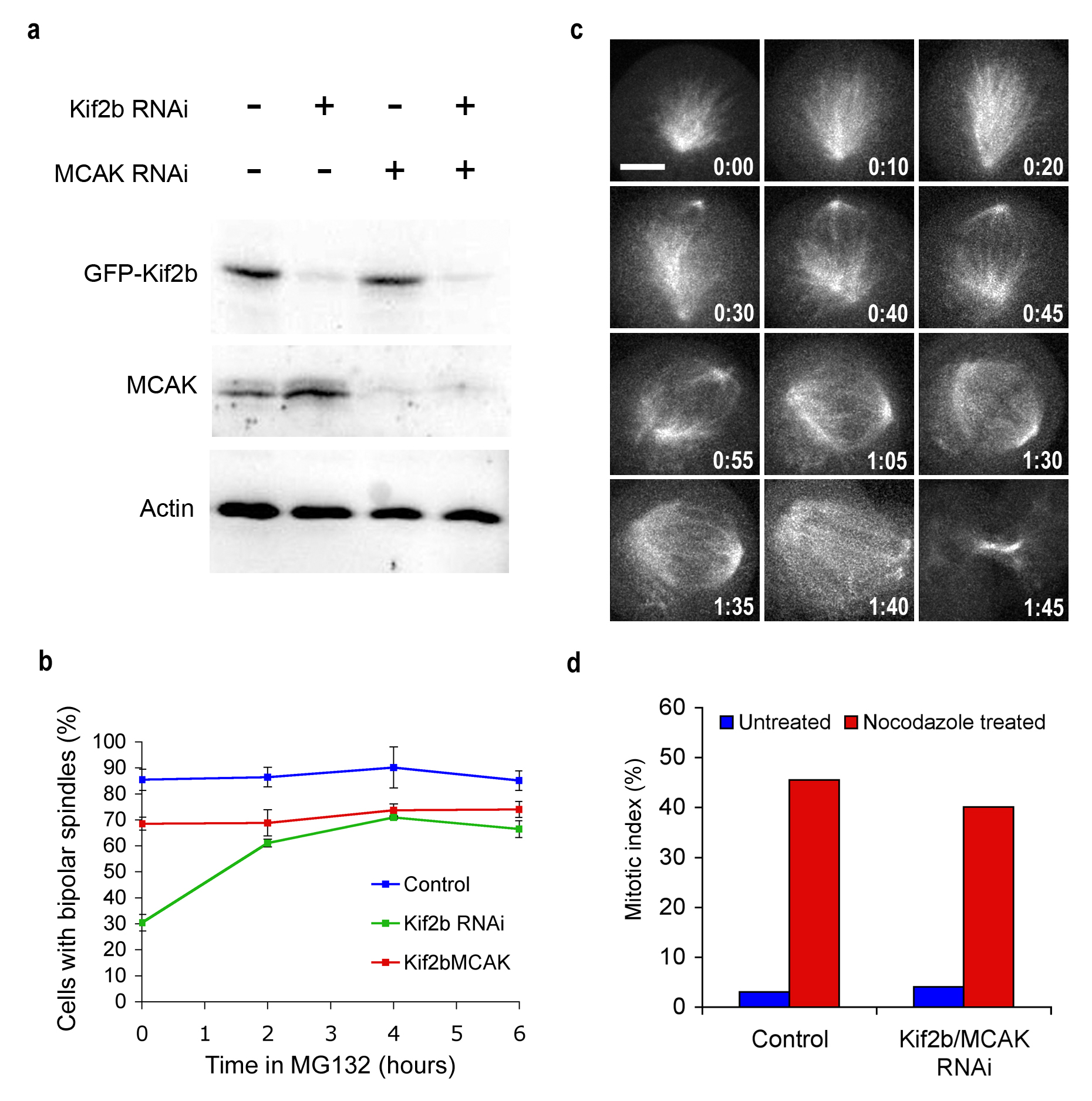
a, Total cell lysates from untreated, Kif2b-deficient, MCAK-deficient and Kif2b/MCAK-deficient U2OS cells overexpressing GFP-Kif2b were immunoblotted with antibodies specific for Kif2b, MCAK, and actin as indicated. b, Percent bipolar spindles of untreated U2OS cells, Kif2b- or MCAK/Kif2b-deficient U2OS cells after treatment in MG132 for various times. Points represent mean ± s.e.m., n = 200 cells, 2 experiments. c, Live cell time-lapse fluorescence of GFP-tubulin in a Kif2b-deficient U2OS cell at the indicated times (hours:minutes) fluorescence images were collected at 0.5-μm stacks over 10-μm and merged. Scale bar 5 μm. d, Mitotic index of untreated (Control) or Kif2b/MCAK-deficient U2OS cells that were either untreated or arrested in nocodazole for 16 hours. n > 200 cells for each condition.
{kind=link}
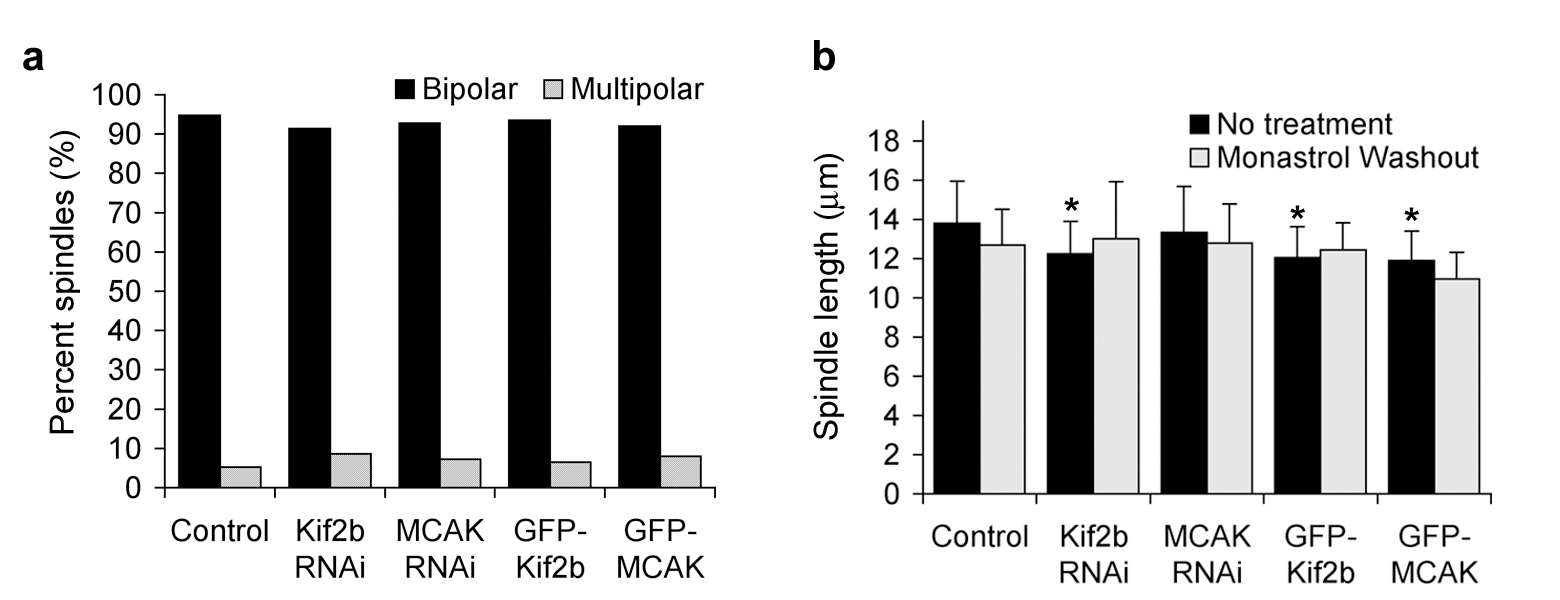
a, Percent bipolar and multipolar spindles in control, Kif2b, or MCAK depleted, or in GFP-Kif2b or GFP-MCAK overexpressing U2OS cells. n = 100 spindles. b, Average spindle length in control, Kif2b, or MCAK depleted, or in GFP-Kif2b, or GFP-MCAK overexpressing U2OS cells. n = 50 spindles, p < 0.05.
{kind=link}
a, Fluorescent images of monopolar spindles induced by monastrol treatment in control or Kif2b-deficient U2OS cells that were exposed to high concentrations of nocodazole for different times indicated in minutes. b, Intensities of total microtubule fluorescence in cells from part (a) above as well as in cells overexpressing GFP-Kif2b normalized to t = 2 min. in nocodazole. Images were collected at 0.2-μm stacks over 10-μm depth and fluorescence was quantified by merging images and subtracting background fluorescence in a region of interest of equal area outside the cell periphery. Bars represent mean ± s.e.m., n = 20 cells. c, Fluorescent images of monopolar spindles induced by monastrol in U2OS cells overexpressing different levels of GFP-Kif2b that were treated in nocodazole for 2 min showing a qualitative inverse correlation between kMT stability and GFP-Kif2b expression level. Scale bars 5 μm.
{kind=link}
Fluorescent images of microtubules (red), DNA (blue) and the overexpressed GFP-tagged proteins (green) as indicated showing the expected targeting of the overexpressed proteins. Right panels represent immunoblots of the total cell lysate of untreated U2OS cells and U2OS cells overexpressing various GFP-MCAK (a), GFP-Kif2a (b), GFP-Kif2b (c), and GFP-α-tubulin (d) blotted with anti-MCAK antibody (a), anti-Kif2a antibody (b), anti-Kif2b antibody (c), and anti-α-tubulin antibody (d). Scale bars 5 μm. e, total cell lysates blotted with anti-GFP antibody. Lanes represents lysates from control cells (lane1), cell populations expressing mixed levels of GFP-Kif2a (lane2), GFP-Kif2b (lane3), GFP-MCAK (lane4), as well as clonal cell populations expressing GFP-Kif2b (lane5; clone 5 from Table 1), GFP-MCAK (lane6; clone 8 from Table 1), and GFP-α-tubulin (lane7).
{kind=link}
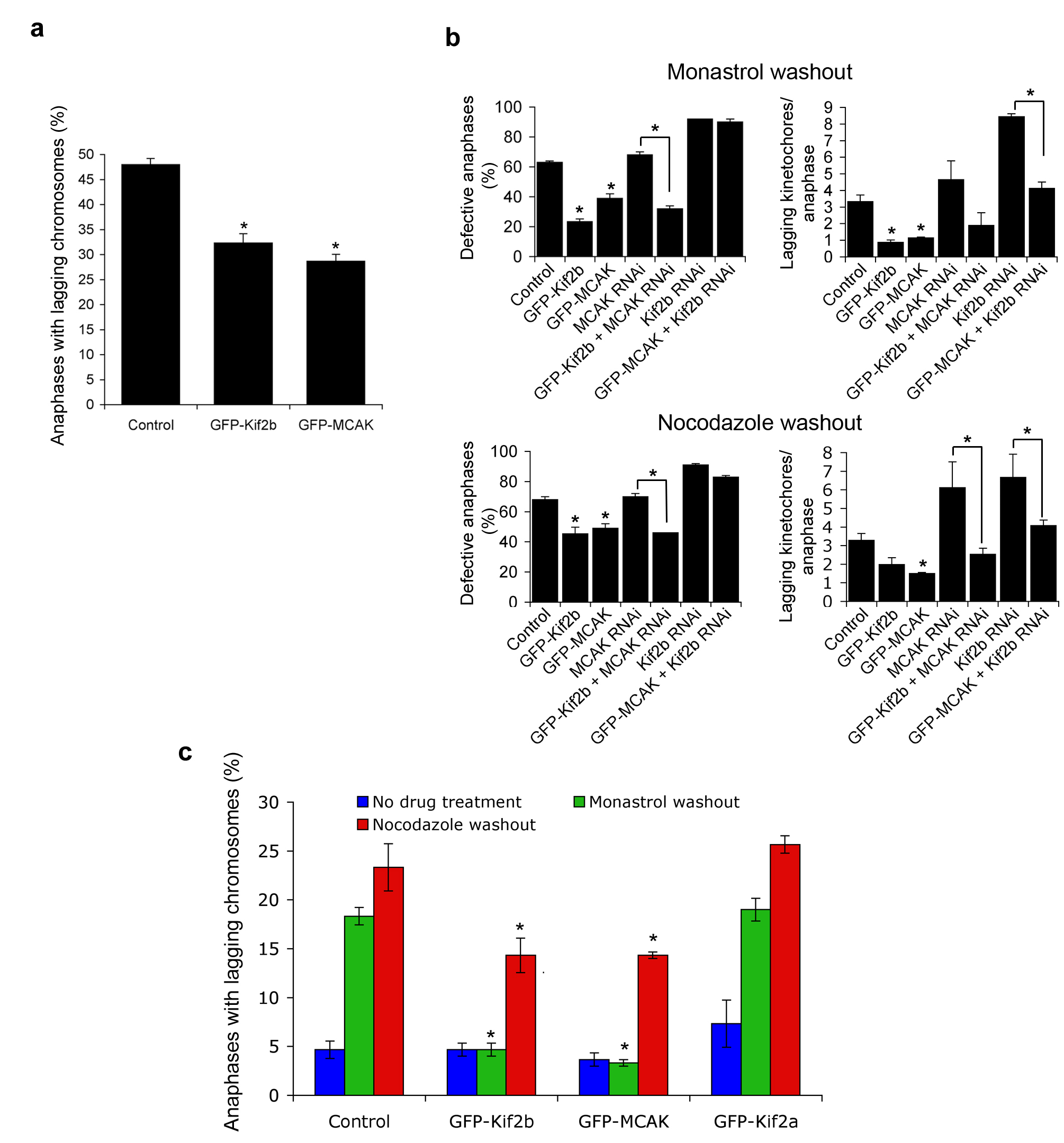
a, Percent of anaphase cells with lagging chromosomes in untreated MCF-7 cells and MCF-7 cells overexpressing GFP-MCAK or GFP-Kif2b as indicated. Bars represent mean ± s.e.m, n = 300 cells, 3 experiments. *, p < 0.05, Chi-square test. b, Percent of anaphase cells with lagging kinetochores and average numbers of lagging chromosomes per anaphase of U2OS cells after monastrol washout (a) and nocodazole washout (b). Cells were either untreated (Control) or depleted of Kif2b (Kif2b RNAi), MCAK (MCAK RNAi), and/or overexpressing GFP-Kif2b or GFP-MCAK as indicated. Bars represent mean ± s.e.m, n = 100 cells, 2 experiments. *, p < 0.05, t-test. c, Percent of anaphase cells with lagging chromosomes in untreated RPE1 cells (blue), or RPE1 cells recovering from monastrol (green) or nocodazole (red). Bars represent mean ± s.e.m, n = 300 cells, 3 experiments. *, p < 0.05, Chi square test.
{kind=link}
Acknowledgements
We thank Linda Wordeman (U. Washington) for providing plasmids encoding GFP-tagged proteins, Tarun Kapoor for providing Hesperadin, and Kevin Sullivan for providing the CenpB-GFP U2OS cells. This work was supported by National Institutes of Health grants GM51542 to D.A.C. and GM008704 to S.L.T. The authors claim no competing financial interest.
References
- 1. http://cgap.nci.nih.gov/Chromosomes/Mitelman.
- 2.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 3.Storchova Z, Pellman D. From polyploidy to aneuploidy, genome instability and cancer. Nat. Rev. Mol. Cell Biol. 2004;5:45–54. doi: 10.1038/nrm1276. [DOI] [PubMed] [Google Scholar]
- 4.Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancer. Nature. 1997;386:623–627. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- 5.Gao C, et al. Chromosome instability, chromosome transcriptome, and clonal evolution of tumor cell populations. Proc. Natl. Acad. Sci. USA. 2007;104:8995–9000. doi: 10.1073/pnas.0700631104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuukasjarvi T, et al. Genetic heterogeneity and clonal evolution underlying development of asynchronous metastasis in human breast cancer. Cancer Res. 1997;57:1597–1604. [PubMed] [Google Scholar]
- 7.Houlston R, Tomlinson I. Association between chromosomal instability and prognosis in colorectal cancer: a meta-analysis. Gut. 2008 doi: 10.1136/gut.2007.135004. d.o.i. 10.1136/gut.2007.135004. [DOI] [PubMed] [Google Scholar]
- 8.Cheeseman IM, Desai A. Molecular architecture of the kinetochore-microtubule interface. Nat. Rev. Mol. Cell Biol. 2008;9:33–46. doi: 10.1038/nrm2310. [DOI] [PubMed] [Google Scholar]
- 9.Loncarek J, et al. The centromere geometry essential for keeping mitosis error free is controlled by spindle forces. Nature. 2007;450:745–749. doi: 10.1038/nature06344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Indjeian VB, Murray AW. Budding yeast mitotic chromosomes have an intrinsic bias to biorient on the spindle. Curr. Biol. 2007;17:1837–1846. doi: 10.1016/j.cub.2007.09.056. [DOI] [PubMed] [Google Scholar]
- 11.McEwen BF, Heagle AB, Cassels GO, Buttle KF, Rieder CL. Kinetochore fiber maturation in PtK1 cells and its implication for the mechanisms of chromosome congression and anaphase onset. J. Cell Biol. 1997;137:1567–1580. doi: 10.1083/jcb.137.7.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cimini D, Degrassi F. Aneuploidy: a matter of bad connections. Trends Cell Biol. 2005;15:442–451. doi: 10.1016/j.tcb.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 13.Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 2007;8:379–383. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- 14.Pinsky BA, Kung C, Shokat KM, Biggins S. The Ipl-1-Aurora protein kinase activates the spindle checkpoint by creating unattached kinetochores. Nat. Cell Biol. 2006;8:78–83. doi: 10.1038/ncb1341. [DOI] [PubMed] [Google Scholar]
- 15.Cahill DP, et al. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–303. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- 16.Barber TD, et al. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc. Natl. Acad. Sci. USA. 2008;105:3443–3448. doi: 10.1073/pnas.0712384105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thompson SL, Compton DA. Examining the link between chromosomal instability and aneuploidy in human cells. J. Cell Biol. 2008;180:665–672. doi: 10.1083/jcb.200712029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cimini D, et al. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J. Cell Biol. 2001;153:517–527. doi: 10.1083/jcb.153.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andrews PD, et al. Aurora B regulates MCAK at the mitotic centromere. Dev. Cell. 2004;6:253–268. doi: 10.1016/s1534-5807(04)00025-5. [DOI] [PubMed] [Google Scholar]
- 20.Lan W, et al. Aurora B phosphorylates centromeric MCAK and regulates its localization and microtubule depolymerization activity. Curr. Biol. 2004;14:273–286. doi: 10.1016/j.cub.2004.01.055. [DOI] [PubMed] [Google Scholar]
- 21.Knowlton AL, Lan W, Stukenberg PT. Aurora B is enriched at merotelic attachment sites, where it regulates MCAK. Curr. Biol. 2006;16:1705–1710. doi: 10.1016/j.cub.2006.07.057. [DOI] [PubMed] [Google Scholar]
- 22.Kline-Smith SL, Khodjakov A, Hergert P, Walczak CE. Depletion of centromeric MCAK leads to chromosome congression and segregation defects due to improper kinetochore attachments. Mol. Biol. Cell. 2004;15:1146–1159. doi: 10.1091/mbc.E03-08-0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maney T, Hunter AW, Wagenbach M, Wordeman L. Mitotic centromere-associated kinesin is important for anaphase chromosome segregation. J. Cell Biol. 1998;142:787–801. doi: 10.1083/jcb.142.3.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manning AL, et al. The kinesin-13 proteins Kif2a, Kif2b, and Kif2c/MCAK have distinct roles during mitosis in human cells. Mol. Biol. Cell. 2007;18:2970–2979. doi: 10.1091/mbc.E07-02-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cimini D, Moree B, Canmen JC, Salmon ED. Merotelic kinetochore orientation occurs frequently during early mitosis in mammalian tissue cells and error correction is achieved by two different mechanisms. J. Cell Sci. 2003;116:4213–4225. doi: 10.1242/jcs.00716. [DOI] [PubMed] [Google Scholar]
- 26.Lampson MA, Renduchitala K, Khodjakov A, Kapoor TM. Correcting improper chromosome-spindle attachments during cell division. Nat. Cell Biol. 2004;6:232–237. doi: 10.1038/ncb1102. [DOI] [PubMed] [Google Scholar]
- 27.Hauf S, et al. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J. Cell Biol. 2003;161:281–294. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nicklas RB, Ward SC. Elements of error correction in mitosis: microtubule capture, release, and tension. J. Cell Biol. 1994;126:1241–1253. doi: 10.1083/jcb.126.5.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cimini D, Wan X, Hirel CB, Salmon ED. Aurora kinase promotes turnover of kinetochore microtubules to reduce chromosome segregation errors. Curr. Biol. 2006;16:1711–1718. doi: 10.1016/j.cub.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 30.Zhai Y, Kronebusch PJ, Borisy GG. Kinetochore microtubule dynamics and the metaphase-anaphase transition. J. Cell Biol. 1995;131:721–734. doi: 10.1083/jcb.131.3.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeLuca JG, et al. Kinetochore microtubule dynamics and attachment stability are regulated by Hec1. Cell. 2006;127:969–982. doi: 10.1016/j.cell.2006.09.047. [DOI] [PubMed] [Google Scholar]
- 32.Fuller BG, et al. Midzone activation of aurora B in anaphase produces an intracellular phosphorylation gradient. Nature. 2008;453:1132–1136. doi: 10.1038/nature06923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Green RA, Kaplan KB. Chromosome instability in colorectal tumor cells is associated with defects in microtubule plus-end attachments caused by dominant mutation of APC. J. Cell Biol. 2003;163:949–961. doi: 10.1083/jcb.200307070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fodde R, et al. Mutations in the APC tumor suppressor gene cause chromosomal instability. Nat. Cell Biol. 2001;3:433–438. doi: 10.1038/35070129. [DOI] [PubMed] [Google Scholar]
- 35.Rajagopalan H, et al. Inactivation of hCDC4 can cause chromosomal instability. Nature. 2001;409:355–359. doi: 10.1038/nature02313. [DOI] [PubMed] [Google Scholar]
- 36.Joukov V, et al. The BRCA1/BARD1 heterodimer modulates Ran-dependent mitotic spindle assembly. Cell. 2006;127:539–552. doi: 10.1016/j.cell.2006.08.053. [DOI] [PubMed] [Google Scholar]
- 37.Cimini D, Tanzarella C, Degrassi F. Differences in malsegregation rates obtained by scoring ana-telophases or binucleate cells. Mutagenesis. 1999;14:563–568. doi: 10.1093/mutage/14.6.563. [DOI] [PubMed] [Google Scholar]
- 38.Mack GJ, Compton DA. Analysis of mitotic microtubule-associated proteins using mass spectrometry identifies astrin, a spindle-associated protein. Proc. Natl. Acad. Sci. USA. 2001;98:14434–14439. doi: 10.1073/pnas.261371298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ganem NJ, Upton K, Compton DA. Efficient mitosis in human cells lacking poleward microtubule flux. Curr. Biol. 2005;15:1827–1832. doi: 10.1016/j.cub.2005.08.065. [DOI] [PubMed] [Google Scholar]
- 40.Ganem NJ, Compton DA. The KinI kinesin Kif2a is required for bipolar spindle assembly through a functional relationship with MCAK. J. Cell Biol. 2004;166:473–478. doi: 10.1083/jcb.200404012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
a, Total cell lysates from untreated, Kif2b-deficient, MCAK-deficient and Kif2b/MCAK-deficient U2OS cells overexpressing GFP-Kif2b were immunoblotted with antibodies specific for Kif2b, MCAK, and actin as indicated. b, Percent bipolar spindles of untreated U2OS cells, Kif2b- or MCAK/Kif2b-deficient U2OS cells after treatment in MG132 for various times. Points represent mean ± s.e.m., n = 200 cells, 2 experiments. c, Live cell time-lapse fluorescence of GFP-tubulin in a Kif2b-deficient U2OS cell at the indicated times (hours:minutes) fluorescence images were collected at 0.5-μm stacks over 10-μm and merged. Scale bar 5 μm. d, Mitotic index of untreated (Control) or Kif2b/MCAK-deficient U2OS cells that were either untreated or arrested in nocodazole for 16 hours. n > 200 cells for each condition.
a, Percent bipolar and multipolar spindles in control, Kif2b, or MCAK depleted, or in GFP-Kif2b or GFP-MCAK overexpressing U2OS cells. n = 100 spindles. b, Average spindle length in control, Kif2b, or MCAK depleted, or in GFP-Kif2b, or GFP-MCAK overexpressing U2OS cells. n = 50 spindles, p < 0.05.
a, Fluorescent images of monopolar spindles induced by monastrol treatment in control or Kif2b-deficient U2OS cells that were exposed to high concentrations of nocodazole for different times indicated in minutes. b, Intensities of total microtubule fluorescence in cells from part (a) above as well as in cells overexpressing GFP-Kif2b normalized to t = 2 min. in nocodazole. Images were collected at 0.2-μm stacks over 10-μm depth and fluorescence was quantified by merging images and subtracting background fluorescence in a region of interest of equal area outside the cell periphery. Bars represent mean ± s.e.m., n = 20 cells. c, Fluorescent images of monopolar spindles induced by monastrol in U2OS cells overexpressing different levels of GFP-Kif2b that were treated in nocodazole for 2 min showing a qualitative inverse correlation between kMT stability and GFP-Kif2b expression level. Scale bars 5 μm.
Fluorescent images of microtubules (red), DNA (blue) and the overexpressed GFP-tagged proteins (green) as indicated showing the expected targeting of the overexpressed proteins. Right panels represent immunoblots of the total cell lysate of untreated U2OS cells and U2OS cells overexpressing various GFP-MCAK (a), GFP-Kif2a (b), GFP-Kif2b (c), and GFP-α-tubulin (d) blotted with anti-MCAK antibody (a), anti-Kif2a antibody (b), anti-Kif2b antibody (c), and anti-α-tubulin antibody (d). Scale bars 5 μm. e, total cell lysates blotted with anti-GFP antibody. Lanes represents lysates from control cells (lane1), cell populations expressing mixed levels of GFP-Kif2a (lane2), GFP-Kif2b (lane3), GFP-MCAK (lane4), as well as clonal cell populations expressing GFP-Kif2b (lane5; clone 5 from Table 1), GFP-MCAK (lane6; clone 8 from Table 1), and GFP-α-tubulin (lane7).
a, Percent of anaphase cells with lagging chromosomes in untreated MCF-7 cells and MCF-7 cells overexpressing GFP-MCAK or GFP-Kif2b as indicated. Bars represent mean ± s.e.m, n = 300 cells, 3 experiments. *, p < 0.05, Chi-square test. b, Percent of anaphase cells with lagging kinetochores and average numbers of lagging chromosomes per anaphase of U2OS cells after monastrol washout (a) and nocodazole washout (b). Cells were either untreated (Control) or depleted of Kif2b (Kif2b RNAi), MCAK (MCAK RNAi), and/or overexpressing GFP-Kif2b or GFP-MCAK as indicated. Bars represent mean ± s.e.m, n = 100 cells, 2 experiments. *, p < 0.05, t-test. c, Percent of anaphase cells with lagging chromosomes in untreated RPE1 cells (blue), or RPE1 cells recovering from monastrol (green) or nocodazole (red). Bars represent mean ± s.e.m, n = 300 cells, 3 experiments. *, p < 0.05, Chi square test.