

Abstract
Conformational changes of Klebsiella aerogenes urease apoprotein (UreABC)3 induced upon binding of the UreD and UreF accessory proteins were examined by a combination of flexibility analysis, mutagenesis, and small-angle x-ray scattering (SAXS). ProFlex analysis of urease provided evidence that the major domain of UreB can move in a hinge-like motion to account for prior chemical cross-linking results. Rigidification of the UreB hinge region, accomplished through a G11P mutation, reduced the extent of urease activation, in part by decreasing the nickel content of the mutant enzyme, and by sequestering a portion of the urease apoprotein in a novel activation complex that includes all of the accessory proteins. SAXS analyses of urease, (UreABC-UreD)3, and (UreABC-UreDF)3 confirm that UreD and UreF bind near UreB at the periphery of the (UreAC)3 structure. This study supports an activation model in which a domain-shifted UreB conformation in (UreABC-UreDF)3 allows CO2 and nickel ions to gain access to the nascent active site.
Keywords: Urease, Activation, Flexibility, Small-angle X-ray scattering
Urease is a nickel-containing enzyme that hydrolyzes urea [1, 2]. Crystallographic analyses of ureases from bacterial and plant sources [3–7] reveal a basic trimeric structure with three active sites, each composed of two nickel ions coordinated by a carboxylated Lys, four His and an Asp. Genetic and biochemical studies carried out with plants, fungi, and bacteria [reviewed in [8–10]] have shown that additional genes encoding accessory proteins are required for proper assembly of the urease metallocenter, with the possible exception of that from Bacillus subtilis [11]. The current model for urease metallocenter assembly (Fig. 1) derives primarily from studies involving expression of the Klebsiella aerogenes ureDABCEFG gene cluster in Escherichia coli [reviewed in [8, 12]]. The active enzyme possesses three copies of each of three subunits (UreA, UreB, and UreC of Mr 11,086, 11,695, and 60,304, respectively)[13]. Deletions within ureD, ureE, ureF, or ureG eliminate urease activity due to production of the inactive (UreABC)3 urease apoprotein1 [14]. Expression of ureDABC produces (UreABC-UreD)3, with UreD (Mr 29,300) in complex with urease apoprotein [15]. Co-expression of ureF (encoding a protein of Mr 25,221) with ureDABC produces (UreABC-UreDF)3 [16]. The soluble protein UreG (Mr 21,943) reversibly binds to (UreABC-UreDF)3 forming (UreABC-UreDFG)3 [17, 18]. Urease activity is generated by incubating these complexes with high concentrations of bicarbonate (to supply the CO2 needed for Lys carboxylation) and nickel ions, but the required levels of these additives (100 mM and 100 µM, respectively) are not physiologically relevant and only a portion of the proteins are activated [19, 20]. In contrast, fully active urease is generated with only 100 µM bicarbonate and 20 µM nickel ions using (UreABC-UreDFG)3 plus UreE (Mr 17,558) and GTP [21]. UreE functions as a nickel-binding protein [22, 23] that delivers the metal ion to (UreABC-UreDFG)3 as GTP is hydrolyzed [24]. Although UreE is often referred to as a metallochaperone [25, 26] and UreDFG has been termed a urease-specific molecular chaperone [9], the mechanism of urease metallocenter assembly has remained obscure.
Fig. 1.
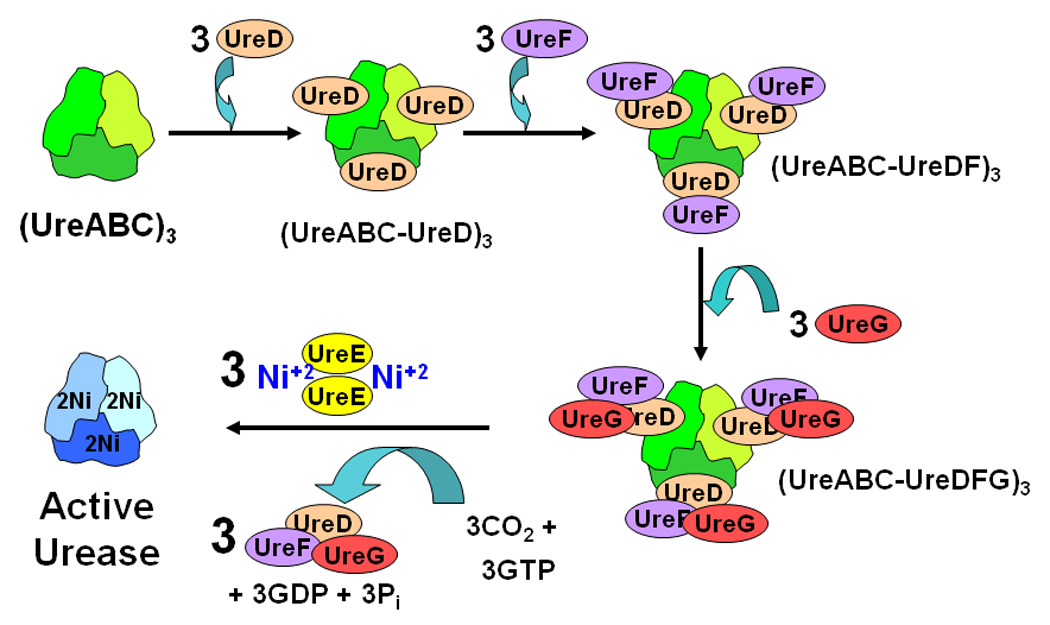
Proposed pathway of urease activation. The K. aerogenes UreA, UreB and UreC urease subunits assemble into the (UreABC)3 apoprotein (depicted simply as a trimeric species since UreA plus UreB or all three subunits are fused together in ureases from some sources). UreD, UreF and UreG sequentially bind to form the (UreABC-UreD)3, (UreABC-UreDF)3, and (UreABC-UreDFG)3 activation complexes. CO2 adds to the active site Lys as Ni+2 ions are delivered to (UreABC-UreDFG)3 by the dimeric UreE metallochaperone in a process that requires GTP hydrolysis, with UreE and (UreDFG)3 being released from the activated urease.
The near identity in structure of the (UreABC)3 apoprotein [27] and the nickel-containing holoenzyme [3] indicate that conformational changes are required to introduce the metal ions and CO2 into the deeply buried nascent active site. Chemical cross-linking of (UreABC-UreDF)3 [28] identified a cross-link between UreB Lys76 and UreC Lys382 that provided evidence for a conformational change of the protein, since UreB Lys76 is positioned far from UreC Lys382 in the (UreABC)3 crystal structure. Here, we use computational flexibility analysis to identify a hinge region that allows the main UreB domain to shift to a position that permits formation of the key intra-urease cross-link. In addition, we show that one of two amino acid changes affecting the hinge region leads to a large reduction in urease activation, partly due to decreasing the extent of nickel incorporation, while also sequestering a large percentage of the urease protein in a complex with the accessory proteins. Finally, the overall shapes of (UreABC-UreD)3 and (UreABC-UreDF)3 were examined by small-angle X-ray scattering (SAXS) [29]. The results derived by this technique demonstrate that UreD and UreF bind together with UreB at the perimeter of the disk formed by (UreAC)3, providing new evidence to confirm interactions between UreB and UreD or UreF derived from previous chemical cross-linking studies [28]. These results are compatible with earlier urease activation studies and suggest that the combined action of UreD and UreF serves to expose the nascent active site of urease.
Materials and methods
Protein Purification
(UreABC-UreD)3, (UreABC-UreDF)3, and urease holoenzyme were produced in E. coli DH5α carrying pKAUD2 [15], E. coli DH5α pKAUD2F+ΔureG [16], or E. coli HMS174(DE3) carrying pKK17 [25] and purified as previously described [30]. HEDG buffer (25 mM HEPES, pH 7.4, 1 mM EDTA, 1 mM DTT, 1% glycerol) was used as a final storage buffer unless noted. The homogeneity of samples was assessed by densitometric analysis (AlphaImager) of Coomassie-stained gels after sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) [31]. The expression level of urease subunits in cell extracts was assessed by SDS-PAGE followed by electroblotting the sample onto Immobilon-P polyvinylidene difluoride membrane, probing with anti-K. aerogenes urease antibodies [32], and visualizing with anti-rabbit immunoglobulin G-alkaline phosphatase conjugates. In a similar manner, the identity of a band in one sample was examined by Western blot with anti-K. aerogenes UreE antibodies [33].
Flexibility Analysis
We used the graph theoretic algorithm ProFlex to analyze the flexibility of urease (Protein Data Bank (PDB) entry 1FWJ). The program identifies the flexible and rigid regions in a given structure (which bonds are constrained and which bonds remain free to rotate) based on analysis of constraints posed by the protein’s network of covalent bonds, hydrogen bonds, salt bridges, and hydrophobic interactions [34]. ProFlex calculations have been shown to predict the conformational flexibility of proteins reliably from a single 3D structure [34–36]. The ProFlex code was modified and extended to enable the program to process structures with a large number of atoms and large number of flexible and rigid regions resulting from hydrogen-bond dilution or an extensive network of interactions. The changes accommodated the size of the urease complex and extend the utility of ProFlex for analysis of other very large proteins, including those with multiple subunits. These changes allowed processing of the very large urease structure (~22000 atoms in the trimer of trimers). The ProFlex software is available to other research groups by request to proflex@sol.bch.msu.edu.
Site-Directed Mutagenesis and Activity Assay
Plasmid pKK17 [25] containing the entire urease gene cluster was cut with BamHI and the smaller of two fragments (3.3 kbp) containing ureB was ligated into BamHI-restricted pUC19 (New England BioLabs), producing pUCB. Mutations of ureB were introduced by PCR using primers 5’-GAA TAT CAC GTT AAG CCC CCA CAG ATA GCC CTG AAT ACC-3’ and its complement to introduce the UreB G11P mutation and 5’- CAG ATA GCC CTG AAT ACC CCA CGG GCA ACC TGT CGC GTG-3’ and its complement for the UreB G18P mutation (the mutated codons are underlined). The PCR reaction (18 cycles of 50 s at 95 °C, 50 s at 50 °C and 8 min at 72 °C) was performed with 12.5 µL of PfuTurbo® Hotstart PCR master mix (Stratagene), 10 µM of each primer, and the pUCB plasmid as template, followed by incubation for 1 h at 37 °C with 0.5 µL of DpnI. DH5α cells were transformed with 5 µL of the digested PCR reaction. Plasmids from putative clones were purified, sequenced to confirm the mutations, and digested with BamHI to recover the 3.3-kbp fragments. These fragments were cloned back into pKK17 to create pKKBG11P and pKKBG18P.
E. coli cells containing pKK17, pKKBG11P, or pKKBG18P were grown in Luria-Bertani medium containing 1 mM NiCl2 for three h and induced overnight with 0.1 mM isopropyl-β-D-thiogalactopyranoside. The stationary phase cells were harvested by centrifugation, sonicated, and clarified by ultracentrifugation. Cell extracts were tested for expression of the urease genes by denaturing gel electrophoresis [31] and subjected to protein analyses [37] and urease activity assays [38] using standard procedures.
Metal Quantification
The nickel content of selected samples was assessed by using inductively coupled plasma-mass spectrometry at the University of Georgia Chemical Analysis Laboratory.
SAXS Measurements and Analysis
SAXS data were obtained using the ORNL Center for Structural Molecular Biology 4m SAXS instrument, described previously [39]. Sample intensity patterns were collected for native urease, (UreABC-UreD)3, and (UreABC-UreDF)3 plus backgrounds consisting of the HEDG buffer solution. Protein concentrations were 3.8 mg/mL for native urease, 5.4 mg/mL for (UreABC-UreD)3, and 2.0 mg/mL for (UreABC-UreDF)3. These concentrations made it impractical to measure a concentration series, but also made it unlikely that interparticle interference effects significantly influenced the data and subsequent analyses. Multiple measurements were averaged together to enable testing for time-dependent aggregation due to radiation damage; none was found. For (UreABC-UreD)3 and (UreABC-UreDF)3, four 4-hour runs were summed together, while five 4-hour runs were summed together for the native urease complex. These measurements included runs with fresh material and runs in which the sample was exposed for an additional 4 hours to check for radiation damage. No artifacts due to radiation damage were observed. Data were reduced, azimuthally averaged and scaled into absolute units (1/cm) according to previously published procedures [39] to provide the 1D intensity profile I(q) vs. q, where q = 4πsin(θ)/λ, 2θ is the scattering angle from the incident beam, and λ is the wavelength of the X-ray radiation (1.542 Å).
Small-Angle X-Ray Scattering Analysis and Modeling
Data were subjected to Guinier analysis [40] for the radius of gyration, Rg, and for the pair-distance distribution function P(r). I(q) and P(r) are related through the Fourier transform shown in Equation 1.
| (1) |
The program GNOM [41] uses an indirect transform method to find P(r) from an input maximum linear dimension, dmax. The optimum dmax is found by trial and error to find a solution that best fits the data and provides an acceptable termination of P(r) at dmax. The P(r) fitting also provides a secondary measure of the Rg, which is the second moment of P(r).
The program ORNL_SAS [42] was employed to compare the scattering profiles calculated from the urease structure and various models of complexes against the measured SAXS profiles of the enzyme, (UreABC-UreD)3 and (UreABC-UreDF)3. To model the (UreABC-UreD)3 and (UreABC-UreDF)3 complexes, ellipsoids were used in place of the unknown structures of UreD and UreF. The structures of the higher-order complexes were built by placing three identical ellipsoids with the (UreABC)3 structure about the same three-fold symmetry axis around which the trimer of trimers is formed. The translation coordinates were chosen randomly from a range of values that made it possible to produce complexes that extended beyond the experimentally determined dmax. To ensure the proper volume for the added proteins, two of the ellipsoidal semiaxes were randomly chosen from a range of 10 Å to 35 Å, and the third was initially picked to produce the correct expected volume based on the amino acid sequence of the subunit. In the event that the third semiaxis was found to be less than 10 Å, a new set of semiaxes was generated. The ellipsoids were placed around the (UreABC)3 structure and the volumes occupied by the ellipsoids that did not overlap with the (UreABC)3, or the (UreABC)3 plus the set of UreD ellipsoids in the case of (UreABC-UreDF)3, were determined. If the non-overlapping volume of the ellipsoid was not within 1% of the expected volume of the UreD or UreF subunit based on the molecular weight, the ellipsoidal semiaxes were scaled to provide the correct volume. As the specific overlap region with the other structures changes as the semiaxes are scaled, an iterative process was employed to rescale the ellipsoidal dimensions until the non-overlapping volumes of ellipsoids were within 1% of the correct volume. Only the portions of the ellipsoid that did not overlap were retained for the intensity calculations. Models found to have Rg values consistent with the experimental data were input into ORNL_SAS for comparison against the experimental data. ORNL_SAS was configured to treat the density of the scattering particle as uniform because no atomic-resolution structures are available for UreD and UreF. A 3 Å thick hydration layer, assumed to be 10% more dense than the surrounding solution, was used for the ORNL_SAS intensity calculation. The thickness and density of the hydration layer were not parameters in the data fitting.
The quality of the fit of the model intensity profiles to the experimental data was evaluated using the reduced χ2 parameter defined in Equation 2.
| (2) |
Npts is the number of data points modelled against in the measured intensity I(q). σ(q) is the experimental uncertainty in the measured intensity I(q). Im(q) is the model intensity profile. Nf is the number of degrees of freedom, and was 2, which accounts for the scaling of the model intensity profile to the data input into ORNL_SAS. ORNL_SAS, being a general intensity calculator [42], does not have a mechanism to account for the ellipsoidal structural parameters in Nf. The number of data points is a great deal larger than the number of degrees of freedom in any of the models tested, so the impact on χ2 is relatively small. Additionally, each model is tested relative to models generated with the same number of free parameters, so the relative comparisons are not affected. In order to judge the range of structures that fit the experimental data collected for (UreABC-UreD)3 and (UreABC-UreDF)3, the best 25 models found were maintained in an ordered list that was updated as better models were found, in a manner similar to previous work [43], making it possible to judge the reproducibility of the modeling. The uncertainties in measured SAXS intensities derive from specific assumptions about the counting statistics. In cases of relatively low count rates, the error propagation can result in uncertainties that overestimate the true uncertainty in the measurement relative to the noise in the data, making it possible to have χ2 significantly less than one. An inspection of the fidelity of the model profile to the data is required to ensure that the quality of the fit is truly excellent. The χ2 parameter suitably serves as a least squared minimization parameter for modeling in such situations.
Results
Flexibility Analysis of Urease
ProFlex [34] was used to analyze the flexibility within the native enzyme trimer of trimers (PDB entry 1FWJ; Fig. 2A and supplementary Fig. S1A), identifying a total of ~3100 hydrogen bonds and ~1500 hydrophobic interactions. The regions of the protein defined as rigid or flexible were found to vary little with the choice of hydrogen-bond energy cutoff in ProFlex (between -1 and -2 kcal/mol), defining the set of hydrogen bonds and salt bridges incorporated in the network. In the crystal structure of urease, UreB is anchored by six N-terminal residues that add to the edge of a beta sheet in UreC (Fig. 2B and 2C, region 1). A salt bridge and at least six hydrophobic interactions between UreB residues 2–8 and UreC residues 6–29 reinforce the attachment (supplementary Tables S1 and S2). ProFlex predicted UreB residues 11–19 to form a flexible hinge (Fig. 2, region 2; Tables S3 and S4) between the N-terminal anchor and the relatively rigid domain formed by UreB residues 20–101. The latter domain includes polar and hydrophobic interactions with UreC (Tables S5 and S6), but these are few in number compared to the interactions with regions 1 and 2 and consistent with the possibility of domain movement. The anchored and hinge residues of the N-terminal region of UreB (residues 1–19) fit into a groove of the N-terminal region of UreC formed by residues C2–C41 (Fig. 2B).
Fig. 2.
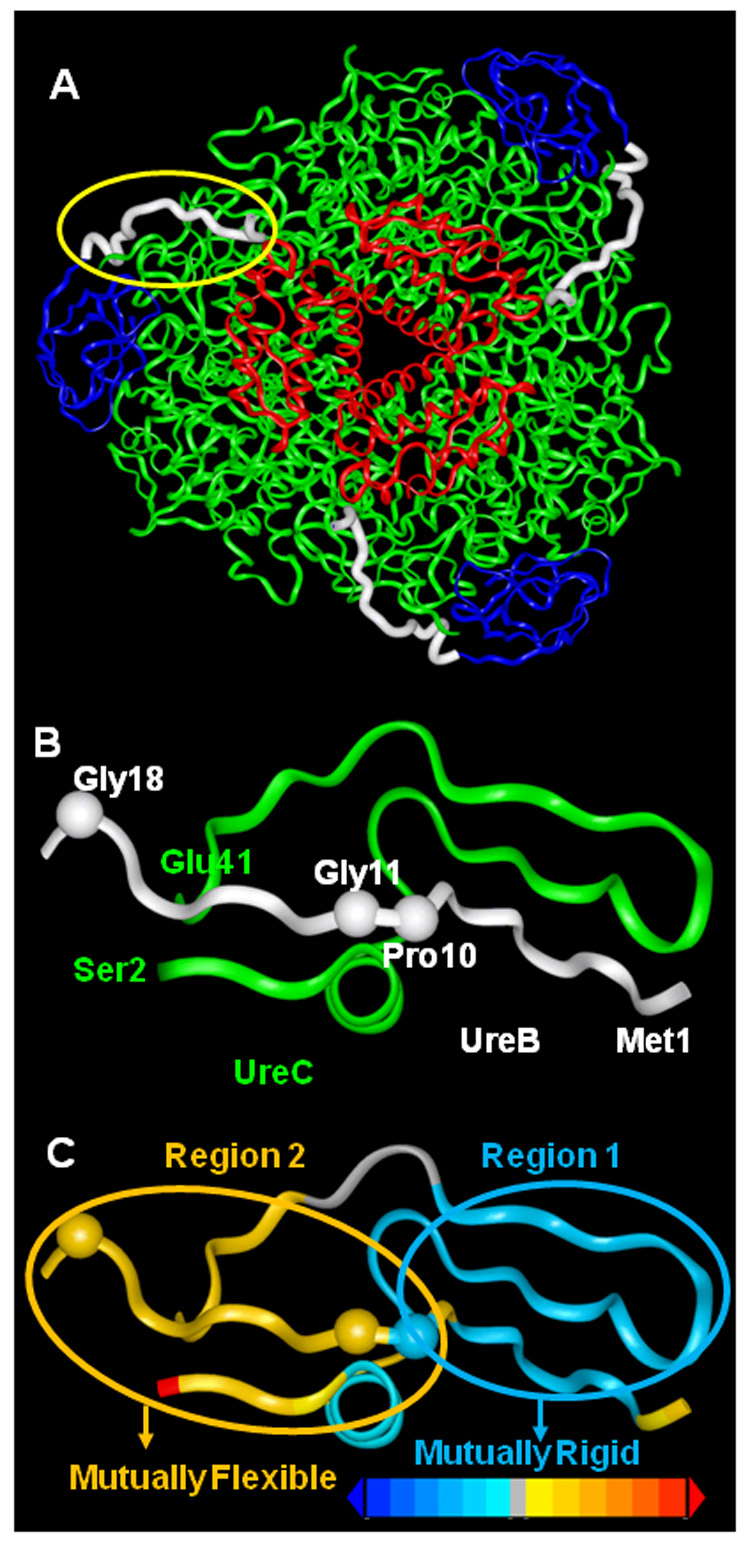
Tether and hinge regions between UreB and UreC from the crystallographic structure of urease. (A) The native urease structure, with ribbons colored red for UreA, blue for UreB (except for its hinge and tether to UreC shown in white), and green for UreC. (B) An expanded view of the region encircled in yellow in panel A. The N-terminus of UreB (residues 2–8) forms the terminal strand of a beta sheet with UreC. UreB residues 11–19 together with UreC residues 2–6 and 13–41 form a flexible linkage between the main domain of UreB (blue ribbons in panel A) and the disk formed by (UreAC)3 (red and green ribbons in panel A). Sites relevant to flexibility probing mutations, UreB Pro10, Gly11 and Gly18, are rendered as beads. (C) The same view as panel B, colored in terms of ProFlex flexibility analysis of the crystal structure (PDB entry 1FWJ). The N-terminus of UreB partitions from a rigid region (colored blue; region 1) to a flexible hinge (colored gold; region 2) which connects to the globular domain of UreB (shown in blue ribbons in panel A). The terminus of UreC is highly flexible (red), whereas residues in UreC that intervene between regions 1 and 2 are isostatic, or barely rigid, as shown in grey.
Chemical modification results [28] indicate that UreB Lys76 and UreC Lys382 can be cross-linked when in the (UreABC-UreDF)3 species. This requires bringing their side chains to within 10 Å, although they are 50 Å apart in the urease crystal structure. Thus, we probed whether the flexibility of UreB residues 11–19 would allow these two Lys residues to move to within cross-linking distance while maintaining favorable packing between UreB and UreC. In the first approach, UreB Gly11 and Gly18 were of special interest due to the prevalence of Gly in flexible regions of proteins. This is because Gly residues have no constraints on main-chain bond rotations (ϕ and ψ angle torsions) due to the absence of side-chain induced steric hindrance. The torsion angles of UreB Gly11 and Gly18 were manually changed to reduce the distance between UreB Lys76 and UreC Lys382 and attain reasonable packing between UreB and UreAC. The resulting distance between the Cα atoms of UreB Lys76 and UreC Lys382 was 19.8 Å, close enough to allow cross-linking of their side chains. This motion involved a rotation of +131 degrees in ϕ and +110 degrees in ψ for Gly11, with 7 degree changes in both ϕ and ψ for Gly18, creating UreB conformation 1 (supplementary Fig. S1B). In a second approach, we cut the tether at UreB Gly11, docked UreB Lys76 within cross-linking distance of UreC Lys282 while maintaining good packing between the subunits, and reconnected the tether. This approach created UreB conformation 2 (supplementary Fig. S1C). A close-up view highlighting the repositioning of UreB to achieve conformation 1 and allow cross-linking is depicted in Figure 3. Both approaches yielded substantially similar placement of UreB at the periphery of (UreAC)3 due to the strong constraints placed by maintaining the anchoring interactions of UreB residues 2–10 while meeting the cross-linking distance between UreB Lys76 and UreC Lys382.
Fig. 3.
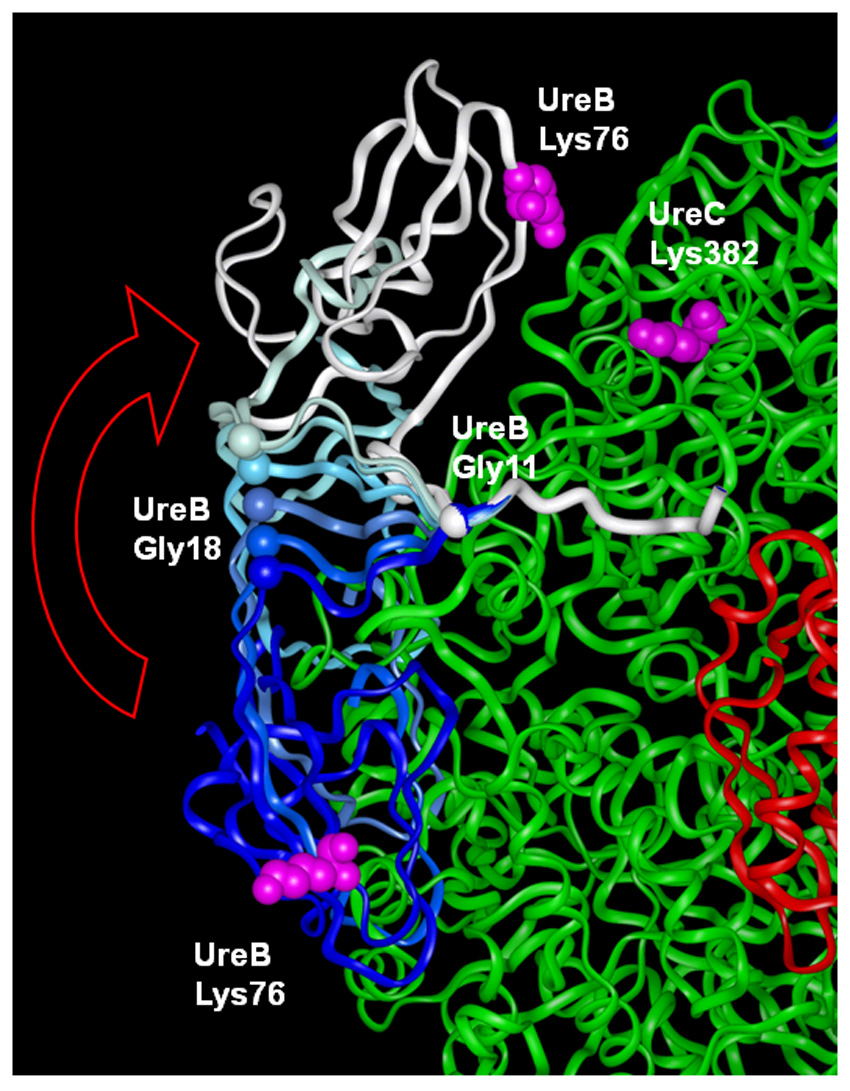
Close-up of the repositioning of UreB. The main domain of UreB is proposed to shift from its crystallographic position (dark blue; PDB 1FWJ) to a position (white) in which UreB Lys76 can cross link with UreC Lys382 (pink CPK spheres), opening access to the active site. The range of motion of UreB hinge residues resulting in this rotation of UreB is shown by the series of blue to lighter blue conformations of residues 11–19 between the UreB crystallographic and cross-linked open positions.
Mutagenesis of Hinge Residues
To directly test the importance of putative UreB hinge region residues Gly11 and Gly18 in urease activation, their codons were independently modified to encode Pro residues that would restrict hinge flexibility. Constructs encoding the G11P and G18P variants of UreB were created and used to substitute for the wild-type sequence in a plasmid containing the complete urease gene cluster. The mutated plasmids were transformed into host E. coli cells, and urease overexpression was shown to be comparable in the control and mutant strains by using Western blots (data not shown). Urease activity in cell extracts containing the G18P variant of UreB was similar to that for extracts containing wild-type enzyme, indicating the flexibility of residue 18 is not critical to urease activation. In contrast, extracts containing the G11P mutant displayed 15–50% (depending on the preparation) of the activity of the control strain. This result suggests that protein dynamics requiring the flexibility of residue 11 are important to the metallocenter assembly process.
Urease containing UreB G11P was purified from the mutant strain and subjected to metal analysis. Whereas control enzyme exhibits a specific activity of 2,200 ± 200 µmol min−1 (mg protein−1) and contains 2.1 ± 0.3 nickel ions per active site [44], the purified UreB G11P variant protein possessed a specific activity of approximately 440 µmol min−1 (mg protein−1) and only contained 1.67 nickel ions per active site (single determination with an estimated error of <10%). For comparison, previous experiments showed that when (UreABC)3 was incubated with nickel plus bicarbonate in vitro, 2.13 to 1.74 nickel ions were present per active site with the activated enzyme yielding specific activities of 0 and 442 µmol min−1 (mg protein)−1 [20], as opposed to 2,200 µmol min−1 (mg protein−1); thus, high nickel content can be associated with inactive protein. These results suggest both a deficiency in nickel incorporation and formation of a less effective dinuclear site in the mutant protein. Significantly, the mutant urease protein was resolved into two fractions during phenyl-Sepharose chromatography (Fig. 4). The highly purified urease analyzed above was obtained by elution with buffer lacking salt, as in the case of wild-type enzyme. In addition, a nearly inactive urease-containing fraction was obtained by subsequent washing of the resin with water. The second pool of urease contained four major contaminating proteins that co-migrated with UreD (Mr 29,807), UreG (Mr 21,943), UreF (Mr 25,221), and UreE (Mr 17,558) (note that the peptides do not migrate precisely according to their known size). A Western blot analysis with anti-UreE antibodies (data not shown) confirmed the identity of UreE in this sample. The finding of this newly identified complex is compatible with the need for flexibility in the hinge region of UreB to achieve accessory protein dissociation. The deleterious effects on urease activity, nickel content, and accessory protein dissociation that come from restricting the motion of UreB Gly11 by Pro substitution are consistent with the observation that large changes in main-chain ϕ and ψ values of UreB Gly11 are needed to place Lys76 of this subunit within cross-linking distance of Lys382 in UreC. The neighboring residue, UreB Pro10, already limits the accessible ϕ angles so the G11P mutant would severely restrict the conformations available to the hinge. These results are consistent with a hinge-like motion of UreB relative to UreC upon binding of UreD and UreF, allowing access to the active site for activation.
Fig. 4.
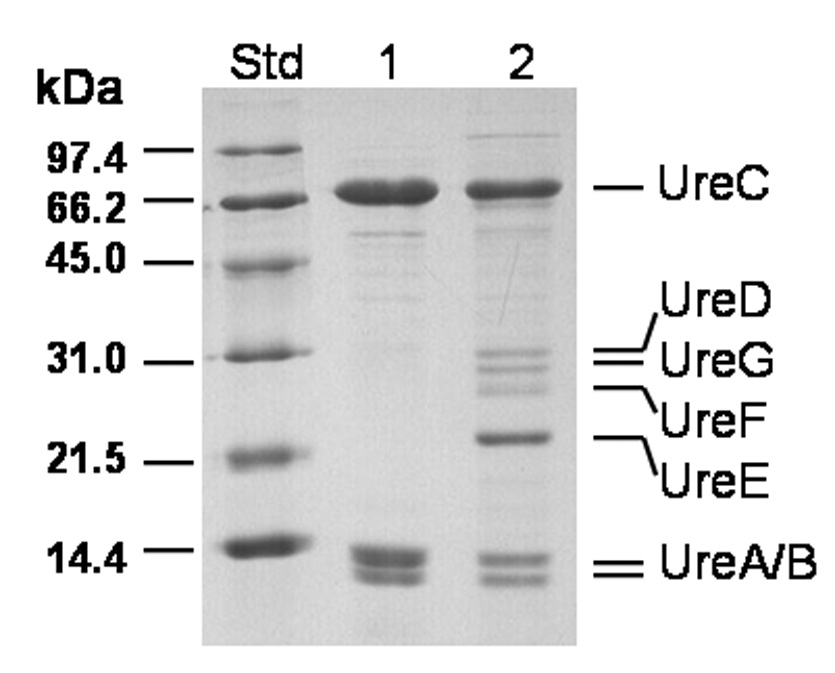
Two pools of the UreB G11P mutant urease resolved by phenyl-Sepharose chromatography. Molecular weight standards (Std), the purified active mutant urease (lane 1), and the very low activity complex containing mutant urease (lane 2) were examined by SDS-PAGE using a 13.5% acrylamide gel and stained with Coomassie brilliant blue.
Small-Angle X-Ray Scattering Measurements and Analyses
SAXS data were collected for native urease, (UreABC-UreD)3, and (UreABC-UreDF)3 (Fig. 5). Instrument stability issues, primarily due to temperature fluctuations in the facility, caused the differences in usable minimum q shown in the graph. The inset curves in Figure 5 are the Guinier regions for the three data sets, and correspond to Rg of 32.7 ± 2.4 Å , 40.3 ± 2.3 Å, and 50.6 ± 2.5 Å for the respective species. In all cases, the Guinier regions are linear, indicative of monodisperse scattering particles. The data do not display artifacts due to interparticle interference, which manifests as a significant downturn at low q-values. The P(r) curves derived from the SAXS data (Fig. 6) indicate increasing size with increasing number of components. The Rg for urease determined from the P(r) fitting was 35.7 ± 0.8 Å, with a dmax of 95 ± 5 Å. The values of Rg for the (UreABC-UreD)3 and (UreABC-UreDF)3 complexes were 44.9 ± 0.7 Å and 53.7 ± 1.4 Å, respectively. The dmax of the (UreABC-UreD)3 complex was 130 ± 8 Å, while that of the (UreABC-UreDF)3 complex was 155 ± 10 Å. The agreement between the Guinier- and GNOM-derived Rg values is reasonable considering the very different methods of obtaining the values and estimating the uncertainties.
Fig. 5.

I(q) curves derived from the scattering data for urease (■), (UreABC-UreD)3 (○) and (UreABC-UreDF)3 (▲). The lines are the model fits to the data using the crystal structure of urease (PDB 1FWJ) (solid line), with UreB Gly11/Gly18 torsionally adjusted to allow cross-linking of UreB Lys76 to UreC Lys382 (dashed line), and UreB docked to UreAC from the crystal structure, allowing cross-linking of UreB Lys76 to UreC Lys382 (dotted line). The curves have been offset by a multiplicative factor for clarity. The inset plot shows the Guinier regions and fit lines for the three measured profiles. Again, the curves have been offset for clarity, and the region of data covered by the line indicates the range of data used for the fitting.
Fig. 6.

P(r) curves derived from the scattering data for urease (■), (UreABC-UreD)3 (○), and (UreABC-UreDF)3 (▲). To simplify comparison, the curves have been scaled to have a value of 1.0 at the peak.
Models of the Activation Complexes
The intensity profile calculated from the wild-type urease crystal structure [3] using the program ORNL_SAS [42] is shown with the data in Figure 5. The agreement between the measured data and the simulated profile is excellent, having a χ2 of 0.493. The fit of the model intensity profile to the data across the entire q-range is excellent.
Models of (UreABC-UreD)3 were generated by adding UreD ellipsoids to the wild-type urease structure and to (UreABC)3 with the two alternative UreB conformations: torsionally-adjusted and docked. Ellipsoids were used because no structure or homology model is available for any UreD. In all cases, the overall structures of the final complexes were very similar. The best models had UreD ellipsoids added to the vertices of (UreABC)3 near the UreB subunit such that the total structure has a planar, triangular character (supplementary Fig. S2). The best three model intensity profiles for the three different starting structures have χ2 values of 0.218, 0.252, and 0.224 when starting with the native structure, torsionally-adjusted UreB, and docked UreB, respectively. In all cases, the fits of the profiles to the data are excellent in light of the experimental uncertainties shown in Figure 5 and suggest that all of the structures are reasonable. It is important to note that the three models all have the same general shape, which is the most reliable result of the modeling considering the method of building the models and the lack of a high-resolution structure of UreD. The addition of UreD results in a planar, triangular structure. The (UreABC-UreD)3 results are in agreement with UreD interacting with UreB as suggested by chemical cross-linking [28].
Models of (UreABC-UreDF)3 were created by adding ellipsoids to represent appropriate molecular volumes of UreD and UreF to the (UreABC)3 crystal structure and the torsionally-adjusted and docked models produced by the flexibility modeling. As above, no high-resolution structure or model is available for UreD; however, a homology model was reported for UreF from Bacillus pasteurii [45]. The 202 residue B. pasteurii protein is 32% identical over only 91 residues of the 224 amino acid K. aerogenes UreF. For this reason ellipsoids were chosen to represent this protein in the modeling, as well. The models produced using the two alternative UreB conformations (torsionally-adjusted and docked) are similar, and in fact resulted in similar placements of UreD and UreF in the best-fitting SAXS models (shown for the docked conformation in Fig. 7). The best UreB conformation 1 (torsionally-adjusted) and UreB conformation 2 (docked) structures fit the scattering data very well, as well as account for the cross-linking results, and have χ2 of 0.093 and 0.094, respectively, as compared to the χ2 of 0.096 observed for the model produced from the native UreB structure. The fit of the model profiles to the data are all excellent, as can be seen in Figure 5, so it is not possible to discriminate between the SAXS models for the reasons provided above. The overall shape of the complex, which can be reliably extracted from the data, is very consistent between the three models (supplementary Fig. S3), having a planar, triangular character with the additional mass corresponding to UreD and UreF located near the vertices, almost coplanar with the rest of the structure. The UreF ellipsoids are near the UreD ellipsoids in all of the models, rather than being spatially separated. The model depicted in Figure 7 appears to build on the models of (UreABC-UreD)3, with the UreD and UreF ellipsoids positioned pair wise at the vertices of the (UreABC)3 structure. In this case UreB, UreD, and UreF essentially add onto the edge of the disk formed primarily by the UreC trimer, in which UreA forms the hub (Fig. 2A). These structures are consistent with immunological results that show anti-UreD antibodies recognize UreD within (UreABC-UreD)3, but not within (UreABC-UreDF)3, suggesting that UreF partially masks UreD [16]. In addition, these results are consistent with cross-linking between UreF and UreB [28].
Fig. 7.
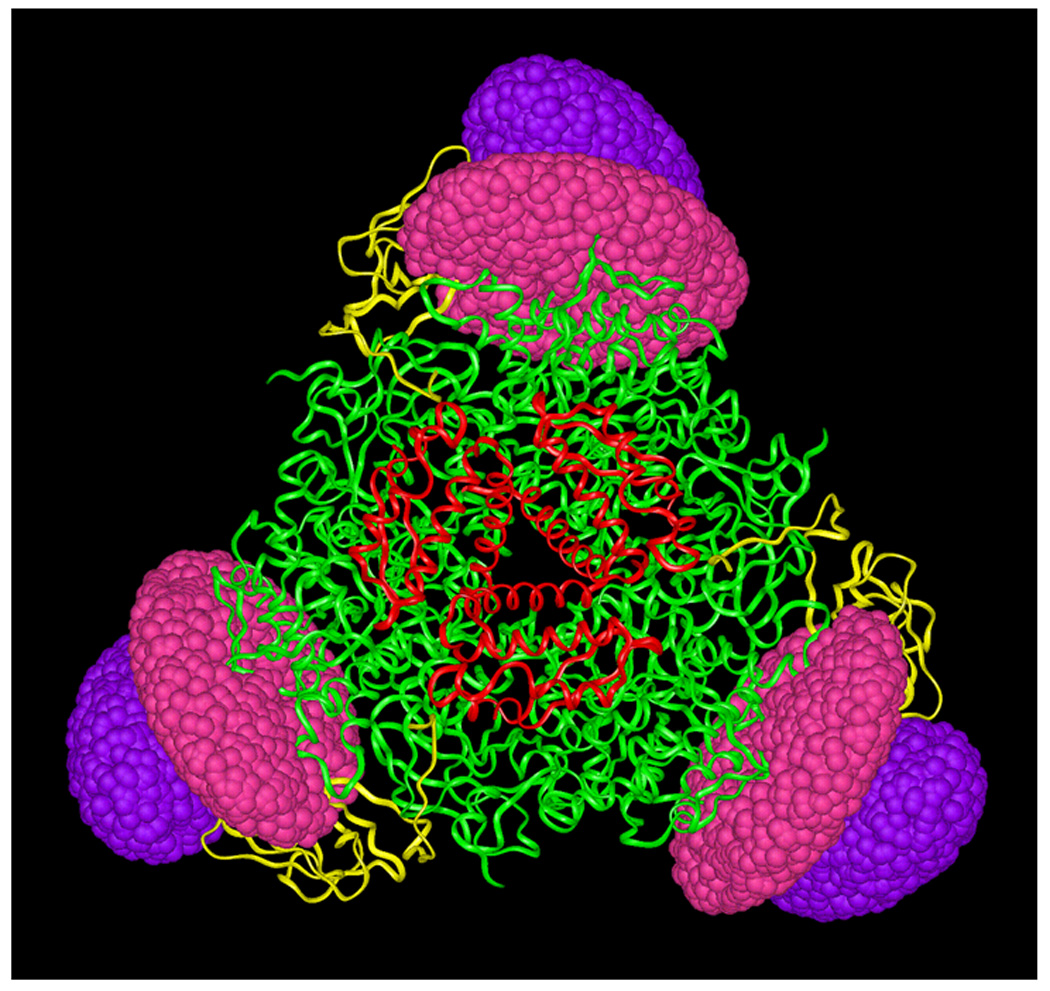
Predicted positioning of UreD and UreF relative to the crystallographic structure of (UreABC)3, based on best-fit models to SAXS data. The best-fit models resulted in packing of UreD and UreF against UreB near a vertex of the (UreAC)3 disk. A representative example is illustrated. UreA, UreB, and UreC are rendered in red, yellow, and green ribbons, respectively. UreD and UreF from SAXS results are rendered as solid ellipsoids colored purple and magenta, respectively. The non-interpenetrating volumes of the UreD and UreF ellipsoids accounts for the appropriate molecular weight of each subunit.
Discussion
In this work we combined multi-scale modeling and sparse experimental constraints to obtain insight into a flexible molecular assembly, the urease activation complex. In particular, we used flexibility analysis to provide evidence that the major domain of UreB can move in a hinge-like motion to allow sufficiently close juxtaposition of UreB Lys76 with UreC Lys382 to form a chemical cross-link between these residues, as previously reported [28]. The UreB G11P variant, which is likely to rigidify the hinge region, was shown to lead to reduced levels of urease activation and lower nickel content while also sequestering a significant portion of the urease apoprotein in an ineffective activation complex that includes all four of the known K. aerogenes accessory proteins. The larger impact observed for the G11P variant compared to the G18P mutant is likely due to the presence of Pro10 which further increases the rigidity of the hinge. These results support the importance of a flexible hinge region in urease activation. Significantly, the predicted structures of (UreABC-UreDF)3 containing UreB rotated away from the active site (Fig. 3) would provide access to the nascent active site and allow urease activation. The SAXS results lack sufficient resolution to address the proposed domain shift of UreB, but they confirm that UreD and UreF bind near UreB. This finding agrees with prior immunological and chemical cross-linking studies [16, 28].
Comparison of the H. pylori urease structure (PDB entry 1E9Z) with that of K. aerogenes urease discussed here provides additional support for the proposed sites of UreD and UreF interaction with UreB, at the periphery of the (UreAC)3 disk. The H. pylori UreA subunit (corresponding to a fusion of UreA and UreB in the K. aerogenes enzyme) contains a fold that matches the K. aerogenes UreB fold, but also contains residues that add to one side of this shared fold in a similar position to where we predict UreD and UreF bind. A viral protein (PDB entry 1C5E) also contains this shared fold, with an additional domain in the same region as the added domain in H. pylori UreA. Thus, both other proteins that share the UreB domain fold with K. aerogenes urease use this domain as a molecular interface, suggesting that this region of UreB has evolved to interact with other domains or proteins. This supports its role in K. aerogenes UreB as a docking interface for UreD or UreF.
Supplementary Material
Acknowledgements
We thank Scott Mulrooney, Kimberly Anderson, and Yiming Mo for their assistance. This work was supported by the National Institutes of Health (DK45686 to R.P.H. and GM67249 to L.A.K.), the MSU Quantitative Biology & Modeling Initiative (Strategic Partnership Grant 71-4841), and Project KP1102010 of the Office of Biological and Environmental Research of the U. S. Department of Energy, under contract No. DE-AC05-00OR22725 with Oak Ridge National Laboratory managed and operated by UT-Batelle, LLC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Abbreviations used: (UreABC)3, urease apoprotein; (UreABC-UreD)3, complex of UreD bound to urease apoprotein; (UreABC-UreDF)3, complex of UreD and UreF bound to urease apoprotein; (UreABC-UreDFG)3, complex formed by UreD, UreF, and UreG bound to urease apoprotein; SAXS, small-angle x-ray scattering; PDB, Protein Data Bank; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis.
References
- 1.Hausinger RP, Karplus PA. In: Handbook of Metalloproteins. Wieghardt K, Huber R, Poulos TL, Messerschmidt A, editors. West Sussex, U.K.: John Wiley & Sons, Ltd.; 2001. pp. 867–879. [Google Scholar]
- 2.Ciurli S, Mangani S. In: Handbook on Metalloproteins. Bertini I, Sigel A, Sigel H, editors. New York, NY: Marcel Dekker; 2001. pp. 669–708. [Google Scholar]
- 3.Jabri E, Carr MB, Hausinger RP, Karplus PA. Science. 1995;268:998–1004. [PubMed] [Google Scholar]
- 4.Pearson MA, Michel LO, Hausinger RP, Karplus PA. Biochemistry. 1997;36:8164–8172. doi: 10.1021/bi970514j. [DOI] [PubMed] [Google Scholar]
- 5.Benini S, Rypniewski WR, Wilson KS, Miletti S, Ciurli S, Mangani S. Structure. 1999;7:205–216. doi: 10.1016/S0969-2126(99)80026-4. [DOI] [PubMed] [Google Scholar]
- 6.Ha N-C, Oh S-T, Sung JY, Cha K-A, Lee MH, Oh B-H. Nature Struct. Biol. 2001;8:505–509. doi: 10.1038/88563. [DOI] [PubMed] [Google Scholar]
- 7.Sheridan L, Wilmont CM, Cromie KD, van der Logt P, Phillips SEV. Acta Crystallographa. 2002;D58:374–376. doi: 10.1107/s0907444901021503. [DOI] [PubMed] [Google Scholar]
- 8.Mulrooney SB, Hausinger RP. FEMS Microbiol. Rev. 2003;27:239–261. doi: 10.1016/S0168-6445(03)00042-1. [DOI] [PubMed] [Google Scholar]
- 9.Quiroz S, Kim JK, Mulrooney SB, Hausinger RP. In: Metal Ions in Life Sciences. Sigel A, Sigel H, Sigel RKO, editors. New York: John Wiley & Sons; 2007. pp. 519–544. [Google Scholar]
- 10.Hausinger RP, Zamble DB. In: Molecular Microbiology of Heavy Metals. Nies DH, Silver S, editors. Springer-Verlag; 2007. pp. 287–320. [Google Scholar]
- 11.Kim JK, Mulrooney SB, Hausinger RP. J. Bacteriol. 2005;187:7150–7154. doi: 10.1128/JB.187.20.7150-7154.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hausinger RP, Colpas GJ, Soriano A. ASM News. 2001;67:78–84. [Google Scholar]
- 13.Mulrooney SB, Hausinger RP. J. Bacteriol. 1990;172:5837–5843. doi: 10.1128/jb.172.10.5837-5843.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee MH, Mulrooney SB, Renner MJ, Markowicz Y, Hausinger RP. J. Bacteriol. 1992;174:4324–4330. doi: 10.1128/jb.174.13.4324-4330.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park I-S, Carr MB, Hausinger RP. Proc. Natl. Acad. Sci. 1994;91:3233–3237. doi: 10.1073/pnas.91.8.3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moncrief MBC, Hausinger RP. J. Bacteriol. 1996;178:5417–5421. doi: 10.1128/jb.178.18.5417-5421.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park I-S, Hausinger RP. J. Bacteriol. 1995;177:1947–1951. doi: 10.1128/jb.177.8.1947-1951.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soriano A, Hausinger RP. Proc. Natl. Acad. Sci. 1999;96:11140–11144. doi: 10.1073/pnas.96.20.11140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park I-S, Hausinger RP. Science. 1995;267:1156–1158. doi: 10.1126/science.7855593. [DOI] [PubMed] [Google Scholar]
- 20.Park I-S, Hausinger RP. Biochemistry. 1996;35:5345–5352. doi: 10.1021/bi952894j. [DOI] [PubMed] [Google Scholar]
- 21.Soriano A, Colpas GJ, Hausinger RP. Biochemistry. 2000;39:12435–12440. doi: 10.1021/bi001296o. [DOI] [PubMed] [Google Scholar]
- 22.Song HK, Mulrooney SB, Huber R, Hausinger RP. J. Biol. Chem. 2001;276:49359–49364. doi: 10.1074/jbc.M108619200. [DOI] [PubMed] [Google Scholar]
- 23.Mulrooney SB, Ward SK, Hausinger RP. J. Bacteriol. 2005;187:3581–3585. doi: 10.1128/JB.187.10.3581-3585.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moncrief MBC, Hausinger RP. J. Bacteriol. 1997;179:4081–4086. doi: 10.1128/jb.179.13.4081-4086.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Colpas GJ, Brayman TG, Ming L-J, Hausinger RP. Biochemistry. 1999;38:4078–4088. doi: 10.1021/bi982435t. [DOI] [PubMed] [Google Scholar]
- 26.Musiani F, Zambelli B, Stola M, Ciurli S. J. Inorg. Biochem. 2004;98:803–813. doi: 10.1016/j.jinorgbio.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 27.Jabri E, Karplus PA. Biochemistry. 1996;35:10616–10626. doi: 10.1021/bi960424z. [DOI] [PubMed] [Google Scholar]
- 28.Chang Z, Kuchar J, Hausinger RP. J. Biol. Chem. 2004;279:15305–15313. doi: 10.1074/jbc.M312979200. [DOI] [PubMed] [Google Scholar]
- 29.Putnam CD, Hammel M, Hura GL, Tainer JA. Q Rev Biophys. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- 30.Todd MJ, Hausinger RP. J. Biol. Chem. 1989;264:15835–15842. [PubMed] [Google Scholar]
- 31.Laemmli UK. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 32.Mulrooney SB, Pankratz HS, Hausinger RP. J. Gen. Microbiol. 1989;135:1769–1776. doi: 10.1099/00221287-135-6-1769. [DOI] [PubMed] [Google Scholar]
- 33.Lee MH, Pankratz HS, Wang S, Scott RA, Finnegan MG, Johnson MK, Ippolito JA, Christianson DW, Hausinger RP. Protein Sci. 1993;2:1042–1052. doi: 10.1002/pro.5560020617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacobs DJ, Rader AJ, Kuhn LA, Thorpe MF. Proteins. 2001;44:150–165. doi: 10.1002/prot.1081. [DOI] [PubMed] [Google Scholar]
- 35.Hespenheide BM, Rader AJ, Thorpe MF, Kuhn LA. J. Molec. Graphics Modeling. 2002;21:195–207. doi: 10.1016/s1093-3263(02)00146-8. [DOI] [PubMed] [Google Scholar]
- 36.Rader AJ, Hespenheide BM, Kuhn LA, Thorpe MF. Proc. Natl. Acad. Sci. 2002;99:3540–3545. doi: 10.1073/pnas.062492699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bradford MM. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 38.Weatherburn MW. Anal. Chem. 1967;39:971–974. [Google Scholar]
- 39.Woodward JD, Pickel JM, Anovitz LM, Heller WT, Rondinone AJ. Journal of Physical Chemistry B. 2006;110:19456–19460. doi: 10.1021/jp062471z. [DOI] [PubMed] [Google Scholar]
- 40.Guinier A, Fournet G. Small-Angle Scattering of X-rays. New York, NY: John Wiley & Sons; 1955. [Google Scholar]
- 41.Svergun DI. J. Appl. Crystallography. 1992;25:495–503. [Google Scholar]
- 42.Tjioe E, Heller WT. J. Appl. Crystallography. 2007;40:782–785. [Google Scholar]
- 43.Heller WT. J. Appl. Crystallography. 2006;39:671–675. [Google Scholar]
- 44.Todd MJ, Hausinger RP. J. Biol. Chem. 1987;262:5963–5967. [PubMed] [Google Scholar]
- 45.Salomone-Stagni M, Zambelli B, Musiani F, Ciurli S. Proteins. 2007;68:749–761. doi: 10.1002/prot.21472. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.